
ABSTRACT
Agents that target angiogenesis have shown limited efficacy for human triple-negative breast cancer (TNBC) in clinical trials. Along with endothelium-dependent vessels, there is also vasculogenic mimicry (VM) in the microcirculation of malignant tumors. The role of VM is not completely understood regarding anti-angiogenic treatment. In this study, human TNBC MDA-MB-231 and Hs578T and non-TNBC MCF-7 and BT474 tumor-bearing mice were treated with sunitinib, an anti-angiogenic drug, using a clinically relevant schedule. The drug was administered for one week and then discontinued. Tumor growth and invasion were observed, and the microcirculation patterns were detected with PAS/endomucin staining. Moreover, hypoxia and VM-associated proteins were evaluated with Hypoxyprobe kits and immunohistochemistry, respectively. Sunitinib significantly inhibited tumor growth in the TNBC and non-TNBC tumors. However, MDA-MB-231 and Hs578T tumors regrew and were more aggressive when the treatment was stopped. The discontinuation had no significant effect on the behavior of the non-TNBC MCF-7 and BT474 tumors. The growth of endothelium-dependent vessels in the TNBC MDA-MB-231 and Hs578T tumors were blocked by sunitinib, during which the number of VM channels significantly increased and resulted in a rebound of endothelium-dependent vessels after sunitinib discontinuation. Moreover, the VM-associated proteins VE-cadherin and Twist1 upregulated in the sunitinib-treated MDA-MB-231 and Hs578T tumors. Furthermore, the clinical significance of this upregulation was validated in 174 human breast cancers. The results from human breast cancer specimens indicated that there were more VM-positive TNBC cases than those in non-TNBC cases. HIF-1α, MMP2, VE-cadherin, and Twist1 were also expressed in a higher level in human TNBC compared with non-TNBC. In aconclusion, sunitinib promoted TNBC invasion by VM. The VM status could be helpful to predict the efficacy of anti-angiogenic therapy in patients with TNBC.
Introduction
Breast cancer is one of the most malignant threats among women worldwide.Citation1 Based on the status of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2), there are 2 subtypes of breast cancer: triple-negative breast cancer (TNBC), which does not express ER, PR, or HER2, and non-TNBC, which expresses at least one of these receptors.Citation2-3 TNBC and non-TNBC differ in behavior, prognosis and treatment. Compared with non-TNBC, the survival of TNBC patients is poor.Citation4 TNBC tumors are more likely to be aggressive and have a higher tendency to metastasize to visceral organs.Citation5 Patients having TNBC do not benefit from either endocrine therapy or anti-HER2 therapy.Citation6 Furthermore, it is not sensitive to chemotherapy. Although clinical trials have been performed to find an efficient drug or therapeutic strategy for TNBC patients, no effective agent or treatment strategy has been developed to date.Citation7
Anti-angiogenic agents such as the vascular endothelial growth factor (VEGF)-neutralizing antibody Avastin (bevacizumab) and VEGF receptor tyrosine kinase inhibitors (sorafenib and sunitinib) have been used to treat TNBC in clinical trials.Citation8-10 Although anti-angiogenic agents have shown some effectiveness in the treatmentof non-small cell lung cancer, renal cell cancer, and hepatocellular carcinoma,Citation11-12 they have shown limited efficacy against TNBC.Citation8-9, 13 This is thought to be in part due to the angiogenesis rebound and tumor regrowth sometimes observed either during drug-free periods or upon treatment discontinuation. Furthermore, Paez-Ribes Met al.have reported that VEGF inhibitors increase tumor aggressiveness and metastasis in a mouse model.Citation14 The mechanism of anti-angiogenic treatment failure remains unclear and remains an important area of investigation for the advancement of TNBC therapy.
Vasculogenic mimicry (VM) involves tumor vascularization in malignant tumors.Citation15 VM channels are formed by tumor cells instead of endothelial cells.Citation16 VM channels connect with endothelium-dependent vessels to provide blood for tumor.Citation17-18 VM has been observed in many aggressive tumors such as melanoma, gastrointestinal stromal tumors and carcinomas including inflammatory breast, prostate, ovarian, and hepatocellulartumors.Citation17,19-23 Tumors that exhibit VM are more aggressive.Citation15,24 Moreover, targeting VEGF, VEGF receptors or endothelial cells shows limited effectiveness on VM formed by tumor cells.Citation25 Previously, we found there is increased VM in TNBC compared within non-TNBC. Based on previous studies, we hypothesize that the VM channels in TNBC are responsible for the tumor rebound and treatment failure of anti-angiogenic agents. In this study, we provide data from human samples and experimental evidence in a mouse model to support this hypothesis.
Results
Discontinuation of sunitinib promoted MDA-MB-231 and Hs578T regrowth and invasion
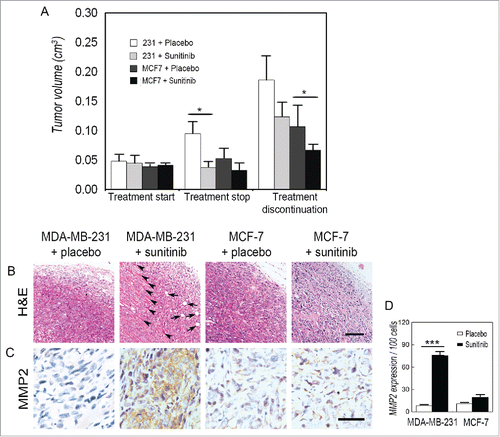
To study the effect of anti-angiogenesis agents on TNBC and non-TNBC, MDA-MB-231- and MCF-7-bearing nude mice were orally administered sunitinib on a clinically relevant schedule. The mice were treated for one week, after which the treatment was stopped. When mice were treated, the tumors grew at a significantly slower rate in the MDA-MB-231- and MCF-7-bearing mice than those in the control groups (). The sunitinib-treated MDA-MB-231 tumors regrew after treatment discontinuation, while the discontinuation had no significant effect on the volume of the MCF-7 tumors (). H&E staining indicated that MDA-MB-231 tumor cells invaded into adipose and skeletal muscle tissue after treatment discontinuation (). Moreover, MDA-MB-231 tumors expressed more invasion-relevant protein MMP2 during the post-treatment period than during the sunitinib treatment ( and ). There was also no increase in aggressive behavior in MCF-7 tumors after sunitinib discontinuation ( to ).
Figure 1. Discontinuation of sunitinib promotes TNBC tumor regrowth and invasion. (A) Effects of sunitinib on the tumor volume of TNBC and non-TNBC cells. There wa sno significant difference in tumor volume when the treatment started. After mice were treated with sunitinib for one week, both the TNBC MDA-MB-231 tumors and the non-TNBC MCF-7 tumors in the treatment groups were smaller than those in the control groups. The sunitinib-treated TNBC MDA-MB-231 tumors regrew after treatment discontinuation, while the discontinuation had no significant effect on the tumor volume of the non-TNBC MCF-7 tumors. (B) H&E staining indicated that the TNBC MDA-MB-231 tumor cells invaded into adipose tissue (arrows) and skeletal muscle tissue (arrow heads) after treatment discontinuation. There was no aggressive behavior in the non-TNBC MCF-7 tumors. (C) IHC for MMP2 staining shows that TNBC MDA-MB-231 tumors after treatment was stopped expressed a higher level of MMP2 than tumors during the sunitinib treatment. There was nodifferenceinMMP2 expression in the non-TNBC MCF-7 tumors between the sunitinib treatment and after sunitinib was discontinued. (D) Quantification of MMP2 expression in the different groups. The scale bar represents 100 μm, and the error bar indicates the standard deviation (SD). *P < 0.05, ** P < 0.01, *** P < 0.001.
To verify the effect of sunitinib on TNBC tumor-bearing mice, human TNBC Hs578T and non-TNBC BT474-bearing mice were administered sunitinib on the same schedule. The growth of both tumor types was inhibited by sunitinib (Fig. S1). Similar to the MDA-MB-231 tumors, sunitinib-treated Hs578T tumors regrew upon treatment discontinuation; however, discontinuation did not influence the BT474 tumors (Fig. S1A). Sunitinib treatment discontinuation also promoted the aggressiveness of Hs578T tumors (Fig. S1B).
Effects of sunitinib treatment on microcirculation patterns in TNBC tumors
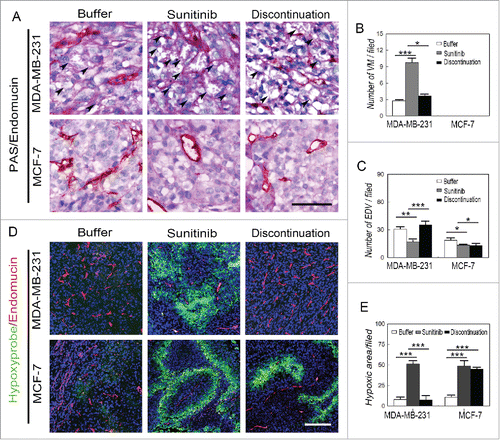
To examine the possible mechanism of the regrowth and aggressiveness of MDA-MB-231 and Hs578T tumors after discontinuation of sunitinib treatment, double staining for endomucin and PAS as well as immunofluorescent staining (IF) for endomucin and hypoxia were performed to investigate the microcirculation patterns and hypoxic regions in the MDA-MB-231, Hs578T, MCF-7, and BT474 tumors. VM channels formed by PAS-positive deposits and tumor cells were observed in the human MDA-MB-231 and Hs578T tumors, whereas no VM channels were observed in the human MCF-7 tumors (). The development of endothelium-dependent vessels in the MDA-MB-231 and Hs578T tumors were blocked by sunitinib, during which the number of VM channels was significantly increased ( and Fig. S2A to S2C). The number of endothelium-dependent vessels rebounded in the MDA-MB-231 and Hs578T tumors after treatment discontinuation, but there was no significant difference in the number of endothelium-dependent vessels during sunitinib treatment and after discontinuation in the MCF-7 and BT474 tumors ( to ). IF double staining for hypoxia and endomucin showed that the sunitinib-treated MDA-MB-231 and Hs578T tumors had more hypoxic regions compared with the other groups when the endothelium-dependent vessels were inhibited ( to and Fig. S2C to S2D). The hypoxic area in the MDA-MB-231 and Hs578T tumors was decreased after endothelium-dependent vessels rebounded upon treatment discontinuation ( to and Fig. S2D to S2E). The hypoxic area in the MCF-7 and BT474 tumors was similar during sunitinib treatment after discontinuation ( and and Fig. S2D and S2E).
Figure 2. Effects of sunitinib treatment on the microcirculation patterns in TNBC tumors. (A) PAS and endomucin double staining in TNBCMDA-MB-231 and non-TNBC MCF-7 tumors. There were VM channels formed by PAS-positive molecules and tumor cells in the human TNBCMDA-MB-231 tumors, whereas no VM channels were observed in the human non-TNBC MCF-7 tumors. Growth of endothelium-dependent vessels in the TNBC MDA-MB-231 tumors was blocked by sunitinib, during which the number of VM channels significantly increased. Endothelium-dependent vessel growth rebounded in MDA-MB-231 tumors after treatment discontinuation, but there was no significant difference in the number of endothelium-dependent vessels between sunitinib-treated and post-treatment non-TNBC MCF-7 tumors. (B)Quantification of VM channels in the different groups. (C)Quantification of endothelium-dependent vessels in the different groups. (D)Hypoxia and endomucin double staining inTNBCMDA-MB-231 and non-TNBC MCF-7 tumors. There were more hypoxic regions in the sunitinib-treatedMDA-MB-231 tumors compared with those in the other groups when the endothelium-dependent vessels are inhibited. After treatment discontinuation, the hypoxic area in the MDA-MB-231 tumors decreased after the endothelium-dependent vessels rebounded. The hypoxic area in the MCF-7 tumors was similar in the sunitinib-treated and discontinued groups. (E)Quantification of the hypoxic area in the different groups. The scale bar indicates 100 μm, and the error bar indicates the standard deviation (SD). * P < 0.05, **P < 0.01, ***P < 0.001.
Effects of sunitinib treatment on EMT-associated transcription factors in breast cancer
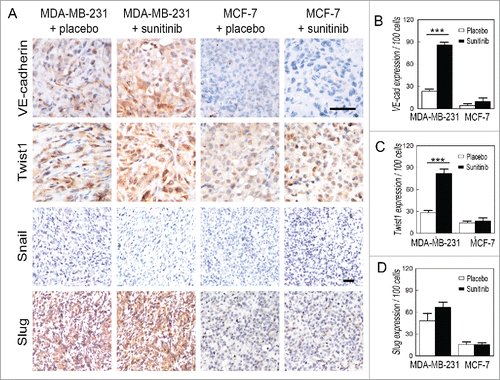
To investigate the possible molecular mechanism of VM in TNBC after discontinuation of sunitinib treatment, VM-associated proteins and EMT-associated transcription factors were detected via immunohistochemistry. The expression of VE-cadherin, a VM-associated molecule, was upregulated in the sunitinib-treated MDA-MB-231 tumors compared with the placebo-treated MDA-MB-231 tumors ( and ). Moreover, sunitinib increased Twist1 expression in the MDA-MB-231 tumors, and Twist1 was localized into the nuclei of tumor cells in the treatment group ( and ). There was no change observed in the MCF-7 groups. The MDA-MB-231 and MCF-7 tumors did not express Snail (). The MDA-MB-231 tumors expressed higher levels of Slug than the MCF-7 tumors. However, sunitinib did not influence the expression of Slug in the MDA-MB-231 tumors ( and ). The results of the Hs578T and BT474 tumors were consistent with those of the MDA-MB-231 and MCF-7 (Fig. S3).
Figure 3. Effects of sunitinib treatment on the expression of EMT-associated transcription factors in TNBC tumors. (A) IHC staining for VE-cadherin, Twist1, Snail and Slug. VE-cadherin is upregulated in the sunitinib-treated MDA-MB-231 tumors compared with the placebo-treated MDA-MB-231 tumors. Sunitinib increased Twist1 expression in the MDA-MB-231 tumors, and Twist1 localized to the nucleus in the treatment group. There was no change observed in the MCF-7 groups. Sunitinib did not affect the expression of Snail and Slug in the TNBC MDA-MB-231 tumors and the non-TNBC MCF-7 tumors. (B) Quantification of VE-cadherin expression in the different groups. (C)Quantification of Twist1 expression in the different groups. (D) Quantification of Slug expression in the different groups. The scale bar indicates 100 μm, and the error bar indicates the standard deviation (SD). * P < 0.05, ** P < 0.01, *** P < 0.001.
Twist1 expression and VM in human TNBC and non-TNBC
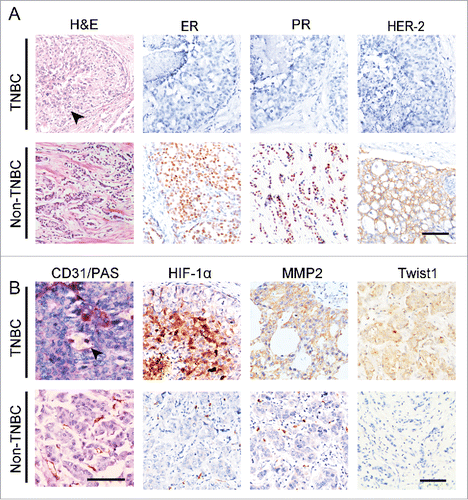
To validate the clinical relevance of sunitinib-induced Twist1 expression and VM, 67 TNBC tumors were collected, and 107 non-TNBC specimens were used as controls. The morphological characteristics and genotypes of the human TNBC and non-TNBC samples are shown in . TNBC tumor nests comprised poorly differentiated small tumor cells, and there was necrosis in the center of the tumor nest. Human TNBC tumors did not express ER, PR, or HER2. The PAS and CD31 double staining confirmed that the VM channels were formed by a PAS-positive matrix and tumor cells (). There were more VM-positive tumors in the TNBC group than in the non-TNBC group ( and ). IHC staining indicated that human TNBC tumors express higher levels of HIF-1α, MMP2, VE-cadherin, and Twist1 than human non-TNBC tumors (). The differences in MMP2, VE-cadherin, and Twist1 expression between the 2 groups were statistically significant (). The correlation analysis revealed that the invasion-associated protein MMP2 was positively correlated with HIF-1α and Twist1 expression (). The presence of VM showed a significantly positive correlation with high levels of HIF-1α, MMP2, VE-cadherin, and Twist1 expression in TNBC ().
Figure 4. The expression of Twist1 in human TNBC and non-TNBC. (A) Morphological characteristics and genotypes of human TNBC and non-TNBC. H&E staining indicates that TNBC tumor nests comprise poorly differentiated small tumor cells, and there is necrosis in the center of a tumor nest (black arrow). Human TNBC tumors do not express ER, PR, orHER2. (B) PAS and CD31double staining showed that TNBC has more VM channels compared with non-TNBC. The arrow indicates a VM channel that is formed by PAS-positive matrix and tumor cells in TNBC. IHC staining indicates that TNBC tumors express higher levels of HIF-1α, MMP2 and Twist1 than non-TNBC tumors.
Discussion
Anti-angiogenic therapy is based on the theory that blocking new blood vessel formation in tumors will either slow or stop tumor growth. Currently, several molecular-targeted drugs that act by disrupting the VEGF signaling pathway have been approved by the FDA.Citation26 Though these anti-angiogenic drugs can affect disease stabilization and improve progression free survival or overall survival, a sunitinib study in patients with renal cell carcinoma reported tumor and blood vessel rebound following treatment discontinuation.Citation27 Moreover, blocking the VEGF pathway leads to increased invasive behavior or dissemination as well as metastasis in some clinical and preclinical models.Citation28-29 In this study, sunitinib was administered to murine models with TNBC (MDA-MB-231 and Hs578T cell lines) and non-TNBC (MCF-7 and BT474 cell lines) tumor burden. The mice were treated for one week and observed for one week following sunitinib discontinuation. During the drug-free period, the primary tumors from the TNBC cell lines MDA-MB-231 and Hs578T regrew with decreased survival, while no significant change was observed in MCF-7- and BT474-bearing mice.
The potential molecular mechanisms of resistance to anti-angiogenic treatment have been investigated; these reports indicated that the disease progression changes in response to therapy, including the tumor and host responses.Citation30 Disruption of the VEGF pathway could increase the expression of prometastatic proteins, upregulate the secretion of exosomalproteolytic enzymes, create ‘premetastatic niches' and acute hypoxic stress, and activate alternative angiogenic pathways.Citation30-31 These effects may influence eventual tumor progression and invasion.Citation32 In addition, different microcirculation patterns may lead to resistance to anti-VEGF therapy in malignant tumors. VM channels, mosaic blood vessels, and endothelium-dependent vessels can be seen in a variety of malignancies.Citation17 VM channels are formed by tumor cells rather than endothelial cells, and importantly, VM channel formation is VEGF-independent, which make anti-VEGF signaling treatments ineffective.Citation33 Similar to tumor angiogenesis, VM channels can provide blood circulation for tumor growth. Furthermore, hypoxia in tumors caused by anti-angiogenic therapy can induce VM. Accordingly, in this study, an increased number of VM channels was observed in sunitinib-treated mice with MDA-MB-231 and Hs578T tumors in association with areas of increased hypoxia. VM channels can support tumor growth and transform to endothelium-dependent vessels, thereby resulting in an angiogenic rebound after sunitinib discontinuation. As the non-TNBC breast cancer cell lines MCF-7 and BT474 have no ability to form VM channels, halted sunitinib treatment did not lead to endothelium-dependent vessel regrowth. Therefore, our findings provide evidence that VM mediates TNBC resistance in the setting of anti-VEGF signaling therapy.
Inhibition of endothelium-dependent vessel growth by anti-angiogenic agents results in acute hypoxic stress, a known promoter of tumor invasion and metastasis.Citation34 Low levels of oxygen block the degradation of hypoxia-inducing factors (HIF), which allows HIF-1α or HIF-2α to translocate into the nucleus and bind to the initiator or enhancer hypoxia-response elements of their respective target genes.Citation15 HIF-1α, a transcriptional activator, upregulates the expression of tumor progression-mediated genes such as matrix metalloproteinases (MMPs), epithelial-mesenchymal transition (EMT) inducers and stemness-associated genes.Citation35 In TNBC, nuclear HIF-1α expression is associated with MMP-2 and Twist1, both of which are implicated in TNBC invasion. Moreover, sunitinib treatment upregulates MMP-2 and Twist1 expression in MDA-MB-231 and Hs578T cells but not in MCF-7 or BT474 cells. MMP-2 can degrade the extracellular matrix (ECM) to induce tumor infiltration. Twist1 is an EMT transcriptional activator, and its downstream genes can accelerate tumor cell progression toward EMT and result in increased aggressiveness and metastasis.Citation36
Hypoxia in the tumor microenvironment is the most important trigger of VM.Citation15 HIF-1α-induced MMP-2 expression and EMT play significant roles in VM.Citation37 MMP-2 digests laminin-5γ2-chain into promigratory γ2′ and γ2x fragments, which are PAS-positive components of the VM lumen38. Hypoxia-associated Twist1 upregulation can also induce VE-cadherin expression in TNBC and hepatocellular carcinoma cells.Citation21,25,38 VE-cadherin can activate MMPs via the EphA2 signaling pathway.Citation39 Furthermore, hypoxia-induced Twist1 expression also induces MDA-MB-231 cells to generate more CSCs, which then promote VM channel formationin Matrigel.Citation25 Hence, hypoxia induced by blocking the VEGF pathway not only directly promotes TNBC tumor invasion but also accelerates VM. Snail and Slug belong to the superfamily of EMT transcriptional activators and have been proved to be involved in VM on hepatocellular carcinoma and epithelial ovarian carcinoma.Citation40-42 However, these factors had no significant relation with sunitinib-induced VM in TNBC MDA-MB-231and Hs578T tumors.
Our previous study showed there are 3 stages of tumor microcirculation in malignant tumors.Citation17 During the rapid growth in the early stage, endothelium-dependent vessels cannot provide an adequate blood supply for tumor progression. Therefore, VM channels serve as a primary provider of microcirculation for some tumors. With tumor growth, endothelial cells bud into VM channels, and endothelium-dependent vessels can transition to become the major conduit for tumor microcirculation. This study indicated that this process can be modulated by changing the tumor microenvironment. Hypoxia induced by blocking endothelium-dependent vessels can promote VM, which in turn replaces the endothelium-dependent vessels and supports tumor growth, invasion and metastasis.
To investigate the clinical significance of these findings in an animal model, we used a set of human breast cancer samples to detect the difference of VM and the expression of invasion-related proteins between human TNBC and non-TNBC tumors. The results indicated that there were more VM-positive cases in the TNBC group than in the non-TNBC group. HIF-1α, MMP2, VE-cadherin, and Twist1, which can induce VM, were also upregulated in human TNBC tumors compared with non-TNBC tumors. The presence of VM is a marker for poor prognosis in patients with malignant tumors and also presents a challenge to anti-angiogenic treatments that target endothelial cells. As there is increased VM in TNBC compared with non-TNBC, the VM status should be considered when anti-angiogenic therapy is proposed. In addition, studies are warranted to investigate therapies targeting both VM as well as endothelium-dependent vessels for TNBC and other VM-associated malignancies.
Materials and methods
Cells and drugs
The human breast cancer cell linesMDA-MB-231, MCF-7, Hs578T, and BT474 were cultured in RPMI-1640 medium supplemented with 10% FBS, 4 mM L-glutamine, and 1% penicillin-streptomycin. Matrigel (BD Bioscience, USA) was diluted with RPMI-1640 medium for cell transplantation. The VEGF receptor tyrosine kinase inhibitor sunitinib malate (S-8803) was purchased from LC Laboratories (Boston, USA). The Hypoxyprobe-1 Kit (HP1–1000Kit) was purchased from HPI Hypoxyprobe, Inc. (Burlington, USA). Information regarding the primary antibodies used in this study are listed in . All secondary antibodies were provided by Zhongshan Golden Bridge Biotechnology Co., Ltd. (Beijing, China).
Table 1. Information of primary antibodies used in this study.
Table 2. Comparison of VM and VM-associated proteins between triple-negative and non-triple-negative breast cancer cases.
Table 3. The correlation of VM and VM-associated proteins between triple-negative and non-triple-negative breast cancer cases.
Tumor-bearing Nude mice models and sunitinib treatment
The animal experiments were approved by the Tianjin Medical University Ethics Committee. All steps were carefully followed to protect the welfare of the animals and prevent any suffering. Forty female Balb/c nude mice (6 weeks old) were purchased from the Beijing HFK Bioscience Company. Approximately 1–2 × 106 of MDA-MB-231, MCF-7, Hs578T, and BT474 breast cancer cells were subcutaneously injected into the hindlimb mammary glands of mice (N = 13–20/group, respectively). Tumors were measured every day, and the tumor volume was calculated using a standard formula (length × widthCitation2 × 0.52). Sunitinib malate was used in a clinic-related schedule, one week on and one week off (Fig. S4). Tumor-bearing mice were administeredwith sunitinib daily when the tumor size reached up to 0.05 cm3. Mice in the treatment group were gavaged with sunitinib for 7 days at a dose of 60 mg/kg. Distilled water was used as the placebo. All mice were killed one week after treatment discontinuation. Pimonidazole HCl was i.p. injected (60 mg/kg) 60 min before the mice were killed. The tumors and organs were collected, weighted, and fixed with 4% paraformaldehyde (PFA). All organs and tumors were embedded in paraffin and sliced into 4-μm-thick sections.
Immunohistochemistry and immunofluorescence of formalin-fixed, paraffin-embedded tissue
Formalin-fixed, paraffin-embedded tissue was sectioned, dewaxed, and rehydrated using graded alcohol dilutions. Endogenous peroxidase was blocked using 5% goat serum at room temperature for 10 min. Antigens in the sections were retrieved bymicrowaving the samples in citrate buffer for 20 min. The slides were incubated with primary antibodies overnight at 4°C, washed with PBS, and individually incubated with either biotin-labeled or FITC-labeled secondary antibodies. The color was developed using DAB. The sections were counterstained with hematoxylin or DAPI and observed using an epifluorescence microscope (80i, Nikon) or a confocal laser scanningmicroscope (A1, Nikon).
Immunohistochemical staining for endomucinand histochemical double staining for CD31 and Periodic Acid Schiff (PAS)
After immunohistochemical staining for endomucin, the sections were exposed to 1% sodium periodate for 10 min and incubated for 15 min at 37°C with PAS after washing with distilled water for 5 min. Afterward, the sections were counterstained with hematoxylin and observed under a microscope (80i, Nikon).
Patient samples
We collected samples from 174 breast cancer patients who underwent surgeryat Tianjin General Hospital from 1997 to 2004. All clinical investigations were conducted according to the principles of the Declaration of Helsinki. All studies involving human samples were approved by the Tianjin General Hospital Ethics Committee. The participating patients were clearly oriented about the aims, methods, and other details of the study. All patients had detailed pathological and clinical information. The median age of the cohort was 51.0 y (31 y to 74 years). All patients had invasive breast cancer, and 76 cases of axillary node metastasis were reported. The diameter of the primary tumor in 31 of these cases was less than 2 cm, whereas the diameter in 24 cases was over 5 cm. The follow-up period ranged from the time of the surgery to December 2008.
Tissue microarrays and counting methods
Formalin-fixed, paraffin-embedded tissues from the patients were analyzed after H&E staining. Specific tissue samples were chosen for tissue microarrays with 1 mm cores (with 1.5 mm between each core). IHC was performed on the tissue microarray sections by following a standard protocol.Citation43 Protein expression was quantified according to the method of Sun et al..Citation43 In terms of staining intensity, “0” indicated no staining, “1” indicated weakly positive staining, “2” indicated moderate staining, and “3” indicated strong staining. The number of positive cells per 100 tumor cells per field was visually evaluated and scored as follows: “0” for < 10% positive cells, “1” for < 25%, “2” for < 50%, and “3” for > 50%. The staining index, or the sum of the staining intensity and the positive cell score, was used to determine the final result for each sample. A sample was defined as positive when the staining index was greater than 1. The VM and endothelium-dependent vessels were counted at 400× magnification, and the number of each vessel type per sample was defined as the average of 10 randomly selected fields.
Statistical methods
SPSS version 17.0 (Chicago, Illinois, USA) was used to evaluate the data in this study. The x2 test was performed to assess the pathological and clinical characteristics of the TNBC and non-TNBC groups. The rank-sum test was used to compare tumor size among all the groups. The 2-tailed Student's t-test was performed to compare the protein expression between 2 groups. The significance level was set to P < 0.05.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplemental_Data.zip
Download Zip (9.1 MB)Funding
This study was supported by the key project of the National Nature Science Foundation of China (No. 81230050), the project of the National Nature Science Foundation of China (Nos. 81172046, 81572872, and 81101724), and the Student's Platform for Innovation and Entrepreneurship Training Program (No. 201510062001).
References
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin 2009; 59:225-49; PMID:19474385; http://dx.doi.org/10.3322/caac.20006
- Kalimutho M, Parsons K, Mittal D, Lopez JA, Srihari S, Khanna KK. Targeted therapies for triple-negative breast cancer: Combating a stubborn disease. Trends Pharmacol Sci 2015; 36:822-46; PMID:26538316; http://dx.doi.org/10.1016/j.tips.2015.08.009
- Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al. Molecular portraits of human breast tumours. Nature 2000; 406:747-52; PMID:10963602; http://dx.doi.org/10.1038/35021093
- Chacon RD, Costanzo MV. Triple-negative breast cancer. Breast Cancer Res 2010; 12(2):S3; PMID:21050424; http://dx.doi.org/10.1186/bcr2574
- Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P, Narod SA. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res 2007; 13:4429-34; PMID:17671126; http://dx.doi.org/10.1158/1078-0432.CCR-06-3045
- Stoeck A, Lejnine S, Truong A, Pan L, Wang H, Zang C, Yuan J, Ware C, MacLean J, Garrett-Engele PW, et al. Discovery of biomarkers predictive of GSI response in triple-negative breast cancer and adenoid cystic carcinoma. Cancer Discov 2014; 4:1154-67; PMID:25104330; http://dx.doi.org/10.1158/2159-8290.CD-13-0830
- Ibrahim YH, Garcia-Garcia C, Serra V, He L, Torres-Lockhart K, Prat A, Anton P, Cozar P, Guzmán M, Grueso J, et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov 2012; 2:1036-47; PMID:22915752; http://dx.doi.org/10.1158/2159-8290.CD-11-0348
- Dieras V, Campone M, Yardley DA, Romieu G, Valero V, Isakoff SJ, Koeppen H, Wilson TR, Xiao Y, Shames DS, et al. Randomized, phase II, placebo-controlled trial of onartuzumab and/or bevacizumab in combination with weekly paclitaxel in patients with metastatic triple-negative breast cancer. Ann Oncol 2015; 26:1904-10; PMID:26202594; http://dx.doi.org/10.1093/annonc/mdv263
- Curigliano G, Pivot X, Cortes J, Elias A, Cesari R, Khosravan R, Collier M, Huang X, Cataruozolo PE, Kern KA, et al. Randomized phase II study of sunitinib versus standard of care for patients with previously treated advanced triple-negative breast cancer. Breast 2013; 22:650-6; PMID:23958375; http://dx.doi.org/10.1016/j.breast.2013.07.037
- Yardley DA, Shipley DL, Peacock NW, Shastry M, Midha R, Priego VM, Hainsworth JD. Phase I/II trial of neoadjuvant sunitinib administered with weekly paclitaxel/carboplatin in patients with locally advanced triple-negative breast cancer. Breast Cancer Res Treat 2015; 152:557-67; PMID:26155975; http://dx.doi.org/10.1007/s10549-015-3482-4
- Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, Lilenbaum R, Johnson DH. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med 2006; 355:2542-50; PMID:17167137; http://dx.doi.org/10.1056/NEJMoa061884
- Rini BI, Halabi S, Rosenberg JE, Stadler WM, Vaena DA, Archer L, Atkins JN, Picus J, Czaykowski P, Dutcher J, et al. Phase III trial of bevacizumab plus interferon alfa versus interferon alfa monotherapy in patients with metastatic renal cell carcinoma: final results of CALGB 90206. J Clin Oncol 2010; 28:2137-43; PMID:20368558; http://dx.doi.org/10.1200/JCO.2009.26.5561
- Sikov WM, Berry DA, Perou CM, Singh B, Cirrincione CT, Tolaney SM, Kuzma CS, Pluard TJ, Somlo G, Port ER, et al. Impact of the addition of carboplatin and/or bevacizumab to neoadjuvant once-per-week paclitaxel followed by dose-dense doxorubicin and cyclophosphamide on pathologic complete response rates in stage II to III triple-negative breast cancer: CALGB 40603 (Alliance). J Clin Oncol 2015; 33:13-21; PMID:25092775; http://dx.doi.org/10.1200/JCO.2014.57.0572
- Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, Inoue M, Bergers G, Hanahan D, Casanovas O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009; 15:220-31; PMID:19249680; http://dx.doi.org/10.1016/j.ccr.2009.01.027
- Zhang S, Zhang D, Sun B. Vasculogenic mimicry: current status and future prospects. Cancer Lett 2007; 254:157-64; PMID:17306454; http://dx.doi.org/10.1016/j.canlet.2006.12.036
- Hendrix MJ, Seftor EA, Meltzer PS, Gardner LM, Hess AR, Kirschmann DA, Schatteman GC, Seftor RE. Expression and functional significance of VE-cadherin in aggressive human melanoma cells: role in vasculogenic mimicry. Proc Natl Acad Sci U S A 2001; 98:8018-23; PMID:11416160; http://dx.doi.org/10.1073/pnas.131209798
- Zhang S, Guo H, Zhang D, Zhang W, Zhao X, Ren Z, Sun B. Microcirculation patterns in different stages of melanoma growth. Oncol Rep 2006; 15:15-20; PMID:16328029; http://dx.doi.org/10.3892/or.15.1.15
- Hess AR, Seftor EA, Gardner LM, Carles-Kinch K, Schneider GB, Seftor RE, Kinch MS, Hendrix MJ. Molecular regulation of tumor cell vasculogenic mimicry by tyrosine phosphorylation: role of epithelial cell kinase (Eck/EphA2). Cancer Res 2001; 61:3250-5; PMID:11309274; http://cancerres.aacrjournals.org/content/61/8/3250
- Sun B, Qie S, Zhang S, Sun T, Zhao X, Gao S, Ni C, Wang X, Liu Y, Zhang L. Role and mechanism of vasculogenic mimicry in gastrointestinal stromal tumors. Hum Pathol 2008; 39:444-51; PMID:18261629; http://dx.doi.org/10.1016/j.humpath.2007.07.018
- Shirakawa K, Kobayashi H, Sobajima J, Hashimoto D, Shimizu A, Wakasugi H. Inflammatory breast cancer: vasculogenic mimicry and its hemodynamics of an inflammatory breast cancer xenograft model. Breast Cancer Res 2003; 5:136-9; PMID:12793894; http://dx.doi.org/10.1186/bcr585
- Sun T, Zhao N, Zhao XL, Gu Q, Zhang SW, Che N, Wang XH, Du J, Liu YX, Sun BC. Expression and functional significance of Twist1 in hepatocellular carcinoma: its role in vasculogenic mimicry. Hepatology 2010; 51:545-56; PMID:19957372; http://dx.doi.org/10.1002/hep.23311
- Sun T, Sun BC, Zhao XL, Zhao N, Dong XY, Che N, Yao Z, Ma YM, Gu Q, Zong WK, et al. Promotion of tumor cell metastasis and vasculogenic mimicry by way of transcription coactivation by Bcl-2 and Twist1: a study of hepatocellular carcinoma. Hepatology 2011; 54:1690-706; PMID:21748764; http://dx.doi.org/10.1002/hep.24543
- Liu X, Wang X, Du W, Chen L, Wang G, Cui Y, Liu Y, Dou Z, Wang H, Zhang P, et al. Suppressor of fused (Sufu) represses Gli1 transcription and nuclear accumulation, inhibits glioma cell proliferation, invasion and vasculogenic mimicry, improving glioma chemo-sensitivity and prognosis. Oncotarget 2014; 5:11681-94; PMID:25373737; http://dx.doi.org/10.18632/oncotarget.2585
- Orecchia P, Conte R, Balza E, Pietra G, Mingari MC, Carnemolla B. Targeting Syndecan-1, a molecule implicated in the process of vasculogenic mimicry, enhances the therapeutic efficacy of the L19-IL2 immunocytokine in human melanoma xenografts. Oncotarget 2015; 6:37426-42; PMID:26460958; http://dx.doi.org/10.18632/oncotarget.6055
- Zhang D, Sun B, Zhao X, Ma Y, Ji R, Gu Q, Dong X, Li J, Liu F, Jia X, et al. Twist1 expression induced by sunitinib accelerates tumor cell vasculogenic mimicry by increasing the population of CD133+ cells in triple-negative breast cancer. Mol Cancer 2014; 13:207; PMID:25200065; http://dx.doi.org/10.1186/1476-4598-13-207
- Wang Z, Dabrosin C, Yin X, Fuster MM, Arreola A, Rathmell WK, Generali D, Nagaraju GP, El-Rayes B, Ribatti D, et al. Broad targeting of angiogenesis for cancer prevention and therapy. Semin Cancer Biol 2015; 35:S224-43; PMID:25600295; http://dx.doi.org/10.1016/j.semcancer.2015.01.001
- Vasudev NS, Goh V, Juttla JK, Thompson VL, Larkin JM, Gore M, Nathan PD, Reynolds AR. Changes in tumour vessel density upon treatment with anti-angiogenic agents: relationship with response and resistance to therapy. Br J Cancer 2013; 109:1230-42; PMID:23922108; http://dx.doi.org/10.1038/bjc.2013.429
- Conley SJ, Gheordunescu E, Kakarala P, Newman B, Korkaya H, Heath AN, Clouthier SG, Wicha MS. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc Natl Acad Sci U S A 2012; 109:2784-9; PMID:22308314; http://dx.doi.org/10.1073/pnas.1018866109
- Soeda A, Park M, Lee D, Mintz A, Androutsellis-Theotokis A, McKay RD, Engh J, Iwama T, Kunisada T, Kassam AB, et al. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha. Oncogene 2009; 28:3949-59; PMID:19718046; http://dx.doi.org/10.1038/onc.2009.252
- McIntyre A, Harris AL. Metabolic and hypoxic adaptation to anti-angiogenic therapy: a target for induced essentiality. EMBO Mol Med 2015; 7:368-79; PMID:25700172; http://dx.doi.org/10.15252/emmm.201404271
- Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer 2008; 8:592-603; PMID:18650835; http://dx.doi.org/10.1038/nrc2442
- Vasudev NS, Reynolds AR. Anti-angiogenic therapy for cancer: current progress, unresolved questions and future directions. Angiogenesis 2014; 17:471-94; PMID:24482243; http://dx.doi.org/10.1007/s10456-014-9420-y
- Seftor RE, Hess AR, Seftor EA, Kirschmann DA, Hardy KM, Margaryan NV, Hendrix MJ. Tumor cell vasculogenic mimicry: from controversy to therapeutic promise. Am J Pathol 2012; 181:1115-25; PMID:22944600; http://dx.doi.org/10.1016/j.ajpath.2012.07.013
- Singleton DC, Rouhi P, Zois CE, Haider S, Li JL, Kessler BM, Cao Y, Harris AL. Hypoxic regulation of RIOK3 is a major mechanism for cancer cell invasion and metastasis. Oncogene 2015; 34:4713-22; PMID:25486436; http://dx.doi.org/10.1038/onc.2014.396
- Chaturvedi P, Gilkes DM, Takano N, Semenza GL. Hypoxia-inducible factor-dependent signaling between triple-negative breast cancer cells and mesenchymal stem cells promotes macrophage recruitment. Proc Natl Acad Sci U S A 2014; 111:E2120-9; PMID:24799675; http://dx.doi.org/10.1073/pnas.1406655111
- Hwang-Verslues WW, Chang PH, Jeng YM, Kuo WH, Chiang PH, Chang YC, Hsieh TH, Su FY, Lin LC, Abbondante S, et al. Loss of corepressor PER2 under hypoxia up-regulates OCT1-mediated EMT gene expression and enhances tumor malignancy. Proc Natl Acad Sci U S A 2013; 110:12331-6; PMID:23836662; http://dx.doi.org/10.1073/pnas.1222684110
- Kirschmann DA, Seftor EA, Hardy KM, Seftor RE, Hendrix MJ. Molecular pathways: vasculogenic mimicry in tumor cells: diagnostic and therapeutic implications. Clin Cancer Res 2012; 18:2726-32; PMID:22474319; http://dx.doi.org/10.1158/1078-0432.CCR-11-3237
- Seftor RE, Seftor EA, Koshikawa N, Meltzer PS, Gardner LM, Bilban M, Stetler-Stevenson WG, Quaranta V, Hendrix MJ. Cooperative interactions of laminin 5 gamma2 chain, matrix metalloproteinase-2, and membrane type-1-matrix/metalloproteinase are required for mimicry of embryonic vasculogenesis by aggressive melanoma. Cancer Res 2001; 61:6322-7; PMID:11522618; http://cancerres.aacrjournals.org/content/61/17/6322.long
- Wang H, Sun W, Zhang WZ, Ge CY, Zhang JT, Liu ZY, Fan YZ. Inhibition of tumor vasculogenic mimicry and prolongation of host survival in highly aggressive gallbladder cancers by norcantharidin via blocking the ephrin type a receptor 2/focal adhesion kinase/paxillin signaling pathway. PLoS One 2014; 9:e96982; PMID:24811250; http://dx.doi.org/10.1371/journal.pone.0096982
- Sun D, Sun B, Liu T, Zhao X, Che N, Gu Q, Dong X, Yao Z, Li R, Li J, et al. Slug promoted vasculogenic mimicry in hepatocellular carcinoma. J Cell Mol Med 2013; 17:1038-47; PMID:23815612; http://dx.doi.org/10.1111/jcmm.12087
- Du J, Sun B, Zhao X, Gu Q, Dong X, Mo J, Sun T, Wang J, Sun R, Liu Y. Hypoxia promotes vasculogenic mimicry formation by inducing epithelial-mesenchymal transition in ovarian carcinoma. Gynecol Oncol 2014; 133:575-83; PMID:24589413; http://dx.doi.org/10.1016/j.ygyno.2014.02.034
- Qi H, Sun B, Zhao X, Du J, Gu Q, Liu Y, Cheng R, Dong X. Wnt5a promotes vasculogenic mimicry and epithelial-mesenchymal transition via protein kinase Calpha in epithelial ovarian cancer. Oncol Rep 2014; 32:771-9; PMID:24898696; http://dx.doi.org/10.3892/or.2014.3229
- Sun B, Zhang S, Zhang D, Li Y, Zhao X, Luo Y, Guo Y. Identification of metastasis-related proteins and their clinical relevance to triple-negative human breast cancer. Clin Cancer Res 2008; 14:7050-9; PMID:18981002; http://dx.doi.org/10.1158/1078-0432.CCR-08-0520