
ABSTRACT
Background: Neuroblastoma (NB) is a common and often lethal cancer of early childhood that accounts for 10% of pediatric cancer mortality. Incidence peaks in infancy and then rapidly declines, with less than 5% of cases diagnosed in children and adolescents ≥ 10 y. There is increasing evidence that NB has unique biology and an chronic disease course in older children and adolescents, but ultimately dismal survival. Methods: We describe a rare constitutional 3p26.3 terminal microdeletion which occurred in an adolescent with NB, with apparently normal phenotype without neurocognitive defects. We evaluated the association of expression of genes involved in the microdeletion with NB patient outcomes using R2 platform. We screened NB patient's tumor cells for CHL1 protein expression using immunofluorescence. Results: Constitutional and tumor DNA were tested by array-comparative genomic hybridization and single nucleotide-polymorphism-array analyses. Peripheral blood mononuclear cells from the patient showed a 2.54 Mb sub-microscopic constitutional terminal 3p deletion that extended to band p26.3. The microdeletion 3p disrupted the CNTN4 gene and the neighboring CNTN6 and CHL1 genes were hemizygously deleted, each of these genes encode neuronal cell adhesion molecules. Low expression of CNTN6 and CNTN4 genes did not stratify NB patients, whereas low CHL1 expression characterized 417 NB patients having worse overall survival. CHL1 protein expression on tumor cells from the patient was weaker than positive control. Conclusion: This is the first report of a constitutional 3p26.3 deletion in a NB patient. Since larger deletions of 3p, indicative of the presence of one or more tumor suppressor genes in this region, occur frequently in neuroblastoma, our results pave the way to the identification of one putative NB suppressor genes mapping in 3p26.3.
Introduction
Neuroblastoma (NB) is a common and often lethal cancer of early childhood that accounts for 10% of pediatric cancer mortality. Incidence peaks in infancy and then rapidly declines, with less than 5% of cases diagnosed in children and adolescents ≥ 10 y. These cases have usually a grim prognosis.Citation2 NB exhibits a remarkably diverse clinical behavior that ranges from spontaneous regression to aggressive disease, reflecting the biologic heterogeneity of this tumor.Citation1,2
Established markers of NB poor prognosis include losses of chromosome 1p, 3p, 4p, 11q and gains of 1q, 2p, and 17q.Citation1 Breakpoints occurring in NB on multiple chromosomes may reflect an underlying defect in DNA maintenance or repair, and/or increased levels of double-stranded DNA breaks. MYCN oncogene amplification is often associated with younger age, unbalanced 17q gain and hemizygous 1p36 loss. 11q loss is associated with older age at diagnosis, absence of MYCN amplification and high chromosomal instability.Citation1 Losses of 3p are rare in MYCN amplified tumors, but are associated with increased metastatic potential and decreased overall survival.Citation1 The 3p region may contain at least one NB suppressor gene.Citation3 Germline structural chromosomal abnormalities, although rare in children with NB, can help in the detection of tumor suppressor genes (TSGs).Citation4
Array-CGH has led to the identification of the new 3p terminal deletion syndrome,Citation5 a rare contiguous gene syndrome caused by 3p26-pter deletions variable in size and mostly occurring de novo. Also the phenotype varies from severe to normal, and chromosomal non-penetrance and gene modification may account for the phenotypic variability.Citation6
We report a sub-microscopic constitutional terminal 3p26.3 deletion, in an adolescent with otherwise apparently normal phenotype without neurocognitive defects, who developed a localized NB. These findings pave the way to the identification of a TSG mapping in 3p26.3.
Case report
The 17-years-old Caucasian male patient, born from non-consanguineous healthy parents, was referred to our Institution with perineal pain, constipation and urinary retention. The physical examination was normal except for reduced reflexes in the lower limbs, and neurocognitive defects were excluded. The CT scan and MR revealed a large pre-sacral mass with intra-spinal (L5-S2) extension. The latter finding was taken as evidence for unresectable tumor and the patient underwent multiple needle biopsies during laparoscopy. Urinary homovanillic and vanillylmandelic acids were normal. I123-methyl-iodo-benzylguanidine (MIBG) scan showed an isolated hyper-fixation at the pelvic mass. No infiltrating neoplastic cells were detected in the bone marrow. The pathological diagnosis was stroma poor, poorly differentiated neuroblastoma. At diagnosis, the patient had no evidence of metastatic disease and his tumor had favorable biologic characteristics. The tumor had a hyperdiploid DNA index, no evidence of 1p36, 4p and 11q deletions, no evidence of unbalanced 1q, 2p24 and 17q gains, and no MYCN amplification. The 3p26.3 hemizygous deletion was the only segmental alteration detected by standard a-CGH analysis.
Constitutional and tumor DNA were tested by comparative genomic hybridization (a-CGH) and SNP-array using the 4 × 180K Kit (Agilent). SNP-array and Oligo-array data were analyzed with Genomic Workbench 7.0.40 software (Agilent). Chromosome positions were determined using GRCh/hg19 (UCSC Genome Browser, http://genome.ucsc.edu, Feb. 2009 release).
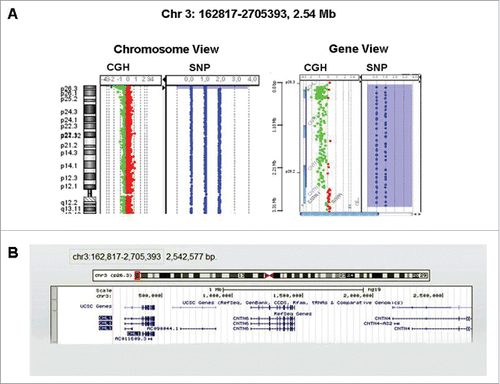
High-resolution a-CGH and SNP-array analyses of DNA from peripheral blood mononuclear cells showed a sub-microscopic constitutional deletion of the short arm of chromosome 3 extended to band p26.3 and encompassing 2.54 Mb in size (). The 3p26.3 region was deleted from oligomer A_16_P16102363 (162,817 bp) (first deleted) to oligomer A_16_P00636404 (2,705,393 bp) (last deleted), containing CHL1 and Contactin 6 (CNTN6) genes (). The proximal breakpoint was located in the Contactin 4 (CNTN4) gene (2,705,393 bp from p telomere). The microdeletion 3p disrupted CNTN4 gene and contained CNTN6 and CHL1 genes, all these genes codified for neuronal cell adhesion molecules. FISH analysis with telomeric probes specific for the 3q and 3p telomeres (ToTel Vysion kit, Vysis, Abbott Molecular, Illinois, USA) confirmed the deletion (data not shown). Karyotype from the mother was normal, unfortunately the father was not available for testing.
Figure 1. (A) CGH/SNP array showed a 2.54 Mb terminal deletion 3p26.3 encompassing the genes CHL1 and CNTN6. The proximal breakpoint was located in the CNTN4 gene 2,705,393 bp from p telomere. (B) Image from UCSC Genome Browser showing the sizes of the terminal 3p deletion extended to band p26.3 and covered 2.54 Mb.
The NB tumor displayed a 3p26.3 microdeletion identical to the constitutional one, as the only segmental alteration. SNP-array was used to confirm and refine the constitutional and the acquired 3p26.3 deletion. No evidence for a homozygous 3p26.3 deletion was obtained.
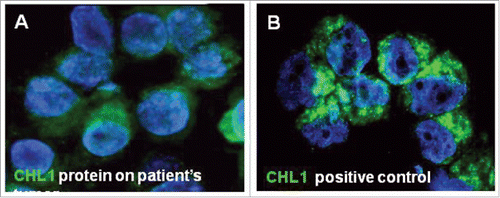
The expression of the CNTN4 gene and the neighboring CNTN6 and CHL1 genes was studied in the 498 NB patients cohort, using online results from microarray analyses of NB tumors obtained from the R2 Genomics Analysis and Visualization Platform (http://r2.amc.nl), the resulting figures and p values were downloaded. Low expression of CNTN6 and CNTN4 genes did not stratify NB patients, whereas low CHL1 expression characterized 417 NB patients having worse overall survival. The optimal cut-off for overall survival analyses was chosen as the expression value where the log-rank statistic for the separation of survival curves reached a maximum. The Kaplan-Meier was highly significant (p<0.0001) indicating that low CHL1 expression was a potential risk factor for NB patients (). Immunofluorescence staining, with specific antibody anti-CHL1 (Abcam Inc.), in the patient's tumor revealed that CHL1 protein expression on tumor cells from the patient was weaker than positive control (HL-60 cell line) ().
Figure 2. Low CHL1 expression in NB is associated with a poor prognosis. Using the neuroblastoma SEQC patient data sets in the R2 Genomics Analysis and Visualization Platform (http://r2.amc.nl), patients were divided into high (blue) and low (red) CHL1 gene expression groups by median-centered Log2 ratios and overall survival curve was generated. Numbers of patients per group are shown between brackets.
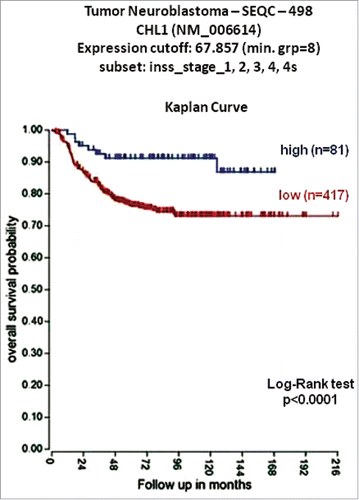
Figure 3. (A) Immunofluorescence staining revealed that CHL1 protein (green) expression on tumor cells from the patient was weaker than positive control. (B) HL-60 cell line (positive control) show strong staining. Nuclei are stained with DAPI (blue). Original magnification 100x.
Discussion
Congenital structural chromosomal aberrations helped the identification of causative TSGs because constitutional deletions or translocations are often the first indicators of the presence of TSGs.Citation4 Cytogenetic and molecular analysis of NB have identified several consistent deletions which may contain one or more TSGs. Furthermore, the numerous chromosome regions involved in constitutional anomaliesCitation7-20 strongly suggest that different pathways may lead to NB. These considerations indicate that the distribution of chromosome constitutional anomalies is not random, and some of them may represent a true predisposition for NB development.
Germline chromosomal abnormalities are rarely observed in children with NB,Citation7-20 but constitutional 1p36 deletions have helped to refine the putative 1p NB suppressor locus.Citation8,10,16,17 There are reports of a constitutional 11q deletion in NB patients,Citation9,13,14,19 although no major NB predisposition gene has been identified in this region.Citation6 Furthermore, the 2p chromosomal region is of primary importance in the NB development. In fact, germinal activating mutations in ALK, an oncogene mapping at chromosome 2p23, have been identified as responsible for most of the familial NB cases, while ALK somatic mutations are present in 8% of sporadic tumors.Citation21 The MYCN gene, whose amplification represents the major unfavorable prognostic factor in NB,Citation4 maps in 2p24.3. However, no clear correlation between constitutional rearrangements involving chromosome 2p and the role of ALK and/or MYCN in NB pathogenesis has been established.Citation18-20 Abnormalities of chromosome 3p in NB tumors have predominantly been reported as deletions.Citation5,6,28 Finally, the constitutional inv(3)(p23p26) seen in some metaphase cells from monozygotic twins with NB might be of some significance.Citation11
Array-CGH has led to the identification of the new 3p terminal deletion syndrome.Citation5 This emerging syndrome is a rare contiguous gene syndrome caused by 3p25-pter deletions variable in size and mostly occurring de novo. Also the phenotype varies from severe to normal, and chromosomal non-penetrance and gene modification have been proposed as possible explanations for the phenotypic variability.Citation22 To our knowledge, 4 familial cases, well documented phenotypically, have been reported until now. Eight more cases have been reported, but the phenotypic features have not been fully described. Tumor predisposition is not part of the 3p deletion syndrome phenotype.
Here, we report a constitutional 3p26.3 terminal microdeletion in a 17-years-old male with localized NB. The deletion breakpoint disrupted the CNTN4 gene and the neighboring CNTN6 and CHL1 genes were hemizygously deleted, each of these genes encode neuronal cell adhesion molecules. The finding of disruption of CNTN4 gene is of particular interest as this gene encodes a neuronal adhesion molecule that is a member of the immunoglobulin (Ig) superfamily of genes.Citation23 Similar to other members of the contactin family, full-length CNTN4 consists of 6 immunoglobulin domains and 4 fibronectin III (FNIII) domains and is anchored to the cell membrane via a glycosylphosphatidyl-inositol (GPI) domain. This family of proteins have been shown to be involved in axon growth, guidance, and fasciculation, and have been proposed to have a role in synaptic plasticity.Citation24 A specific role for CNTN4 in neuronal development has not yet been clarified. However, knockouts of homologous neuronal adhesion molecules in mouse have resulted in viable mutants demonstrating morphological, neurologic, and behavioral abnormalities.Citation25 The 3p telomeric region to the disrupted CNTN4 gene contains CNTN6 and CHL1, both neuronal cell adhesion molecules that also contain Ig and FNIII domains. CNTN6 gene codes for neural cell adhesion proteins that promote neurite outgrowth and synaptogenesis.Citation26 The CHL1 gene is involved in general cognitive activities and some neurologic diseases.Citation27 Recently, CHL1 was silenced to facilitate in situ tumor growth as a putative tumor suppressor.Citation28 Thus these reports suggest that CHL1 is involved in carcinogenesis, not only in neuronal activities. Moreover a frequent decrease of expression of CHL1 was observed in breast, colon, rectum, thyroid, kidney and small intestine cancers, indicating that CHL1 may act as a putative tumor suppressor gene.
The index patient showed apparently normal phenotype without neurocognitive defects or symptoms indicative of 3p deletion syndrome. Of note, a patient with nonspecific mental retardation and a balanced translocation disrupting CHL1 was described elsewhere.Citation27 This patient was not found to have any other physical abnormalities associated with 3p deletion syndrome. In the single reported case of a translocation disrupting CHL1, the patient did not show dysmorphic features consistent with 3p deletion syndrome.Citation27
The patient's tumor showed an identical 3p26.3 deletion and there was no evidence for a homozygous deletion. The maternal constitutional DNA showed no copy number aberrations, paternal sample was not available for testing. The database search for all patients with congenital structural chromosomal aberrations who had NB did not identify any patient with deletions involving chromosome 3p26.3 making our observation the first report of a constitutional 3p26.3 deletion in a NB patient. Since larger deletions of 3p, indicative of the presence of one or more TSGs in this region, occur frequently in NB, our results pave the way to the identification of one putative tumor suppressor genes mapping in 3p26.3. So far, loss-of-function mutations of CHL1, CNTN6 and CNTN4 genes have been reported in a next generation sequencing study in several primary NB tumors.Citation6 Low expression of CHL1 was significantly associated with reduced overall survival rate in 417 NB patient, and low expression of CHL1 protein was observed in the patient's tumor cells by immunofluorescence staining. These results suggested that CHL1 gene expression might contribute to the poor prognosis of NB tumors with 3p deletions. Further studies are necessary to established if 3p26.3 is a NB predisposition locus and provides unambiguous mapping data for future TSGs identification studies.
Disclosure of potential conflicts of interests
No potential conflicts of interests were disclosed.
Acknowledgments
We are grateful to the patient and his family members for giving the consent to publish clinical data. Dr. Martina Morini and Dr. Raffaella Defferrari are fellows of the Fondazione Italiana Neuroblastoma.
Funding
This work was supported by Cinque per mille dell'IRPEF Finanziamento della ricerca sanitaria; Finanziamento Ricerca Corrente, Ministero della Salute; Fondazione Maria Piaggio Casarsa; Associazione Italiana per la Ricerca sul Cancro (AIRC); and Fondazione Italiana Neuroblastoma.
References
- Cheung NK, Dyer MA. Neuroblastoma: developmental biology, cancer genomics and immunotherapy. Nat Rev Cancer 2013; 13:397-11; PMID:23702928; https://doi.org/10.1038/nrc3526
- Mossé YP, Deyell RJ, Berthold F, Nagakawara A, Ambros PF, Monclair T, Cohn SL, Pearson AD, London WB, Matthay KK. Neuroblastoma in older children, adolescents and young adults: a report from the International Neuroblastoma Risk Group project. Pediatr Blood Cancer 2014; 61:627-35; PMID:24038992; https://doi.org/10.1002/pbc.24777
- Ejeskar K, Aburatani H, Abrahamsson J, P Kogner P, Martinsson T. Loss of heterozygosity of 3p markers in neuroblastoma tumours implicate a tumour-suppressor locus distal to the FHIT gene. Br J Cancer 1998; 77:1787-91; PMID:9667647; https://doi.org/10.1038/bjc.1998.297
- van Mater D, Knelson EH, Kaiser-Rogers KA, Armstrong MB. Neuroblastoma in a pediatric patient with a microduplication of 2p involving the MYCN locus. Am J Med Genet 2013; 161:605-10; PMID:23401364; https://doi.org/10.1002/ajmg.a.35766
- Tassano E, Biancheri R, Denegri L, Porta S, Novara F, Zuffardi O, Gimelli G, Cuoco C. Heterozygous deletion of CHL1 gene: detailed array-CGH and clinical characterization of a new case and review of the literature. Eur J Med Genet 2014; 57:626-9; PMID:25451713; https://doi.org/10.1016/j.ejmg.2014.09.007
- Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, Carter SL, Cibulskis K, Hanna M, Kiezun , et al. The genetic landscape of high-risk neuroblastoma. Nat Genet 2013; 45:279-84; PMID:23334666; https://doi.org/10.1038/ng.2529
- Laureys G, Speleman F, Opdenakker G, Benoit Y, Leroy J. Constitutional translocation t(1;17)(p36;q12-21) in a patient with neuroblastoma. Genes Chromosomes Cancer 1990; 2:252-4; PMID:2078517; https://doi.org/10.1002/gcc.2870020315
- Biegel JA, White PS, Marshall HN, Fujimori M, Zackai EH, Scher CD, Brodeur GM, Emanuel BS. Constitutional 1p36 deletion in a child with neuroblastoma. Am J Hum Genet 1993; 52:176-82; PMID:8434586
- Koiffmann CP, Gonzalez CH, Vianna-Morgante AM, Kim CA, Odone-Filho V, Wajntal A. Neuroblastoma in a boy with MCA/MR syndrome, deletion 11q, and duplication 12q. Am J Med Genet 1995; 58:46-9; PMID:7573155; https://doi.org/10.1002/ajmg.1320580110
- Roberts T, Chernova O, Cowell JK. NB4S, a member of the TBC1 domain family of genes, is truncated as a result of a constitutional t(1;10)(p22;q21) chromosome translocation in a patient with stage 4S neuroblastoma. Hum Mol Genet 1998; 7:1169-78; PMID:9618176; https://doi.org/10.1093/hmg/7.7.1169
- Anderson J, Kempski H, Hill L, Rampling D, Gordon T, Michalski A. Neuroblastoma in monozygotic twins: a case of probable twin-to-twin metastasis. Br J Cancer 2001; 85:493-6; PMID:11506485; https://doi.org/10.1054/bjoc.2001.1979
- Yuksel A, Seven M, Karaman B, Yilmaz S, Deviren A, Hacihanefioglu S, Basaran S. Neuroblastoma in a dysmorphic girl with a partial duplication of 2p caused by an unbalanced translocation. Clin Dysmorphol 2002; 11:39-42; PMID:11822704; https://doi.org/10.1097/00019605-200201000-00008
- Satgé D, Moore SW, Stiller CA, Niggli FK, Pritchard-Jones K, Bown N, Bénard J, Plantaz D. Abnormal constitutional karyotypes in patients with neuroblastoma: a report of four new cases and review of 47 others in the literature. Cancer Genet Cytogenet 2003; 147:89-98; PMID:14623457; https://doi.org/10.1016/S0165-4608(03)00203-6
- Mossé Y, Greshock J, King A, Khazi D, Weber BL, Maris JM. Identification and high-resolution mapping of a constitutional 11q deletion in an infant with multifocal neuroblastoma. Lancet Oncol 2003; 4:769-71; PMID:14662434; https://doi.org/10.1016/S1470-2045(03)01283-X
- Dowa Y, Yamamoto T, Abe Y, Kobayashi M, Hoshino R, Tanaka K, Aida N, Take H, Kato K, Tanaka Y, et al. Congenital neuroblastoma in a patient with partial trisomy of 2p. J Pediatr Hematol Oncol 2006; 28:379-82; PMID:16794507; https://doi.org/10.1097/00043426-200606000-00011
- Vandepoele K, Andries V, Van Roy N, Staes K, Vandesompele J, Laureys G, De Smet E, Berx G, Speleman F, van Roy F. A Constitutional Translocation t(1;17)(p36.2;q11.2) in a Neuroblastoma Patient Disrupts the Human NBPF1 and ACCN1 Genes. PlosOne 2008; 3:5 e2207; PMID:18493581; https://doi.org/10.1371/journal.pone.0002207
- Bertrand I, Le Cunff M, Boceno M, Boisseau P, Thomas C, Rival JM, David A, Le Caignec C. Complex constitutional subtelomeric 1p36.3 deletion/duplication in a mentally retarded child with neonatal neuroblastoma. Eur J Med Genet 2008; 51:679e684; PMID:18672103; https://doi.org/10.1016/j.ejmg.2008.06.004
- Lipska BS, Koczkowska M, Wierzba J, Ploszynska A, Iliszko M, Izycka-Swieszewska E, Adamkiewicz-Drozynska E, Limon J. On the significance of germline cytogenetic rearrangements at MYCN locus in neuroblastoma. Mol Cytogenetics 2013; 6:43; PMID:24131700; https://doi.org/10.1186/1755-8166-6-43
- Passariello A, De Brasi D, Defferrari R, Genesio R, Tufano M, Mazzocco K, Capasso M, Migliorati R, Martinsson T, Siani P, et al. Constitutional 11q14-q22 chromosome deletion syndrome in a child with neuroblastoma MYCN single copy. Eur J Med Genet 2013; 56:626-34; PMID:24035971; https://doi.org/10.1016/j.ejmg.2013.08.005
- van Mater D, Knelson EH, Kaiser-Rogers KA, Armstrong MB. Neuroblastoma in a pediatric patient with a microduplication of 2p in1volving the MYCN locus. Am J Med Genet 2013; 161:605-10; PMID:23401364; https://doi.org/10.1002/ajmg.a.35766
- Janoueix-Lerosey I, Lequin D, Brugieres L, Ribeiro A, de Pontual L, Combaret V, Raynal V, Puisieux A, Schleiermacher G, Pierron G, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 2008; 455:967-70; PMID:18923523; https://doi.org/10.1038/nature07398
- Girirajan S, Rosenfeld JA, Coe BP, Parikh S, Friedman N, Goldstein A, Filipink RA, McConnell JS, Angle B, Meschino WS, et al. Phenotypic heterogeneity of genomic disorders and rare copy-number variants. N Engl J Med 2012; 367:1321-31; PMID:22970919; https://doi.org/10.1056/NEJMoa1200395
- Hansford LM, Smith SA, Haber M, Norris MD, Cheung B, Marshall GM. Cloning and characterization of the human neural cell adhesion molecule, CNTN4 (alias BIG-2). Cytogenet Genome Res 2003; 101:17-23; PMID:14571131; https://doi.org/10.1159/000073412
- Yoshihara Y, Kawasaki M, Tamada A, Nagata S, Kagamiyama H, Mori K. Overlapping and differential expression of BIG-2, BIG-1, TAG-1, and F3: four members of an axon associated cell adhesion molecule subgroup of the immunoglobulin superfamily. J Neurobiol 1995; 28:51-69; PMID:8586965; https://doi.org/10.1002/neu.480280106
- Montag-Sallaz M, Schachner M, Montag D. Misguided axonal projections, neural cell adhesion molecule 180 mRNA upregulation, and altered behavior in mice deficient for the close homolog of L1. Mol Cell Biol 2002; 22:7967-81; PMID:12391163; https://doi.org/10.1128/MCB.22.22.7967-7981.2002
- Mercati O, Danckaert A, Andre-Leroux G, Bellinzoni M, Gouder L, Watanabe K, et al. Contactin 4, -5 and -6 differentially regulate neuritogenesis while they display identical PTPRG binding sites. Biol Open 2013; 2:324-34; PMID:23519440; https://doi.org/10.1242/bio.20133343
- Frints SG, Marynen P, Hartmann D, Fryns JP, Steyaert J, Schachner M, Rolf B, Craessaerts K, Snellinx A, Hollanders K, D'Hooge R, De Deyn PP, Froyen G. CALL interrupted in a patient with non-specific mental retardation: gene dosage-dependent alteration of murine brain development and behavior. Hum Mol Genet 2003; 12:1463-74. PMID:12812975; PMID:12812975; https://doi.org/10.1093/hmg/ddg165
- He LH, Ma Q, Shi YH, Ge J, Zhao HM, Li SF, Tong ZS. CHL1 is involved in human breast tumorigenesis and progression. Biochem Biophys Res Commun 2013; 438:433-8; PMID:23906755; https://doi.org/10.1016/j.bbrc.2013.07.093