
ABSTRACT
BCR-ABL kinase domain (KD) mutations are well known for causing resistance against tyrosine kinase inhibitors (TKIs) and disease progression in chronic myeloid leukemia (CML). In recent years, compound BCR-ABL mutations have emerged as a new threat to CML patients by causing higher degrees of resistance involving multiple TKIs, including ponatinib. However, there are limited reports about association of compound BCR-ABL mutations with disease progression in imatinib (IM) sensitive CML patients. Therefore, we investigated presence of ABL-KD mutations in chronic phase (n = 41), late chronic phase (n = 33) and accelerated phase (n = 16) imatinib responders. Direct sequencing analysis was used for this purpose. Eleven patients (12.22%) in late-CP CML were detected having total 24 types of point mutations, out of which 8 (72.72%) harbored compound mutated sites. SH2 contact site mutations were dominant in our study cohort, with E355G (3.33%) being the most prevalent. Five patients (45%) all having compound mutated sites, progressed to advanced phases of disease during follow up studies. Two novel silent mutations G208G and E292E/E were detected in combination with other mutants, indicating limited tolerance for BCR-ABL1 kinase domain for missense mutations. However, no patient in early CP of disease manifested mutated ABL-KD. Occurrence of mutations was found associated with elevated platelet count (p = 0.037) and patients of male sex (p = 0.049). The median overall survival and event free survival of CML patients (n = 90) was 6.98 and 5.8 y respectively. The compound missense mutations in BCR-ABL kinase domain responsible to elicit disease progression, drug resistance or disease relapse in CML, can be present in yet Imatinib sensitive patients. Disease progression observed here, emphasizes the need of ABL-KD mutation screening in late chronic phase CML patients for improved clinical management of disease.
Introduction
The reciprocal translocation of BCR and ABL genes t(9;22)(q34;q11) result in formation of Philadelphia chromosome (ph), the principal cause of CML. Incidence of CML is approximately 1.6 per 100,000 population,Citation1 where estimated new cases in United States alone during year 2016 are 18,960.Citation2 CML is categorized by 3 phases: a clinically treatable chronic phase (CP) and an accelerated phase (AP) and blast phase (BP) with high degrees of drug resistance and treat failures.Citation3 Therefore, finding bio markers of CML progression can help in timely therapeutic interventions to minimize drug resistance, treatment failures and relapses, subsequently reducing morbidities and mortalities associated with CML, significantly.Citation4
Mutations in BCR-ABL1 gene have been found to be a major cause of disease progression and resistance to tyrosine kinase inhibitors in CML patients.Citation5-7 Therefore BCR-ABL mutation detection using capillary sequencing remains to be a gold standard for CML patients exhibiting disease progression and/or drug resistance in European and North American guidelines.Citation8-10 However, clinical significance of detecting BCR-ABL mutations in chronic phase or late CP drug sensitive CML patients remains to be established.
In recent years, emergence of so-called compound mutations, defined by the presence of 2 or more mutated sites in the same BCR-ABL, have posed a new threat to CML patients. Though BCR-ABL compound mutations have been found to be responsible for causing resistance to all tyrosine kinase inhibitors (TKIs) currently in clinical practiceCitation10-15 including ponatinib,Citation16,17 reports about role of compound ABL mutations in CML progression are lacking.Citation5,9
In the present study, we studied BCR-ABL kinase domain mutations in imatinib sensitive patients of CML in CP, late CP, AP and BC of the disease using Sanger sequencing. Our objective was to find out role of BCR-ABL mutations, specifically compound mutations, in disease progression in CML drug responders.
Methods
Patients
Diagnostic peripheral blood was obtained from 90 Imatinib sensitive CP, late CP and accelerated phase Philadelphia positive CML patients attending oncology clinics at 1) Jinnah hospital, Lahore 2) Mayo hospital, Lahore and 3) Institute of nuclear medicine and oncology (INMOL) Lahore, Pakistan. All participants submitted written informed consent and the study were approved by Punjab university research ethics body as well as the ethical committees of concerned hospitals. Disease phases were defined as CP: Blast cells less than 15%, Basophils less than 20%, blast and promyelocytes less than 30%; Late CP: patients in CP for more than 12 months post diagnosis; AP: Blast cells 15–29%, Basophils >20%, blast and promyelocytes >30%.Citation6 Standard response measures were followed; Complete hematologic response (CHR), Complete cytogenetic response (CCyR) and Partial cytogenetic response (PCyR) were defined as described previously,Citation6,7 but molecular response (MR) could not be noted as real time quantitative PCR was not available for BCR-ABL transcript quantification (). Clinical follow up studies were performed up till 2 y to comprehend association of mutations with disease phase and progression.
Table 1. Clinical characteristics of CML patients selected for the analysis of ABL-KD mutations.
Patterns of resistance toward Imatinib were followed from European leukemia net (ELN) guidelines.Citation6 Primary resistance was described as; failure to achieve land mark response i.e. CHR in 3 months, any level of CyR by 6 months, Major cytogenetic response (MCyR) by 12 months, and complete cytogenetic response (CCyR) by 18 months. Whereas, acquired resistance was defined as loss of already achieved hematologic, cytogenetic or molecular response at any stage of treatment.
Imatinib treatment protocol and response monitoring
Chronic phase patients were given an initial dose of 400mg/day after diagnosis. Hematologic response was assessed every 2 weeks until the CHR was achieved, and every 3 months afterwards. Cytogenetic response i.e., conventional metaphase analysis and/or FISH analysis was assessed every 6 months until a CCyR is achieved, and every 12 months afterwards. While molecular response was evaluated every 3 months (if produced by the patient). In case of hematologic resistance (no CHR in 3 months) or cytogenetic resistance (no MCyR within 12 months) to standard-dose imatinib, higher dose of Imatinib was recommended (600–800mg/day). The dose was also increased in case of disease relapse. Accelerated phase and blast phase patients were given an initial dose of 600mg/day Imatinib, and dose was adjusted afterwards according to patient's response.Citation6
Amplification of BCR-ABL fusion oncogene
Genomic RNA was isolated by TRIZOL® LS reagent following manufacturer's instructions.Citation18,19 Spectrophotometric quantification of RNA and its integrity check was performed by 2% agarose gel electrophoresis. cDNA was prepared by reverse transcription of one microgram RNA, using random hexamer primers (1.35mM) and RevertAid Reverse Transcriptase (200u). The integrity of cDNA was evaluated by amplification of housekeeping gene, Glyceraldehyde 3-phosphate dehydrogenase (GAPDH).Citation20 Amplification of BCR-ABL fusion oncogene was performed by using nested RT-PCR. Pre-validated primer sequences and PCR reaction conditions were followed from Willis et al.Citation8 Patient cDNA was amplified using B2A forward (TTCAGAAGCTTCTCCCTGACAT) and Abl 4065 reverse (CCTTCTCTAGCAGCTCATACACCTG). One microliter of the amplicon was used as template in 2nd round with Bcr F4 forward (ACAGCATTCCGCTGACCATCAATA) and U396 reverse (GCCATAGGTAGCAATTTCCC). Negative and positive control reactions were always run with every batch to ensure accuracy of the reaction.
ABL-Kinase domain amplification from healthy individuals
It was indispensable to amplify and sequence the kinase domain of ABL gene from healthy population so that those from CML patients could be compared with them. For this purpose blood samples from 20 healthy individuals were processed and sequenced. Primer sequences and amplification protocols were same as for CML patients.
Mutation analysis by sequencing of PCR products
The amplified PCR products were subjected to purification and direct sequencing analysis for detection of any possible point mutations present in ABL-kinase domain of CML patients. Nested RT-PCR products of processed samples were sequenced by Sanger method. The forward primer (3306F) 5′-TGGTTCATCATCATTCAACGG-3′ and reverse primer (4000R) 5′-GGACATGCCATAGGTAGCA-3′ were used for sequencing reaction.Citation21 The sequenced templates were analyzed using sequencing analysis software Geneious R9. DNA sequences of Abl kinase domain of healthy individuals were aligned against the reference sequence (NCBI GenBank accession number M14752.1). This was to find out any polymorphic variations in Pakistani population which should not be misunderstood with point mutations while analyzing CML patient's sequences. Later on Abl- kinase domain sequences of patients were aligned against the reference sequence M14752.1. The gene sequences showing heterozygous mutations and those with noise in data were repeated for the whole procedure.
Statistical analysis
All variables were checked for normal distribution by the Kolmogorov-Smirnov test. Wilcoxon–Mann–Whitney test was performed for comparison of numeric variables and Chi-square test or Fisher's exact test was used to compare categories, wherever appropriate. Survival probability of patients with respect to clinical features and response outcomes were estimated by Kaplan-Miere method, with differences between groups (Log-rank test) (IBM SPSS version 19, Chicago, IL, USA.). For all analyses, p < 0.05 was considered statistically significant.
Results
BCR-ABL gene was successfully amplified in all samples and among 90 CML patients screened for KD mutations, 11 (12.22%) were detected positive. These patients presented a total of 24 types of amino acid substitutions all together. Disease progression was not observed in CP and AP patients however, out of 33 late-CP patients 5 (15.15%) progressed to AP. All of them were having compound mutations.
Three mutations at position V228, F317 and Y413 were noticed twice and E355G for 3 times in different patients. Their individual clinical response and mutation analysis are mentioned (). Contribution of these mutations in producing suboptimal response against Imatinib, described by earlier studies is also provided (). The electropherogram screen shots, captured during mutation analysis for each patient separately are shown ().
Table 2. Clinical response and mutation data of Imatinib sensitive mutatnt (IMS-m) chronic myeloid leukemia patients (n = 90).
Table 3. An inclusive presentation of all point mutations in the kinase domain of BCR-ABL fusion oncogene detected in chronic myeloid leukemia patients included in the present study (n = 90).
Figure 1. Representative electropherograms showing compound mutations in BCR-ABL kinase domain of IM sensitive CML patients. The nucleotide change and its subsequent amino acid substitution is shown by blue longitudinal indicator along with the nucleotide number. NCBI GenBank accession number: M14752 is the reference sequence.
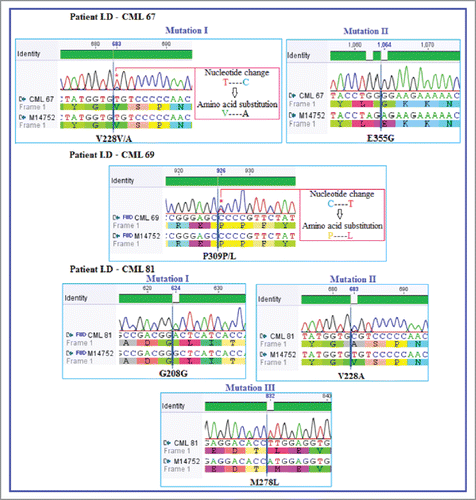
Two discrepant sites in ABL-KD of healthy individuals
The ABL-KD sequences from healthy Pakistani individuals were alligned to the reference sequence, NCBI GenBank accession number: M14752, before analyzing patient data, to ensure any dissimilar sites between the 2 sequences. This sequence has 2 nucleotide positions that differ to other ABL sequences in GenBank (mRNA sequence X16416 and DNA sequence U07563) as well as to the sequence of our healthy population. The discrepant nucleotides were 894 A>G and 1062 A>G (the first nucleotides represent those published in M14752). Hence, these were not considered as mutations while analyzing the sequences of CML patients. Amino acid positions are those of ABL type 1a, which has the following subdomains: P-loop (Phosphate binding loop) 244–256, C-helix 276–290, SH3 contact 292–304, IBS- 311–317, SH2 contact 330–360, A loop (activation loop) 381–411.
Development of compound mutations in late CP predicts CML progression
Out of 11 mutant patients 8 (72.72%) in late CP-CML were observed to harbour compound nucleotide substitutions. Out of which, 5 (45%) progressed to advance phase of disease during follow up studies (). The key residues in native BCR-ABL1 are: M244, G250, Q252, Y253, E255, V299, F311, T315, F317, M351, F359, and H396.Citation9 Two of them i.e., Q252 and F317 were found compound with other mutants in our study, namely Q252R/K291E/K294X/I360V (CML # 85), F317L/E236G/E334G/A350T/T406A (CML # 137) and F317L/T277A/N358D (CML # 110). Patient no. 85, harboring a P-loop mutation progressed to AP much earlier (1.1 year) than the rest of individuals. Rest of compound mutants are also described ().
Incidence of mutations is associated to high platelet count and CML patients of male sex
The presence or absence of mutation was found to be independent of age groups, hematologic response, cytogenetic response and sokal risk score of patients. Likewise, wild type and mutant patients did not differ expressively, in terms of total leukocyte count, levels of hemoglobin and spleen size measures. However, male CML patients manifested more mutations than females (95% confidence interval (CI) 0.047–0.056; p = .049) and mutant patients exhibited high platelet count than normal (95% CI 465.61–688.29; p = 0.037).
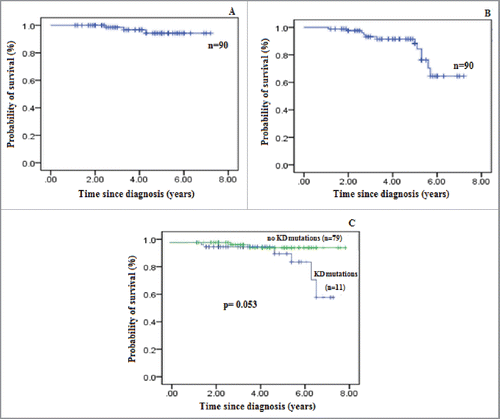
The median overall survival (OS) of CML patients was 6.98 y (95% CI 6.484–7.057) and 7 y overall survival rate was noted to be 94.2% (). To determine event free survival (EFS) following were considered events: progression to advance phase of disease, Loss of complete cytogenetic response and loss of complete hematologic response. The median EFS was 5.8 y (95% CI 4.868–5.998) after a follow up time of 7 y. Five year and 7 y rate of event free survival was noted to be 88.3% and 64.5%, respectively (). There lies a nearly significant difference (p = 0.053) in terms of progression free survival (PFS) among the 2 groups of patients i.e., mutant (95%CI 6.024–6.952) and non-mutant (95% CI 6.345–7.216). The 7 y rate of PFS for non-mutant and mutant patients was 96.3% and 61.7% respectively ().
Figure 2. The Kaplan-Meier curves showing overall (A) and event free survival (B) of Imatinib sensitive CML patients included in this study (C) Progression free survival (PFS) according to the presence of BCR-ABL kinase domain mutations.
Discussion
Employing direct sequencing analysis, we show that KD mutations are detectable in a considerable number of patients in late-CP of CML. Mutant patients progressing to advance phases of disease indicate possible role of compound ABL-KD mutations in impaired Imatinib binding leading to aggressive disease. Very few studies reported types and frequencies of kinase domain mutations in TKI-naïve or TKI-sensitive patients of CML.Citation10 This is the first study from Pakistan, reporting incidence of compound mutants in Imatinib sensitive late CP-CML patients and their possible role in disease progression.
In present investigation, majority of mutations were found clustered in SH2 (Src homology 2) contact site. SH2 contact (outside the kinase domain) holds great importance in ABL kinase domain, as it plays a key role in cellular communication by binding phosphorylated tyrosine residues.Citation11 Mutations in this domain contribute to impaired drug binding.Citation22 A total of 24 mutations were detected out of which 2 were novel silent mutations i.e., G208G and E292E/E, one was nonsense K294X and rest of the mutations were missense in nature. The frequently observed mutant E355E/G (3.33%) has already been reported the most prevelent among Pakistani CML population by our group.Citation23 This mutant is well known to bring about suboptimal response against tyrosine kinase inhibitors.Citation8,13-15 All of our patients (no. 67, 117 and 124) having amino acid substitution E355G compound with other mutations, manifested disease progression along the course of treatment.
Moreover, one patient (no. 85) identified with P-loop mutation Q252R in combination with others, showed advancement to next phase of disease. Mutations within phosphate binding loop hold great importance as in vitro studies suggest that these substitutions are far more oncogenic than other mutations or unmutated BCR-ABL fusion oncogene.Citation24 Nevertheless, the same mutant Q252R/H is reported to cause moderate to complete resistance against Imatinib therapy.Citation14 The P-loop is distorted in ABL-Imatinib interface in such a way that a hydrophobic cage for the drug is formed. Mutations in this region undermine the distorted shape and cause poor drug binding.Citation25 However, the most resistant mutant T315I was not detected in any CML patient with late-CP of disease.
During follow up studies, 5 patients (45%) having compound mutations progressed to advance phase of disease. These mutations span across all the subdomains of ABL-KD. The compound mutations in tyrosine kinase domain are well studied in Imatinib resistant CML patients. Their contribution in establishing resistance against first line tyrosine kinase inhibitors is also reported.Citation26 The presence of compound mutations describe poor-risk group of chronic phase CML patients, with increased chances of disease progression and acquiring resistance with suboptimal response to second generation TKIs.Citation16 However, it is expected that each of compound mutant clone retains its own sensitivity against given treatment.Citation5 Presence of 2 or more mutations in ABL kinase domain impair the binding of Imatinib, resulting in suboptimal treatment outcome and progression of disease. These mutations are know particularly to cause secondary resistance.Citation27
Our study cohort was exposed to TKI therapy before detection of mutations. Two perceptions prevail in this regard; some scientists believe that patients do not have pre-existing mutations, rather they emerge after exposure to Imatinib.Citation28 Whereas, some state that tyrosine kinase inhibitors do not induce mutations, rather they select them.Citation29 Majority of studies suggest that emergance of BCR-ABL kinase domain mutations is a disease evolution process, irrespective of TKI therapy. The chances of occurance of KD mutations is higher in AP/BP as compared with CP.Citation14 Soverini et al.Citation30 mentioned that irrespective of the hematologic response achieved by patients, monitoring of emerging mutations can play a vital role in predicting patients with poor prognosis and revising their therapeutic strategy. Similarly, Khorashad et al. stated presence of KD mutations and achievement of CCyR as 2 independent prognostic factors for PFS in CP-CML patients.Citation31 Reported literature suggests that different muatations instigate varying levels of response alteration against Imatinib therapy in CML patients.Citation32 So, the detection of KD mutations before any change in cytogenetic or molecular response might be a potent indicator for changing therapy.Citation33
Our patients were evaluated as Imatinib sensitive by physicians at the time of mutation analysis. However, some of them in late-CP CML, having compound mutated sites progressed to accelerated phase and blast phase, with 2 P-loop mutant patients reaching Imatinib resistance. So, there is always a greater possibility of change in response due to underlying amino acid substitutions.
Conclusion
Single mutants in BCR-ABL KD are well known for their possible role in disease progression and TKI resistance, while the role of compound mutations in CML progression has not been reported frequently. Our findings however emphasize the need of mutation analysis in late CP-CML patients, to identify early indicators of disease progression and strategize the treatment accordingly. An extended follow up of a larger patient cohort is necessary for confirmation of these intriguing findings.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
Financial assistance provided by Higher Education Commission (HEC) Pakistan (20–1370/R&D/09), Laboratory facilities at Department of Zoology, University of The Punjab and The University of Lahore are gratefully acknowledged. The study was partly supported by the College of Medicine Research Centre, Deanship of Scientific Research, King Saud University, Riyadh.
References
- Jain P, Das VNR, Ranjan A, Chaudhary R, Pandey K. Comparative study for the efficacy, safety and quality of life in patients of chronic myeloid leukemia treated with Imatinib or Hydroxyurea. J Res Pharm Pract 2013; 2:156-61; PMID:24991625; http://dx.doi.org/10.4103/2279-042X.128145
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016; 66:7-30; PMID:26742998; http://dx.doi.org/10.3322/caac.21332
- Chen R, Chen B. The role of dasatinib in the management of chronic myeloid leukemia. Drug Des Devel Ther 2015; 9:773; PMID:25709401; http://dx.doi.org/10.2147/DDDT.S80207
- Miller GD, Bruno BJ, Lim CS. Resistant mutations in CML and Ph+ ALL–role of ponatinib. Biologics 2014; 8:243-54; http://dx.doi.org/10.2147/BTT.S50734
- Khorashad JS, Kelley TW, Szankasi P, Mason CC, Soverini S, Adrian LT, Eide CA, Zabriskie MS, Lange T, Estrada JC, et al. BCR-ABL1 compound mutations in tyrosine kinase inhibitor–resistant CML: frequency and clonal relationships. Blood 2013; 121:489-98; PMID:23223358; http://dx.doi.org/10.1182/blood-2012-05-431379
- Baccarani M, Deininger MW, Rosti G, Hochhaus A, Soverini S, Apperley JF, Cervantes F, Clark RE, Cortes JE, Guilhot F, et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood 2013; 122:872-84; PMID:23803709; http://dx.doi.org/10.1182/blood-2013-05-501569
- Baccarani M, Castagnetti F, Gugliotta G, Rosti G. A review of the European LeukemiaNet recommendations for the management of CML. Ann Hematol 2015; 94 Suppl 2:S141-7; PMID:25814080; http://dx.doi.org/10.1007/s00277-015-2322-2
- Willis SG, Lange T, Demehri S, Otto S, Crossman L, Niederwieser D, Stoffregen EP, McWeeney S, Kovacs I, Park B, et al. High-sensitivity detection of BCR-ABL kinase domain mutations in imatinib-naive patients: correlation with clonal cytogenetic evolution but not response to therapy. Blood 2005; 106:2128-37; PMID:15914554; http://dx.doi.org/10.1182/blood-2005-03-1036
- Zabriskie MS, Eide CA, Tantravahi SK, Vellore NA, Estrada J, Nicolini FE, Khoury HJ, Larson RA, Konopleva M, Cortes JE, et al. BCR-ABL1 compound mutations combining key kinase domain positions confer clinical resistance to ponatinib in Ph chromosome-positive leukemia. Cancer Cell 2014; 26:428-42; PMID:25132497; http://dx.doi.org/10.1016/j.ccr.2014.07.006
- Press RD, Willis SG, Laudadio J, Mauro MJ, Deininger MW. Determining the rise in BCR-ABL RNA that optimally predicts a kinase domain mutation in patients with chronic myeloid leukemia on imatinib. Blood 2009; 114:2598-605; PMID:19625707; http://dx.doi.org/10.1182/blood-2008-08-173674
- Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell 2010; 141:1117-34; PMID:20602996; http://dx.doi.org/10.1016/j.cell.2010.06.011
- Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer 2009; 9:28-39; http://dx.doi.org/10.1038/nrc2559
- Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, Sawyers CL. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2002; 2:117-25; PMID:12204532; http://dx.doi.org/10.1016/S1535-6108(02)00096-X
- O'Hare T, Eide CA, Deininger MW. Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood 2007; 110:2242-9; PMID:17496200; http://dx.doi.org/10.1182/blood-2007-03-066936
- Tokarski JS, Newitt JA, Chang CYJ, Cheng JD, Wittekind M, Kiefer SE, Kish K, Lee FY, Borzillerri R, Lombardo LJ, et al. The structure of Dasatinib (BMS-354825) bound to activated ABL kinase domain elucidates its inhibitory activity against imatinib-resistant ABL mutants. Cancer Res 2006; 66:5790-7; PMID:16740718; http://dx.doi.org/10.1158/0008-5472.CAN-05-4187
- Parker WT, Yeung DT, Yeoman AL, Altamura HK, Jamison BA, Field CR, Hodgson JG, Lustgarten S, Rivera VM, Hughes TP, et al. The impact of multiple low-level BCR-ABL1 mutations on response to ponatinib. Blood 2016; 127(15):1870-80; http://doi.org/10.1182/blood-2015-09-666214
- Cortes JE, Kantarjian H, Shah NP, Bixby D, Mauro MJ, Flinn I, O'Hare T, Hu S, Narasimhan NI, Rivera VM, et al. Ponatinib in refractory Philadelphia chromosome–positive leukemias. N Eng J Med 2012; 367:2075-88; PMID:23190221; http://dx.doi.org/10.1056/NEJMoa1205127
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 1987; 162:156-9; PMID:2440339; http://dx.doi.org/10.1016/0003-2697(87)90021-2
- Liedtke W, Battistini L, Brosnan CF, Raine CS. A comparison of methods for RNA extraction from lymphocytes for RT-PCR. PCR Methods Appl 1994; 4:185-7; PMID:7580904; http://dx.doi.org/10.1101/gr.4.3.185
- Asad S, Ijaz B, Ahmad W, Kausar H, Sarwar MT, Gull S, Shahid I, Khan MK, Hassan S. Development of persistent HCV genotype 3a infection cell culture model in huh-7 cell. Virol J 2012; 9:1; PMID:22214262; http://dx.doi.org/10.1186/1743-422X-9-11
- Willis SG, Lange T, Demehri S, Otto S, Crossman L, Niederwieser D, et al. High-sensitivity detection of BCR-ABL kinase domain mutations in imatinib-naive patients: correlation with clonal cytogenetic evolution but not response to therapy. Blood 2005; 106:2128-37; PMID:15914554; http://dx.doi.org/10.1182/blood-2005-03-1036
- Ray A, Cowan-Jacob SW, Manley PW, Mestan J, Griffin JD. Identification of BCR-ABL point mutations conferring resistance to the Abl kinase inhibitor AMN107 (nilotinib) by a random mutagenesis study. Blood 2007; 109:5011-5; PMID:17303698; http://dx.doi.org/10.1182/blood-2006-01-015347
- Iqbal Z AAM, Akhtar T, Khalid M, Aziz Z, Aleem Aamer, Gill Ammara T, Khalid Ahmad M, Alanazi Abdullah, Shah Ijaz H, Khalid Muhammad, Sabar Muhammad Farooq, Iqbal Mudassar. High frequencies of compound BCR-ABL mutations and their association with imatinib resistant, disease progression and late chronic phase disease in pakistani chronic myeloid leukemia patients necessitate the inclusion of molecular testing in routine clinical settings. Blood 2015; 126:5167.
- Griswold IJ, MacPartlin M, Bumm T, Goss VL, O'Hare T, Lee KA, Corbin AS, Stoffregen EP, Smith C, Johnson K, et al. Kinase domain mutants of Bcr-Abl exhibit altered transformation potency, kinase activity, and substrate utilization, irrespective of sensitivity to imatinib. Molecular and Cell Biol 2006; 26:6082-93; PMID:16880519; http://dx.doi.org/10.1128/MCB.02202-05
- Kimura S, Ando T, Kojima K. Ever-advancing chronic myeloid leukemia treatment. Int J Clin Oncol 2014; 19:3-9; PMID:24258348; http://dx.doi.org/10.1007/s10147-013-0641-7
- Quintás-Cardama A, Kantarjian H, O'Brien S, Jabbour E, Borthakur G, Ravandi F, Verstovsek S, Shan J, Cortes J. Outcome of patients with chronic myeloid leukemia with multiple ABL1 kinase domain mutations receiving tyrosine kinase inhibitor therapy. Haematologica 2011; 96:918-24; PMID:21357704; http://dx.doi.org/10.3324/haematol.2010.039321
- Bixby D, Talpaz M. Mechanisms of resistance to tyrosine kinase inhibitors in chronic myeloid leukemia and recent therapeutic strategies to overcome resistance. ASH Educ Program Book 2009; 2009:461-76; http://dx.doi.org/10.1182/asheducation-2009.1.461
- Trela E, Glowacki S, Błasiak J. Therapy of chronic myeloid Leukemia: twilight of the imatinib era? ISRN Oncol 2014; 2014:596483; PMID:24634785; http://dx.doi.org/10.1155/2014/596483
- Soverini S, Hochhaus A, Nicolini FE, Gruber F, Lange T, Saglio G, Pane F, Müller MC, Ernst T, Rosti G, et al. BCR-ABL kinase domain mutation analysis in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors: recommendations from an expert panel on behalf of European LeukemiaNet. Blood 2011; 118:1208-15; PMID:21562040; http://dx.doi.org/10.1182/blood-2010-12-326405
- Soverini S, Martinelli G, Rosti G, Bassi S, Amabile M, Poerio A, Giannini B, Trabacchi E, Castagnetti F, Testoni N, et al. ABL mutations in late chronic phase chronic myeloid leukemia patients with up-front cytogenetic resistance to imatinib are associated with a greater likelihood of progression to blast crisis and shorter survival: a study by the GIMEMA Working Party on Chronic Myeloid Leukemia. J Clin Oncol 2005; 23:4100-9; PMID:15867198; http://dx.doi.org/10.1200/JCO.2005.05.531
- Khorashad JS, de Lavallade H, Apperley JF, Milojkovic D, Reid AG, Bua M, Szydlo R, Olavarria E, Kaeda J, Goldman JM, et al. Finding of kinase domain mutations in patients with chronic phase chronic myeloid leukemia responding to imatinib may identify those at high risk of disease progression. J Clin Oncol 2008; 26:4806-13; PMID:18645191; http://dx.doi.org/10.1200/JCO.2008.16.9953
- Elias MH, Baba AA, Azlan H, Rosline H, Sim GA, Padmini M, Fadilah SA, Ankathil R. BCR-ABL kinase domain mutations, including 2 novel mutations in imatinib resistant Malaysian chronic myeloid leukemia patients—Frequency and clinical outcome. Leukemia Res 2014; 38:454-9; PMID:24456693; http://dx.doi.org/10.1016/j.leukres.2013.12.025
- de Lavallade H, Apperley JF, Khorashad JS, Milojkovic D, Reid AG, Bua M, Szydlo R, Olavarria E, Kaeda J, Goldman JM, et al. Imatinib for newly diagnosed patients with chronic myeloid leukemia: incidence of sustained responses in an intention-to-treat analysis. J Clin Oncol 2008; 26:3358-63; PMID:18519952; http://dx.doi.org/10.1200/JCO.2007.15.8154
- Jiang X, Saw KM, Eaves A, Eaves C. Instability of BCR-ABL gene in primary and cultured chronic myeloid leukemia stem cells. J Natl Cancer Inst 2007; 99:680-93; PMID:17470736; http://dx.doi.org/10.1093/jnci/djk150
- Hochhaus A, Lee FY, Müller M. Methods Of identifying and treating individuals exhibiting mutant Bcr/Abl kinase polypeptides. Google Patents 2007.
- Sawyers CL, Gorre ME, Shah NP, Nicoll J. Mutations in the bcr-abl tyrosine kinase associated with resistance to sti-571. Google Patents 2010
- Azam M, Latek RR, Daley GQ. Mechanisms of autoinhibition and STI-571/imatinib resistance revealed by mutagenesis of BCR-ABL. Cell 2003; 112:831-43; PMID:12654249; http://dx.doi.org/10.1016/S0092-8674(03)00190-9
- Gorre M, Nicoll J, Sawyers C, Shah N. Mutations in the Bcr-Abl tyrosine kinase associated with resistance to STI-571. Google Patents 2002.
- Elias MH, Baba AA, Azlan H, Rosline H, Sim GA, Padmini M, Fadilah SA, Ankathil R. BCR-ABLkinase domain mutations, including 2 novel mutations in imatinib resistant Malaysian chronic myeloid leukemia patients—Frequency and clinical outcome. Leukemia Res 2014; 38:454-9; PMID:24456693; http://dx.doi.org/10.1016/j.leukres.2013.12.025
- Takahashi N, Miura M, Kuroki J, Mitani K, Kitabayashi A, Sasaki O, Kimura H, Imai K, Tsukamoto N, Noji H, et al. Multicenter phase II clinical trial of nilotinib for patients with imatinib-resistant or -intolerant chronic myeloid leukemia from the East Japan CML study group evaluation of molecular response and the efficacy and safety of nilotinib. Biomarker Res 2014; 2:6; PMID:24650752; http://dx.doi.org/10.1186/2050-7771-2-6
- Azam M, Latek RR, Daley GQ. Mechanisms of Autoinhibition and STI-571/Imatinib Resistance Revealed by Mutagenesis of BCR-ABL. Cell 2003; 112:831-43; PMID:12654249; http://dx.doi.org/10.1016/S0092-8674(03)00190-9
- Soverini S, Vitale A, Poerio A, Gnani A, Colarossi S, Iacobucci I, Cimino G, Elia L, Lonetti A, Vignetti M, et al. Philadelphia-positive acute lymphoblastic leukemia patients already harbor BCR-ABL kinase domain mutations at low levels at the time of diagnosis. Haematologica 2011; 96:552-7; PMID:21193419; http://dx.doi.org/10.3324/haematol.2010.034173
- Sawyers CL, Gorre ME, Shah NP, Nicoll J. Mutations in the Bcr-Abl tyrosine kinase associated with resistance to STI-571. Google Patents 2014.