
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Pancreatic cancer presents with a dismal mortality rate and is in urgent need of methods for early detection with potential for timely intervention. All living cells, including cancer cells, generate exosomes. We previously discovered double stranded genomic DNA in exosomes derived from the circulation of pancreatic cancer patients, which enabled the detection of prevalent mutations associated with the disease. Here, we report a proof-of-concept study that demonstrates the potential clinical utility of circulating exosomal DNA for identification of KRASG12D and TP53R273H mutations in patients with pancreas-associated pathologies, including pancreatic ductal adenocarcinoma (PDAC), chronic pancreatitis (CP) and intraductal papillary mucinous neoplasm (IPMN), and in healthy human subjects. In 48 clinically annotated serum samples from PDAC patients, digital PCR analyses of exosomal DNA identified KRASG12D mutation in 39.6% of cases, and TP53R273H mutation in 4.2% of cases. KRASG12D and TP53R273H mutations were also detected in exosomal DNA from IPMN patients (2 out of 7 with KRASG12D, one of which also co-presented with TP53R273H mutation). Circulating exosomal DNA in 5 out of 9 CP patients enabled the detection of KRASG12D mutation. In 114 healthy subject-derived circulating exosomal DNA, 2.6% presented with KRASG12D mutation and none with TP53R273H mutation. This study highlights the value of circulating exosomal DNA for a rapid, low-cost identification of cancer driving mutations. The identification of mutations in IPMN patients and healthy subjects suggests that liquid biopsies may allow potential assessment of cancer risk but with a cautionary note that detection of clinical cancer cannot be assumed.
Introduction
Despite current treatment modalities, pancreatic ductal adenocarcinoma (PDAC) has a dismal prognosis with a median overall survival between 6–12 months for patients with metastatic disease, and a 7.7% 5-year survival.Citation1,2 The genetics of PDAC indicate that about 50% of the patients exhibit KRASG12D mutation and about 12% possess TP53 mutation at codon 273, with R273H being the most common point mutation.Citation3-7 Similarly, KRAS mutations, at lower prevalence, have been observed in patients with chronic pancreatitis (CP)Citation8-10 and intraductal papilliary mucinous neoplasm (IPMN).Citation11,12 In recent years, many different studies used cell-free DNA (cfDNA) isolated from the blood of pancreatic and colorectal cancer patients to identify KRAS mutations.Citation5,13-17 Circulating cfDNA originates from dying cells in injured tissue or accumulates as a consequence of physiologic cell turnover, thus possibly contributing to a decreased sensitivity and making it more challenging to identifying cancer specific mutations.Citation18,19
Exosomes are 40–150 nm extracellular vesicles that contain DNA, RNA and proteins.Citation20,21 All living cells, including cancer cells, generate exosomes and cancer cells generate higher levels of exosomes than normal cells.Citation22,23 Recently, genomic DNA was discovered in serum-derived exosomes from healthy donors and cancer patients, with utility to identify cancer specific mutations.Citation24 This original finding was subsequently validated by other research groups.Citation25-27 Circulating exosomal DNA may offer a superior advantage to circulating cfDNA as exosomes contain larger fragments of DNA, making this source advantageous for PCR based detection of mutations. Moreover, exosomes originate from live cells. The ease of exosomes isolation from the blood has led to a strong push to explore the cancer diagnostic utility of circulating exosomes. This proof-of-concept study here highlights the potential use of circulating exosomal DNA in detection of cancer specific mutations.
Results
Defining digital PCR parameters using exosomal DNA derived from cancer cells
We previously reported the detection of a heterozygous KRAS mutation at codon 12 (KRASG12D c35G>A) and a homozygous TP53 mutation at codon 273 (TP53R273H c.818G>A) using the genomic DNA from PDAC cell line, Panc-1, derived exosomes.Citation24 To determine the detection limit of KRASG12D and TP53R273H mutations in exosomes by digital PCR, we used exosomal DNA derived from Panc-1 cells as well as HMLE cells (wild-type for KRAS at codon 12 and wild-type for TP53 at codon 273). Nanoparticle tracking analysis showed that the exosomes enriched from cell culture supernatant displayed the characteristic particle size distribution that peaks between 100 and 200nm (). Exosomal DNA from HMLE cells (n = 3 repeats) indicated a 0 ± 0% rate of detection for KRASG12D mutation and 0 ± 0.05% rate of detection for TP53R273H mutation, validating their wild-type status at both loci. In contrast, exosomal DNA from Panc-1 cells (n = 3 repeats) indicated a 59.62 ± 0.89% rate of detection for KRASG12D mutation and 99.67 ± 0.13% rate of detection for TP53R273H mutation, validating their reported genotype for each mutation. Known amounts of HMLE exosomal DNA were used to determine the false positive detection rates upon digital PCR analyses for KRASG12D and TP53R273H mutations. A stringent threshold of 8800 A.U. (arbitrary units, referring to the intensity of mutant allele (), for each mutation was used, reflecting an average false positive rate of 0.040% and 0.034% for KRASG12D and TP53R273H mutation, respectively (). Importantly, the wild-type HMLE exosomal DNA concentration range in these experiments reflects the range of exosomal DNA concentration obtained from serum samples of patients (vide infra). Whether relatively low or high concentrations of exosomal DNA were used, the average false positive rate remained consistent (), supporting the reliability of the chosen limit set for defining false positives (< 8800 A.U.). To define the sensitivity of detection of KRASG12D and TP53R273H mutations, we next performed a titration experiment, in which different amounts of Panc-1 mutant exosomal DNA were mixed with different amounts of wild-type HMLE exosomal DNA. These mixtures reflected the ‘spiked-in’ mutation at an expected frequency of 0.05, 0.1, 0.25, 0.5, 1.0, 5.0 and 10% (). The titration experiment revealed that the sensitivity of detection for both mutations by digital PCR was 0.25% (), below which mutant alleles could not be detected reliably, especially at low concentrations (0.5ng in ). We reasoned that detection was limited by the low concentration of exosomal DNA, and decided that a functional abundance above 0.25% was a conservative and reliable threshold to accurately define samples as ‘positive’ or ‘negative’ for the 2 mutations. This threshold was previously reported when using droplet digital PCR to detect low-prevalence somatic mutation.Citation28 Taking into consideration the respective false positive rates for each mutation, we defined a sample as ‘positive’ for a given mutation if 1) the intensity of mutant allele was higher than 8800 A.U. () and 2) the functional abundance, with the average false positive rate deducted (as described in experimental procedures), was greater than 0.25%.
Figure 1. Definition of digital PCR parameters using cell line-derived exosomal DNA. Exosomes were pelleted from HMLE and Panc-1 cell culture supernatant with sequential filtration and ultracentrifugation, and DNA was extracted from purified exosomes. (A) Concentration and size distribution of HMLE (left) and Panc-1 (right) cell-derived exosome were analyzed using nanoparticle tracking analysis. (B) Determination of the average false positive rate using HMLE exosomal DNA. ng: nanograms. (C) Determination of the sensitivity threshold by measuring the relative functional abundance of titrated mixture of Panc-1 exosomal DNA spiked in HMLE exosomal DNA samples. Defined percentages of spiked Panc-1 exosomal DNA (with KRASG12D mutation and TP53R273H mutation) with HMLE exosomal DNA were expressed as an expected percentage of spiked mutation, and dPCR analyses were run using the listed amount of DNA input (n≥ 2 for all other points except n = 1 for 5% in KRASG12D and 10% in TP53R273H). The sensitivity threshold was set to 0.25% to reliably detect either KRASG12D (left) or TP53R273H (right) mutations.
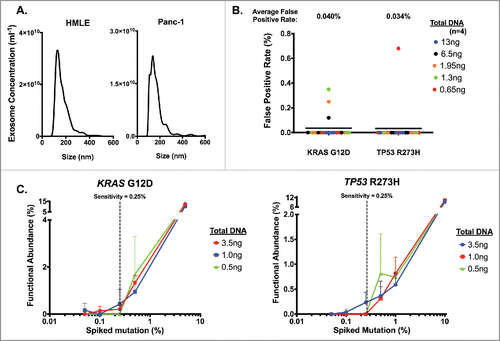
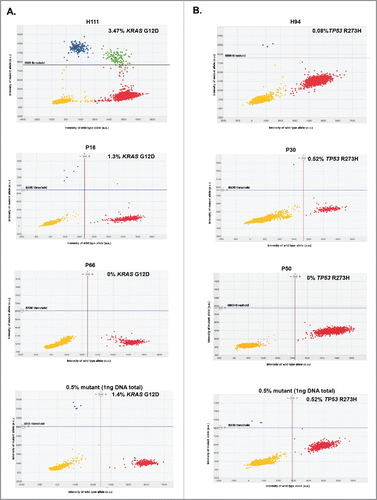
Figure 2. Representative 2D intensity scatter plot of wild-type and mutant amplicon for KRASG12D (A) KRASG12D and (B) TP53R273H, PDAC patients (P16, P50), an IPMN patient (P30) and a CP patient (P66), and control spiked-in reactions (0.5% mutant). Threshold for mutant was set to 8800. Yellow, no DNA; blue, mutant; red, wild-type; green, mutant and wild-type, ng: nanograms.
Detection of KRASG12D and TP53R273H mutations from circulating exosomal DNA of patients and healthy individuals
Exosomes from the serum of healthy subjects and patients (PDAC, IPMN, CP or others) were enriched following sequential size exclusion filtration and ultracentrifugation as described in the accompanying experimental procedures. Demographic and, when pertinent, cancer related characteristics of subjects are listed in Table S1. Our analyses included other patients with non-pancreas related disease and non-cancer pancreas pathologies distinct from chronic pancreatitis and IPMN (classified as ‘others’ and including autoimmune pancreatitis, common bile duct cancer, pancreatic cystadenoma, pancreatic neuroendocrine tumor (NET), duodenal adenoma, and uterine sarcoma, Table S2). Nanoparticle tracking analyses indicated that the exosome preparations contained particles with a size distribution characteristic of exosomes for both healthy donors and patients (). Despite the low volume of some of the serum samples (as low as 150 µl, Table S2 and S3), sufficient amount of exosomal DNA, as defined by the Qubit detection sensitivity, was obtained and with a circulating exosomal DNA concentration range of 0.102 – 1.35 ng/µl for patients (0.11 – 1.22 ng/µl for PDAC patients, 0.106–0.584 for IPMN, 0.13–1.35 for CP, 0.102–0.476 for others), and of 0.212 – 19.7 ng/µl for healthy subjects (). The minimal amount of exosomal DNA used for digital PCR analyses was thus 0.663 ng. Samples with DNA concentration greater than 8 ng/µl were diluted 1:3, equating to a 42.68 ng maxima.
Figure 3. Identification of KRASG12D and TP53R273H mutation in exosomal DNA from serum samples of cancer patients and healthy donors. (A) Representative concentration and size distribution of exosome using nanoparticle tracking analysis, from a healthy donor (left) and a PDAC patient (right). (B) Serum derived exosomal DNA concentration in healthy donors and patients. (C) Functional abundance of KRASG12D (top) and TP53R273H (bottom) mutations for the indicated groups. (D) Percent distribution of KRASG12D and TP53R273H mutations in the indicated groups with overlapping circles indicating individuals with both mutations. For PDAC: n = 48 patients, IPMN: n = 7, CP: n = 9, Others: n = 12, Healthy: n = 114.
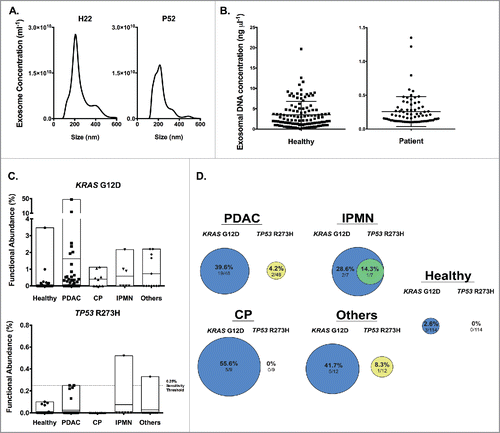
Informed by the thresholds established by our control experiments ( and experimental procedures), defined clusters of mutant and wild-type populations of positive wells were identified by digital PCR () and the data informed on the positivity for detection of KRASG12D and TP53R273H mutations. For PDAC patients, KRASG12D and TP53R273H mutations were detected in 39.6% (19/48) and 4.2% (2/48) of the samples, respectively ( and Table S2). The highest functional abundance observed was 47.45% (patient P8) and 0.25% (patients P27 and P65) for KRASG12D and TP53R273H mutations, respectively (, Table S2). We found that 3 out of 7 IPMN patients harbored the KRASG12D mutation with the highest observed functional abundance at 2.17%, and one of these patients also co-harbored the TP53R273H mutation (0.52% functional abundance) (). In CP patients, KRASG12D mutation was found in 5 out of 9 of the serum samples (highest functional abundance of 1.12%), but none with the TP53R273H mutation (). Five out of 12 (41.7%) patients diagnosed with other diseases (autoimmune pancreatitis, common bile duct cancer, pancreatic cystadenoma, pancreatic neuroendocrine tumor, duodenal adenoma, and uterine sarcoma, Table S2) harbored the KRASG12D mutation (highest functional abundance of 2.20%), and only 1 had the TP53R273H mutation (functional abundance of 0.33%). In healthy subjects, the KRASG12D mutation was observed in 2.6% (3/114) of individuals, with highest functional abundance of 3.47% ( and Table S3). TP53R273H mutation was not detected in the exosomal DNA from healthy subjects.
Discussion
The use of circulating cfDNA as a diagnostic and monitoring tool for cancer management, and also perhapsas as a screening tool for early detection of cancer is suggested by emerging studies. Such potential could likely be achieved for cancer types with high mutation frequency in specific loci.Citation19 This study indicates that even small amount of serum, as low as 150 microliters, can be used to isolate exosomes and purify DNA to identify cancer specific mutations. We identified, in 48 serum samples from PDAC patients, KRASG12D mutation in 39.6% of the samples and TP53R273H mutation in 4.2% of them, leaving 27 samples without these 2 specific mutations. These results are relevant as the prevalence of KRASG12D mutation identified using PDAC tumor tissue is approximately 40–50%.Citation7 Therefore, this exosomal DNA study likely captures most of the anticipated KRASG12D mutations in PDAC patients. This also appears to be the case for TP53R273H mutation, with an estimated 7% of patients possibly presenting with a c.818G>A substitution reflecting the R273H mutation. Previous studies have used serum or plasma-derived cfDNA from PDAC patients to define a detection rate of specific mutation in tumor tissue and corresponding (matched) serum/plasma samples, and in some cases, also evaluated a limited number of healthy donors.Citation13,29 Using droplet digital PCR analyses of cfDNA derived from 1 mL of serum, a 36.3% detection rate of KRASG12D mutation was reported in a cohort of 66 patients with pancreatic cancer, and in 5% (1 out of 20) of healthy donors.Citation16 Our results using circulating exosomal DNA appear to provide similar sensitivity for mutation detection. Future studies using larger cohorts will address whether cfDNA or exosomal DNA offer better sensitivity and/or specificity, or be of equal value. Nonetheless, our study shows that circulating exosomal DNA can be used to identify mutations using high-throughput techniques such as digital PCR. Importantly, while KRASG12D mutations were noted in 2.6% of our large cohort of healthy donors (n = 114), TP53 mutation (here, R273H associated with c.818G>A mutation) was not detected in healthy subjects. Detection of such mutations in healthy subjects.Citation16,30
Our study, using a large number of healthy subjects to detect cancer associated mutations in liquid biopsy, leads to a cautionary note that identification of driver mutations may not signify presence of disease. On the other hand, patients with CP (at higher risk of developing PDAC lesions) appear to present with a greater rate of KRAS mutation in our study; and this is also supported by other studies using cfDNA (5–20%)Citation16,29 as well as in analyses of pancreatic tissue.Citation8,9 In previous studies, KRAS mutations in IPMN patients were identified using tissue samplesCitation11,12,31-34 and pancreatic juiceCitation31,35; however, KRAS mutations (G12D and G12V) were not detected in cfDNA of IPMN patients.Citation36 With regards to TP53 mutation, immunostaining and PCR analyses of pancreas from CP patients failed to detect them,Citation10,37 supporting our results. TP53 mutations, typically associated with disease progression, were detected in tissue samplesCitation32,34 and pancreatic juiceCitation35 of IPMN patients. However, both mutations (KRASG12D and TP53R273H) in the same IPMN patient had not been reported but was identified in our study for one IPMN patient. We noted in our analyses that 4 of the 5 highest functional abundances for the KRASG12D mutation were in PDAC exosomal DNA samples, supporting previous studies in which higher concentration of KRAS mutation in pancreatic juice was able to distinguish PDAC and IPMN patients.Citation38
Using multiple stringent criteria for the false positive rate, intensity and sensitivity, we noted that there were patients with sensitivity values just above or below 0.25% (Table S2). As reported by Uchiyama et al.,Citation28 these samples may be in the “gray zone” of detection due to low exosomal DNA concentration, which may be a potential limitation for liquid biopsy; however, we expect that improvement in exosomal DNA isolation techniques will increase sensitivity of digital PCR, and thus, improve detection of DNA with low mutation rates. Our study, in combination with other studies, suggests that the use of circulating exosomal DNA analyses, together with other nucleic acid analyses might offer a more reliable screening tool for early detection of pancreatic neoplasia. Nucleic acids, including DNA and RNA species, are presumably protected from degradation in the blood stream by exosomes encapsulation; hence, exosomal nucleic acid analyses may refine the sensitivity of the assay for early detection of cancer. Indeed, while identification of highly prevalent mutations in circulating exosomal nucleic acids by itself may not offer a specific diagnosis, this approach, when combined with imaging modalities and other diagnostic procedure, may enhance personalized care of high-risk individuals. Circulating exosomal nucleic acid analyses may allow us to further develop precision medicine techniques for tailoring personalized care, with early detection and cancer prevention in mind.
Materials and methods
Healthy individual and PDAC patient serum samples
The study was performed on 171 patients who underwent major surgical pancreatic resection due to several underlying diagnosis at the Department of General, Visceral and Thoracic surgery, University Medical Center of Hamburg, Germany between 2003 and 2013. The diagnosis was confirmed upon histopathological analyses of the resected material. The study was approved by the Ethics Committee of the Chamber of Physicians in Hamburg, Germany (PVN1045). Written consent for using the samples for research purposes was obtained from all patients before surgery or blood drawing. Blood samples of patients with suspected PDAC were obtained from central venous catheter from each patient directly before surgery. After collection of the blood samples, serum was separated from the blood by centrifugation at 3400×g for 10 minutes and these serum samples were kept frozen at −80°C. Samples were chosen randomly from the pre-existing tumor bank unaware of the underlying disease at the time of exosome analysis. Histopathologically proven diagnoses (PDAC, IPMN, chronic pancreatitis and in a few cases autoimmune pancreatitis, common bile duct cancer, pancreatic cystadenoma, pancreatic neuroendocrine tumor, duodenal adenoma, and uterine sarcoma) were revealed after digital PCR analyses of serum-derived exosomal DNA. None of the patients with malignant disease received preoperative cancer related therapies.
Blood samples of healthy individuals were collected by MD Anderson Blood Bank. Serum samples were collected in Vacutainer Plus plastic serum tube (Becton Dickinson) and stored at room temperature until the end of the day. Samples were then spun at 3000xg for 20 minutes and then stored at 4°C overnight. The serum was then extracted the next day and the samples were stored at −80°C.
Cell lines
A pancreatic cancer cell line, Panc-1 (CRL-1469) was purchased from ATCC and cultured in RPMI 1640 (Corning) completed with 10% fetal bovine serum and 1% Penicillin/Streptomycin (Gibco). HMLE cell line was a gift from Dr. Sendurai Mani at MD Anderson and cultured in DMEM/Ham's F12 50/50 media (Corning) completed with 5% horse serum, 5 µg/ml insulin, 1 µg/ml hydrocortisone, 10 ng/ml epidermal growth factor (Gibco) and 100 ng/ml cholera toxin (Sigma Aldrich). All cells were cultured in 37°C and 5% CO2. The cells routinely tested negative for mycoplasma. The KRAS and TP53 genotypes of the cells were confirmed by Sanger sequencing.
Exosome isolation
For cell culture supernatant derived exosomes, the cell lines were cultured until 80% confluence, washed with phosphate buffered saline (PBS), and 20ml of serum free media was added to the cells for 48 hours. This conditioned medium was then collected for exosome isolation (vide infra). Serum samples from healthy donors or PDAC patients were thawed at 37°C before exosomes extraction. The starting volumes of plasma are listed in Table S2. Serum samples and cell line-conditioned medium were centrifuged at 800xg for 5 minutes, then 2000xg for 10 minutes. The supernatant was filtered using a 0.2µm syringe filter (Corning or Whatman). For serum samples, the filter was washed with PBS to maximize sample recovery, and samples were brought up to 11ml with PBS. Exosomes were pelleted by ultracentrifugation (Beckman Coulter Optima L-90K) at 198,000xg for 4 to 8 hours for serum samples (SW-41 Ti rotor), and at 134,000xg for 2 to 3 hours for cell lines (SW-32 Ti rotor). Exosome pellets were resuspended in 200µl PBS and stored at −80°C. The enrichment of exosomes in our preparations was verified using nanoparticle tracking analysis (Malvern NanoSight LM10-HS) and manufacturer's software (v3.1 with camera level set to 9 for serum and 13 for cell line, and detection threshold to 5).
DNA extraction
DNA was extracted from exosomes using QIAamp DNA Micro kit (Qiagen) according to the manufacturer's instructions with the following modifications: exosomes lysis was performed at 56°C for 1 hour, and DNA was eluted with 25µl of elution buffer. DNA concentration was quantified using Qubit dsDNA high sensitivity assay according to manufacturer's instructions (Thermo Fisher). The samples with detectable DNA concentration were subsequently used for KRAS and TP53 mutation analysis by digital PCR. Sufficient amount of DNA was obtained from 49% (76/156) of patient and 66% (114/171) of healthy serum samples.
Digital PCR
We aimed to detail the experimental procedures following the guidelines proposed for reporting digital PCR dataCitation39 and proceeded with analyses of our results keeping in mind reported considerations on sensitivity and specificity.Citation40 To identify KRASG12D and TP53R273H mutations in human sample exosomal DNA, digital PCR was performed using Taqman SNP Genotyping assays on the Quantstudio 3D system (Thermo Fisher) according to manufacturer's instructions. The probe assay ID used are AH6R5PI (KRAS G12D c35G>A (WT:C —> MUT:T) and AHWSLEX (TP53 R273H c.818G>A (WT: C —> MUT:T). For human samples, 6.5µl of DNA was added regardless of DNA concentration. Samples with DNA concentration greater than 8 ng/µl was diluted 1:3 in elution buffer to ensure proper cluster separation during analysis. The functional abundance was calculated and reported as a percentage using the formula below:
The analysis of the digital PCR data was performed with the manufacturer's software (QuantStudio 3D Analysis Suite). The detection limit of the assay was determined using exosomal DNA extracted from cell lines. Exosomal DNA extracted from HMLE cell lines were used as wild-type DNA, while that extracted from Panc-1 was used as mutant DNA for KRAS (heterozygous G12D) and TP53 (homozygous R273H). False positive rate and threshold (intensity) limit for mutant alleles were determined using wild-type DNA at concentrations representative of that of samples - 4 replicates at 0.1, 0.2 and 0.3 ng/µl for patients (totaling 0.65, 1.3, 1.95ng of DNA), and 1 and 2 ng/µl for healthy donors (totaling 6.5 and 13ng of DNA). The average false positive rate was calculated from all replicates (), and used to calculate the reported functional abundance. A titration of mutant exosomal DNA from 10% to 0.01% with a total of 0.5, 1 and 3ng DNA was performed (), determining the functional abundance threshold at 0.25%. We also conducted a ‘no template’ control (n = 1) and did not detect any mutant or wild-type alleles.
Disclosure of potential conflicts of interest
MD Anderson Cancer Center and R. Kalluri hold patents in the area of exosomes biology, which are licensed to Codiak Biosciences Inc. MD Anderson Cancer Center and R. Kalluri are stock equity holders in Codiak Biosciences Inc. R. Kalluri receives research support from Codiak Biosciences and serves as a member of the board of directors.
Supplementary_Data.zip
Download Zip (270.4 KB)Acknowledgments
We thank Lisa Norberg for her managerial assistance in sample logistics and annotations and for obtaining the healthy donor samples, Shruti Malasi and Yang Chen for assisting with the digital PCR analyses, and Qian Peng for assisting with sample preparation for sequencing. The Sequencing and Microarray Facility at MD Anderson Cancer Center is supported by the Core grant CA016672 (SMF).
Funding
The work on exosomes in the Kalluri laboratory is supported by Cancer Prevention and Research Institute of Texas; and the Metastasis Research Center. The LeBleu laboratory is supported by the UT MDACC Khalifa Bin Zayed Al Nahya Foundation. LBG is funded by Norwegian Cancer Society.
References
- Polireddy K, Chen Q. Cancer of the Pancreas: Molecular Pathways and Current Advancement in Treatment. J Cancer 2016; 7(11):1497-1514; PMID:27471566; http://dx.doi.org/10.7150/jca.14922
- Howlader NNA, Krapcho M, Miller D, Bishop K, Altekruse SF, Kosary CL, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, Feuer EJ, Cronin KA. (eds). SEER Cancer Statistics Review, 1975–2013, National Cancer Institute. Bethesda, MD; 2016 Apr [accessed 2016 Nov 8]. https://seer.cancer.gov/csr/1975_2013/
- Ying H, Dey P, Yao W, Kimmelman AC, Draetta GF, Maitra A, DePinho RA. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev 2016; 30:355-385; PMID:26883357; http://dx.doi.org/10.1101/gad.275776.115
- Bouaoun L, Sonkin D, Ardin M, Hollstein M, Byrnes G, Zavadil J, Olivier M. (2016) TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum Mutat 2016; 37:865-876; PMID:27328919; http://dx.doi.org/10.1002/humu.23035
- Sausen M, Phallen J, Adleff V, Jones S, Leary RJ, Barrett MT, Anagnostou V, Parpart-Li S, Murphy D, Kay Li Q, et al. Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat Comm 2015; 6:7686; PMID:26154128; http://dx.doi.org/10.1038/ncomms8686
- Chang DK, Grimmond SM, Biankin AV. Pancreatic cancer genomics. Curr Opin Genet Dev 2014; 24:74-81; PMID:24480245; http://dx.doi.org/10.1016/j.gde.2013.12.001
- Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov 2014; 13(11):828-851; PMID:25323927; http://dx.doi.org/10.1038/nrd4389
- Popovic Hadzija M, Korolija M, Jakic Razumovic J, Pavkovic P, Hadzija M, Kapitanovic S. K-ras and Dpc4 mutations in chronic pancreatitis: case series. Croat Med J 2007; 48(2):218-224; PMID:17436386
- Lohr M, Kloppel G, Maisonneuve P, Lowenfels AB, Luttges J. Frequency of K-ras mutations in pancreatic intraductal neoplasias associated with pancreatic ductal adenocarcinoma and chronic pancreatitis: a meta-analysis. Neoplasia 2005; 7(1):17-23; PMID:15720814; http://dx.doi.org/10.1593/neo.04445
- Luttges J, Diederichs A, Menke MA, Vogel I, Kremer B, Kloppel G. Ductal lesions in patients with chronic pancreatitis show K-ras mutations in a frequency similar to that in the normal pancreas and lack nuclear immunoreactivity for p53. Cancer 2000; 88(11):2495-2504; PMID:10861425; http://dx.doi.org/10.1002/1097-0142(20000601)88:11%3c2495::AID-CNCR10%3e3.0.CO;2-B
- Jang JY, Park YC, Song YS, Lee SE, Hwang DW, Lim CS, Lee HE, Kim WH, Kim SW. Increased K-ras mutation and expression of S100A4 and MUC2 protein in the malignant intraductal papillary mucinous tumor of the pancreas. J Hepatobiliary Pancreat Surg 2009; 16(5):668-674; PMID:19412570; http://dx.doi.org/10.1007/s00534-009-0105-7
- Z'Graggen K, Rivera JA, Compton CC, Pins M, Werner J, Fernandez-del Castillo C, Rattner DW, Lewandrowski KB, Rustgi AK, Warshaw AL. Prevalence of activating K-ras mutations in the evolutionary stages of neoplasia in intraductal papillary mucinous tumors of the pancreas. Ann Surg. 1997; 226(4):491-500; PMID:9351717; http://dx.doi.org/10.1097/00000658-199710000-00010
- Brychta N, Krahn T, von Ahsen O. Detection of KRAS Mutations in Circulating Tumor DNA by Digital PCR in Early Stages of Pancreatic Cancer. Clin Chem 2016; 62(11):1482-1491; PMID:27591291; http://dx.doi.org/10.1373/clinchem.2016.257469
- Olmedillas Lopez S, Garcia-Olmo DC, Garcia-Arranz M, Guadalajara H, Pastor C, Garcia- Olmo D. KRAS G12V Mutation Detection by Droplet Digital PCR in Circulating Cell-Free DNA of Colorectal Cancer Patients. Int J Mol Sci 2016; 17(4):484; PMID:27043547; http://dx.doi.org/10.3390/ijms17040484
- Yamada T, Iwai T, Takahashi G, Kan H, Koizumi M, Matsuda A, Shinji S, Yamagishi A, Yokoyama Y, Tatsuguchi A, et al. Utility of KRAS mutation detection using circulating cell-free DNA from patients with colorectal cancer. Cancer Sci 2016; 107(7):936-943; PMID:27116474; http://dx.doi.org/10.1111/cas.12959
- Kinugasa H, Nouso K, Miyahara K, Morimoto Y, Dohi C, Tsutsumi K, Kato H, Matsubara T, Okada H, Yamamoto K. Detection of K-ras gene mutation by liquid biopsy in patients with pancreatic cancer. Cancer 2015; 121(13):2271-2280; PMID:25823825; http://dx.doi.org/10.1002/cncr.29364
- Taly V, Pekin D, Benhaim L, Kotsopoulos SK, Le Corre D, Li X, Atochin I, Link DR, Griffiths AD, Pallier K, et al. Multiplex picodroplet digital PCR to detect KRAS mutations in circulating DNA from the plasma of colorectal cancer patients. Clin Chem 2013; 59(12):1722-1731; PMID:23938455; http://dx.doi.org/10.1373/clinchem.2013.206359
- Haber DA, Velculescu VE. Blood-based analyses of cancer: circulating tumor cells and circulating tumor DNA. Cancer Discov 2014; 4(6):650-661; PMID:24801577; http://dx.doi.org/10.1158/2159-8290.CD-13-1014
- Schwarzenbach H, Hoon DS, Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer 2011; 11:426-437; PMID:21562580; http://dx.doi.org/10.1038/nrc3066
- Kalluri R. The biology and function of exosomes in cancer. J Clin Invest 2016; 126(4):1208-1215; PMID:27035812; http://dx.doi.org/10.1172/JCI81135
- Kowal J, Tkach M, Thery C. Biogenesis and secretion of exosomes. Curr Opin in Cell Biol 2014; 29:116-125; PMID:24959705; http://dx.doi.org/10.1016/j.ceb.2014.05.004
- Milane L, Singh A, Mattheolabakis G, Suresh M, Amiji MM. Exosome mediated communication within the tumor microenvironment. J Control Release 2015; 219:278-294; PMID:26143224; http://dx.doi.org/10.1016/j.jconrel.2015.06.029
- Kahlert C, Kalluri R. Exosomes in tumor microenvironment influence cancer progression and metastasis. J Mol Med (Berl) 2013; 91(4):431-437; PMID:23519402; http://dx.doi.org/10.1007/s00109-013-1020-6
- Kahlert C, Melo SA, Protopopov A, Tang J, Seth S, Koch M, Zhang J, Weitz J, Chin L, Futreal A, Kalluri R. Identification of double-stranded genomic DNA spanning all chromosomes with mutated KRAS and p53 DNA in the serum exosomes of patients with pancreatic cancer. J Biol Chem 2014; 289(7):3869-3875; PMID:24398677; http://dx.doi.org/10.1074/jbc.C113.532267
- San Lucas FA, Allenson K, Bernard V, Castillo J, Kim DU, Ellis K, Ehli EA, Davies GE, Petersen JL, Li D., et al. Minimally invasive genomic and transcriptomic profiling of visceral cancers by next-generation sequencing of circulating exosomes. Ann Oncol 2016; 27(4):635-641; PMID:26681674; http://dx.doi.org/10.1093/annonc/mdv604
- Thakur BK, Zhang H, Becker A, Matei I, Huang Y, Costa-Silva B, Zheng Y, Hoshino A, Brazier H, Xiang J, et al. Double-stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res 2014; 24(6):766-769; PMID:24710597; http://dx.doi.org/10.1038/cr.2014.44
- Lee TH, Chennakrishnaiah S, Audemard E, Montermini L, Meehan B, Rak J. Oncogenic ras-driven cancer cell vesiculation leads to emission of double-stranded DNA capable of interacting with target cells. Biochem Biophys Res Commun 2014; 451(2):295-301; PMID:25086355; http://dx.doi.org/10.1016/j.bbrc.2014.07.109
- Uchiyama Y, Nakashima M, Watanabe S, Miyajima M, Taguri M, Miyatake S, Miyake N, Saitsu H, Mishima H, Kinoshita A, et al. Ultrasensitive droplet digital PCR for detecting a low-prevalence somatic GNAQ mutation in Sturge-Weber syndrome. Sci Rep 2016; 6:22985; PMID:26957145; http://dx.doi.org/10.1038/srep22985
- Castells A, Puig P, Mora J, Boadas J, Boix L, Urgell E, Sole M, Capella G, Lluis F, Fernandez-Cruz L, et al. K-ras mutations in DNA extracted from the plasma of patients with pancreatic carcinoma: diagnostic utility and prognostic significance. J Clin Oncol. 1999; 17(2):578-584; PMID:10080602; http://dx.doi.org/10.1200/jco/1999.17.2.578
- Lüttges J, Reinecke-Lüthge A, Möllmann B, Menke MAOH, Clemens A, Klimpfinger M, Sipos B, Klöppel G. Duct changes and K-ras mutations in disease-free pancreas: analysis of type, age relation and spatial distribution. Virchows Arch 1999; 435:461-468; PMID:10592048; http://dx.doi.org/10.1007/s004280050428
- Kondo H, Sugano K, Fukayama N, Hosokawa K, Ohkura H, Ohtsu A, Mukai K, Yoshida S. Detection of K-ras gene mutations at codon 12 in the pancreatic juice of patients with intraductal papillary mucinous tumors of the pancreas. Cancer 1997; 79(5):900-905; PMID:18813127; http://dx.doi.org/10.1002/(SICI)1097-0142(19970301)79:5%3c900::AID-CNCR5%3e3.0.CO;2-F
- Chadwick B, Willmore-Payne C, Tripp S, Layfield LJ, Hirschowitz S, Holden J. Histologic, immunohistochemical, and molecular classification of 52 IPMNs of the pancreas. Appl Immunohistochem Mol Morphol 2009; 17(1):31-39; PMID:18813127; http://dx.doi.org/10.1097/PAI.0b013e31817c02c6
- Wada K. p16 and p53 gene alterations and accumulations in the malignant evolution of intraductal papillary-mucinous tumors of the pancreas. J hepatobiliary Pancreat Surg 2002; 9(1):76-85; PMID:12021900; http://dx.doi.org/10.1007/s005340200007
- Lubezky N, Ben-Haim M, Marmor S, Brazowsky E, Rechavi G, Klausner JM, Cohen Y. High-throughput mutation profiling in intraductal papillary mucinous neoplasm (IPMN). J Gastrointest Surg 2011; 15(3):503-511; PMID:21225475; http://dx.doi.org/10.1007/s11605-010-1411-8
- Kaino M, Kondoh S, Okita S, Hatano S, Shiraishi K, Kaino S, Okita K. Detection of K-ras and p53 gene mutations in pancreatic juice for the diagnosis of intraductal papillary mucinous tumors. Pancreas 1999; 18(3):294-299; PMID:10206488; http://dx.doi.org/10.1097/00006676-199904000-00011
- Berger AW, Schwerdel D, Costa IG, Hackert T, Strobel O, Lam S, Barth TF, Schroppel B, Meining A, Buchler MW, et al. Detection of Hot-Spot Mutations in Circulating Cell-Free DNA From Patients With Intraductal Papillary Mucinous Neoplasms of the Pancreas. Gastroenterology 2016; 151(2):267-270; PMID:27343369; http://dx.doi.org/10.1053/j.gastro.2016.04.034
- Casey G, Yamanaka Y, Friess H, Kobrin MS, Lopez ME, Buchler M, Beger HG, Korc M. p53 mutations are common in pancreatic cancer and are absent in chronic pancreatitis. Cancer Lett. 1993; 69(3):151-160; PMID:8513440; http://dx.doi.org/10.1016/0304-3835(93)90168-9
- Yu J, Sadakari Y, Shindo K, Suenaga M, Brant A, Almario JA, Borges M, Barkley T, Fesharakizadeh S, Ford M, et al. Digital next-generation sequencing identifies low-abundance mutations in pancreatic juice samples collected from the duodenum of patients with pancreatic cancer and intraductal papillary mucinous neoplasms. Gut 2016:0:1-11; PMID:27432539; http://dx.doi.org/10.1136/gutjnl-2015-311166
- Huggett JF, Foy CA, Benes V, Emslie K, Garson JA, Haynes R, Hellemans J, Kubista M, Mueller RD, Nolan T, et al. The digital MIQE guidelines: Minimum Information for Publication of Quantitative Digital PCR Experiments. Clin Chem 2013; 59(6):892-902; PMID:23570709; http://dx.doi.org/10.1373/clinchem.2013.206375
- Zonta E, Garlan F, Pecuchet N, Perez-Toralla K, Caen O, Milbury C, Didelot A, Fabre E, Blons H, Laurent-Puig P, et al. Multiplex Detection of Rare Mutations by Picoliter Droplet Based Digital PCR: Sensitivity and Specificity Considerations. PloS one 2016; 11:e015909; PMID:27416070; http://dx.doi.org/10.1371/journal.pone.0159094