
ABSTRACT
Pharmacologic inhibition of the cytotoxic T lymphocyte antigen 4 (CTLA4) and the programmed death receptor-1 (PD1) has resulted in unprecedented durable responses in metastatic melanoma. However, resistance to immunotherapy remains a major challenge. Effective immune surveillance against melanoma requires 4 essential steps: activation of the T lymphocytes, homing of the activated T lymphocytes to the melanoma microenvironment, identification and episode of melanoma cells by activated T lymphocytes, and the sensitivity of melanoma cells to apoptosis. At each of these steps, there are multiple factors that may interfere with the immune surveillance machinery, thus allowing melanoma cells to escape immune attack and develop resistance to immunotherapy. We provide a comprehensive review of the complex immune surveillance mechanisms at play in melanoma, and a detailed discussion of how these mechanisms may allow for the development of intrinsic or acquired resistance to immunotherapeutic modalities, and potential avenues for overcoming this resistance.
Introduction
The concept of immunotherapy in metastatic melanoma is not new. Scientists and physicians used vaccines, bacterial toxins, and T cell growth factors to stimulate the immune system to fight cancer.Citation1 The success rate has been variable but these different techniques have helped 5 to 10 percent of patients with metastatic melanoma achieve long-term remission. The recent introduction of checkpoint inhibitors has revolutionized the treatment of metastatic melanoma.Citation1
Unquestionably, recent advances in melanoma drugs that target the host immune response have led to growing interest in the role of immunotherapy. Ipilimumab, pembrolizumab, and nivolumab, are immune checkpoint inhibitors approved by the FDA to treat metastatic melanoma.Citation1,2 Ipilimumab is a fully humanized monoclonal antibody directed against CTLA-4. Patients with metastatic melanoma treated with this drug have a 2 y overall survival rate of 24%.Citation2,3 The 10-year overall survival rate of patients treated with ipilimumab is approximately 22% in a pooled analysis of overall survival data from 12 clinical trials, suggesting a sustained response rate.Citation4 Pembrolizumab is a selective anti-PD-1 humanized monoclonal antibody. It received an accelerated FDA approval based on the results of a phase I study in which it was used in the setting of ipilimumab-resistant melanoma with a median follow-up of 18 months.Citation3 The response rate of 34% was maintained in 81% of patients who responded to therapy with a median overall survival was 25.9 months.Citation3 Nivolumab is another selective anti-PD-1 humanized monoclonal antibody. In a phase III trial, the objective response to nivolumab was significantly better than chemotherapy (32% vs 10%) in patients with metastatic melanoma who progressed after ipilimumab and BRAF inhibitor therapy (if BRAF was mutated).Citation5 The combination of ipilimumab and nivolumab significantly improved progression free survival compared with ipilimumab alone [HR 0.40 (95% CI: 0.22, 0.71); p-value < 0.002] in a subsequent study.Citation6
Unfortunately, not all patients with metastatic melanoma respond to immunotherapy. Many patients fail to respond to immunotherapy (i.e. primary resistant) while others develop initial responses which can be later lost due to development of acquired resistance (i.e., secondary resistant) leading to disease relapse.Citation7,8 Primary resistance to anti-PD1 therapies is common, affecting up to 60% of patients in some cancer types.Citation9 In primary resistance there are intrinsic- host, tumor, or microenvironment-related factors that prevent the mounting of an effective T cell response. We speculate that one or more of these factors will prevent patients with metastatic melanoma from responding to immunotherapy upfront. In secondary resistance, patients initially respond to immunotherapy; however, this response is lost due to acquired tumor or microenvironment-related changes. We speculate that the T cell activation process begins intact in cases of secondary resistance; however, acquired changes at the level of the melanoma cell (epigenetic or oncogenic changes) or its microenvironment result in the formation of a new clone that is resistant to immunotherapy. It is essential to have an awareness of the different potential mechanisms of resistance so that we can identify strategies to target these tumors and enhance the efficacy of immunotherapy as an effective treatment modality in metastatic melanoma. This article will review the concepts of immune surveillance in melanoma and potential mechanisms adopted by melanoma tumor cells to escape it.
Immune surveillance in melanoma
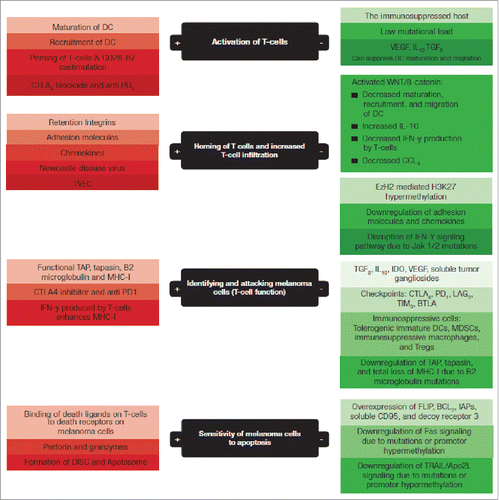
In order for the immune system to mount an effective immune response against melanoma several steps must occur (). These include 1) activation of T cells, 2) homing of the activated T cells to the melanoma microenvironment, 3) identification and episode of melanoma cells by activated T lymphocytes, and 4) sensitivity of melanoma cells to programmed cell death (apoptosis).
Figure 1. The 4 steps of immune surveillance in black boxes; factors that enhance the efficacy and contribute to an effective immune surveillance in green boxes; factors that impair an effective immune surveillance and help melanoma escape immune attack in red boxes. DC: dendritic cells, TVEC: Talimogene laherparepvec, IFN-y: interferon gamma, CCL4: chemokine CCL4, TAP: Transporter associated with antigen processing, PD1: programmed death receptor 1, CTLA4: cytotoxic lymphocyte antigen 4, LAG3: lymphocyte-activation gene-3, TIM3: T-cell immunoglobulin mucin-3, BTLA: B- and T-lymphocyte attenuator, FLIP: FLICE-inhibitory proteins, IAPs: inhibitor of apoptosis proteins, CD95: death receptor also known as Fas, TRAIL/Apo2L: TNF-related apoptosis-inducing ligand, DISC: death-inducing signaling complex.
Activation of the T cells
There are 2 types of T cells involved in adaptive immunity; CD4+ T helper and cytotoxic CD8+ T cells. The CD4+ T helper cell type 1 (Th1) cells provide “help” for the activation of CD8+ T cells while CD4+ Th2 cells stimulate a humoral immune response and suppress the development of a Th1 response. CD4+ T cells can also display cytotoxic activity in some situations.Citation10 Both cell types are derived from the thymocytes (bone marrow-derived progenitors) in the thymus where they undergo a process of rearrangement and selection.Citation11 This process is important in selecting appropriate T cell subsets that can detect the major histocompatibility complex (MHC) and react to non-self0peptides rather than self-peptides.Citation11
The process of T cell activation starts with the recruitment of dendritic cells (DCs), the most specialized of the antigen presenting cells (APCs), to the tumor microenvironment where they initiate phagocytosis (), a process by which tumor antigens are processed into peptides and loaded onto the MHC.Citation12 Danger signals, originating from the tumor microenvironment, stimulate Toll-like receptors (TLRs) on DCs, leading to their maturation and migration to regional lymph nodes, where they present the MHC-bound tumor-specific antigens to the T-cell receptors on lymphocytes –– a process called T cell priming.Citation13 The binding of the CD28 receptors on the T cell with B7 molecules on the APCs generates a stimulatory signal from the APCs assuring the T cells that the presented tumor antigen is non-self and an appropriate target for destruction. If co-stimulation does not occur, the primed T cells will become hypoactive (i.e., T cell anergy) ().
Figure 2. Phagocytosis of tumor antigens by dendritic cells (DCs) and the antigen-presenting machinery that helps load the tumor peptides onto MHC-II molecules on the DC surface; 1) Endocytosis of tumor specific antigen (TSA), 2) Formation of endocytic vesicle that transports the tumor specific antigens into the cytosol, 3) the tumor specific antigens are degraded in the cytosol by proteasomes that degrades the proteins into peptides, 4) The tumor peptides are transported to the endoplasmic reticulum (ER) by transporter associated with antigen processing (TAP), 5) In the ER the tumor peptides are loaded on the MHC I or MHC II molecules to form complexes that (6) that moves through the Golgi apparatus and got displayed on the dendritic cell wall where they interact with the T cell receptors.
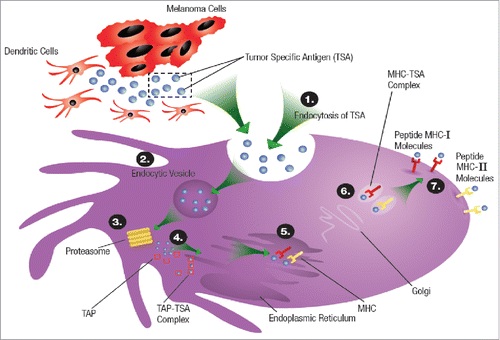
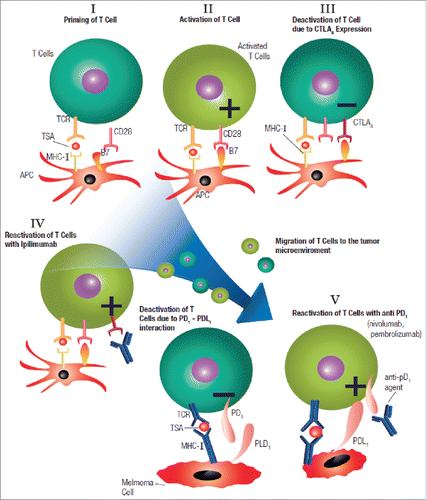
Figure 3. T cell activation and the mechanism of actions of both PD1 and CTLA4 inhibitors; PD1: programmed death 1 receptor, CTLA4: cytotoxic lymphocyte antigen-4, MHC: major histocompatibility complex, TCR: T cell receptor, TSA: tumor specific antigen, APS: antigen presenting cell, PDL1: programmed death ligand-1.
Homing of the activated T cells
Once activated, the T cells are now prepared to recognize and “licensed” to attack melanoma cells, but they first need to home to the melanoma microenvironment. This “homing” mechanism by which activated T-cells return to the tumor microenvironment, may be variably successful. We know that patients with high CD8+ T cell infiltration within their melanoma have the best clinical outcomes,Citation14 however, diffuse CD8+ T cell infiltration into the melanoma microenvironment is observed in less than 10% of melanoma metastases.Citation14 Several mechanisms facilitate homing of activated T-cells to the tumor microenvironment. The expression of cell adhesion molecules by the activated T cells and both adhesion molecules and chemokines by the vascular endothelium will enable homing of the activated T cells to the tumor microenvironment.Citation15,16 Murine models of vaccination for melanoma have shown that homing of the activated T cells appears to be integrin-dependent.Citation17,18 In humans, chemokine immune signatures of metastatic melanoma have correlated with CD8+ T cell infiltration.Citation15 Specifically, Salerno et al. revealed a significant role for retention integrins α1β1, α2β1, and αEβ7 in maintaining intratumoral T cells. A site-specific homing to bowel metastases was mediated by CCR9 and increased expression of retention integrins.Citation19 Hence, successful T cell infiltration is associated with immune signatures that include many genes associated with T cell homing such as adhesion molecules, retention integrins, and the chemokine ligands for CCR5, CCR9, and CXCR3.Citation20,21 These immune signatures may identify patients who are likely to respond to immunotherapy.
Identification and episode of melanoma cells
Once the activated T cells are in the tumor microenvironment, they will scan the area searching for melanoma cells that express and share the same tumor-specific antigens that initiated the T cells activation. The tumor specific-antigen processing machinery includes molecules such as the transporter protein TAP, the linker protein tapasin, and β2-microglobulin. These molecules ensure that the tumor peptides are processed and loaded onto the MHC-I on the melanoma cell surface.Citation22,23 Once the activated T cells are in contact with the tumor antigen-MHC I complex that is expressed on melanoma cells, they initiate a cytotoxic signal, leading to destruction and lysis of the tumor cells.Citation22,23
Sensitivity of melanoma cells to programmed cell death
There are 2 major mechanisms by which T cells and natural killer (NK) cells kill melanoma: the apoptosis pathway and the granule exocytosis pathway.Citation24
Apoptosis
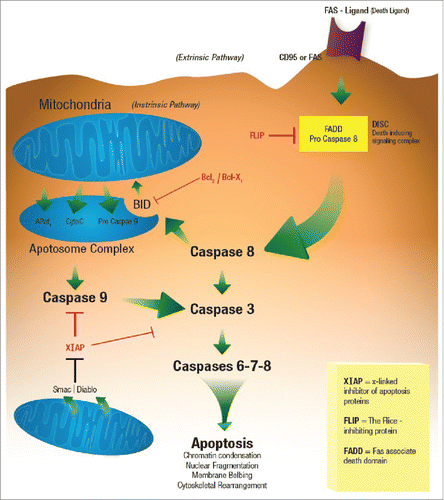
Cytotoxic ligands, which generally belong to the tumor necrosis factor (TNF) family of ligands, include tumor necrosis factor α (TNF-α), Fas ligand (FasL or CD95L) and TRAIL/Apo2L. These ligands transmit a death signal upon binding to their cognate receptors.Citation25 The binding of the death ligand FasL (CD95L on lymphocytes) to the death receptor Fas (CD95 on melanoma cells) attracts adaptor proteins to the intracellular domain of the death receptor, which in turn recruits procaspase-8 ().Citation26-29 The resulting protein complex is called death-inducing signaling complex (DISC), in which procaspase-8 gets activated to “the initiator” caspase-8. In some cells, the amount of active caspase-8 is sufficient to initiate apoptosis directly, while in others the amount is too small and requires mitochondrial activation to amplify the apoptotic signal.Citation30 The Bcl−2 family member Bid, which is cleaved by active caspase-8 and translocated to the mitochondria, mediates mitochondrial activation. After activation, mitochondria release cytochrome c, an apoptosis-inducing factor, and other apoptogenic factors into the cytosol.Citation31,32 Cytochrome c, Apaf-1, adenosine 5′-triphosphate, and procaspase-9 form a complex called the apoptosome, which activates caspase-9. Activated “initiator” caspases (caspase-8 and −9) activate “executioner” caspases (caspase-3, −6, and −7), which cleave cellular substrates, known as the “death substrates,” leading to the biochemical and morphological changes characteristic of apoptosis.Citation31,32 ()
Figure 4. Apoptosis.
The granule exocytosis pathway
Perforin and granzymes are located within cytotoxic lymphocyte (CTL) granules. In the presence of calcium, perforin polymerization induces changes in the melanoma cell membrane which allow the passing of granzymes from the CTL into the melanoma cells.Citation33,34 Granzymes are neutral serine proteases that can activate caspases in the target cell.Citation35,36 In addition, granzyme B may directly cleave the Bcl−2 family member Bid, initiating the mitochondrial death pathway as described above.Citation37
Mechanisms of immune escape in melanoma
As we have now described, there are 4 complex steps that are required to ensure effective immune surveillance against melanoma (). Moving forward, we discuss factors that may interfere with each step of this immune surveillance and may thus contribute to melanoma escaping from the immune attack.
Factors that interfere with T cell activation
The immunosuppressed host
Recent studies have documented poor survival of melanoma among immunocompromised individuals, such as those with HIV or those who have received an organ transplant, presumably because these individuals are unable to elicit sufficient immune anti-tumor responses. However, further investigation is needed as there is a substantial lack of immunotherapy-outcome data among these groups of patients as they are almost always excluded from clinical trials of immunotherapy in metastatic melanoma.Citation38
Elderly individuals are another population with decreased diversity of the total T cell subset pool, and in theory, might be less able to mount an effective immune response against melanoma.Citation39,40 However, this may not be an accurate assumption as many of our elderly patients with metastatic melanoma respond to immunotherapy as well, if not better, than younger patients and with fewer adverse events
Mutational load
Genomic and RNA-based studies in melanoma suggest that tumor-specific mutational load and neoantigen signature as well as cytolytic activity are significantly associated with clinical benefit and increased overall survival.Citation41 Tumors with a high mutational load respond better to immunotherapy because these mutations give rise to a variety of neoantigens that are truly non-self and can more easily be recognized by the immune system.Citation42 Mutations or translocations of normal cellular genes in a tumor (i.e., melanoma) generate tumor-specific antigens that are identified by T cells as non-self-antigens.Citation10,43,44 Tumor-associated antigens, on the other hand, are expressed by tumor cells as well as normal cells.Citation10 Differentiation antigens (e.g. MART-1/Melan-A, tyrosinase, gp100) are examples of tumor-associated antigens present on both normal/benign melanocytes and melanoma cells, against which reactive T lymphocytes have been identified.Citation45 Melanoma, lung cancer, and renal cell carcinoma are examples of tumors with high mutational loads that derive benefit from checkpoint inhibitors.Citation46,47 Conversely, tumors with low mutational loads such as pediatric tumors are more likely to evade immune surveillance.Citation48
As discussed earlier, once the T cells are activated, they home to the tumor microenvironment and eliminate melanoma cells, which share the same tumor-specific antigens that caused the T cell activation. Melanoma recurrences seen in some patients after an initial response phase to immunotherapy could be related to the development of further mutations that result in the formation of new and/or the loss of original tumor-specific antigens to which the T cells were activated.Citation49-51 Thus acquiring new mutations help melanoma cells escape immune surveillance and could contribute to disease progression.Citation52,53
Maturation and migration of dendritic cells
Cytokines, present in the tumor microenvironment, may help melanoma cells evade immune surveillance.Citation8 The vascular endothelial growth factor (VEGF), a cytokine commonly produced by both tumor and associated immune cells, via stimulation of VEGF receptor 1 (VEGFR1), can suppress dendritic cells maturation and migration via a nuclear factor kB (NFкB)-dependent pathway.Citation8,54 Elevated tumor-derived transforming growth factor β (TGFβ) and IL-10 produced by immunosuppressive cells such as regulatory T-cells (Tregs) can suppress dendritic cells maturation and migration to lymph node basin by reducing expression of class II MHC and costimulatory machinery. Moreover, VEGF and IL-10 can affect antigen presentation and prevents T-cell activation.Citation55
The activated WNT/β-catenin pathway in melanoma cells impairs maturation of DCs at least in part through increased production of IL-10, and it inhibited IFN-α production by melanoma-specific CTLs.Citation56 This activated pathway also results in failure of CD103+ DC recruitment due to defective CCL4 chemokine production.Citation57 Failure of DCs to be recruited and migrate to lymph nodes prevents T cell activation and hence is one method that can lead to melanoma immune escape. Thus, patients with metastatic melanoma and an activated WNT/β-catenin pathway may represent one group of patients who will not respond up front to immunotherapy.
Factors that interfere with homing of activated T cells
Oncogenic and epigenetic changes
Chemokines play an important role in the homing of activated T cells to the tumor microenvironment. Specific oncogenic or epigenetic alterations may result in defective production of chemokines needed for T cell homing.Citation50 IFN-y increases the production of important chemokines CXCL9, 10, and 11 important for T cell migration and infiltration into the tumor microenvironment.Citation58 The activated WNT/β-catenin pathway in melanoma cells impairs homing of T cells to the tumor microenvironment by inhibiting IFN-y production by melanoma-specific CTLs.Citation56 Moreover, lack of interferon gamma signaling due to Jak1/2 loss of function mutations or perhaps epigenetic silencing of the Jak1/2, results in decreased production of chemokines required for T cell homing and may represent a mechanism of resistant to immune surveillance.Citation58-60 It is entirely possible and even likely that other oncogenic pathways in melanoma also mediate immune suppression.Citation61
Epigenetic alterations including DNA methylation patterns, post-translational modification of histones, and chromatin remodeling play a large role in melanoma progression and metastasis.Citation62,63 There is growing interest in epigenetic changes and their role in the formation of resistance to immunotherapy in melanoma.Citation63 EZH2 is a histone modifier that functions as the catalytic component of the Polycomb Repressive Complex 2 (PRC2).Citation62,63 EZH2 mediated H3K27 hypermethylation has been shown to repress the production of Th1- type chemokines CXCL9 and CXCL10 in the tumor resulting in low tumor infiltrating CD8+ cells and poor patient outcomes in ovarian cancer, but this deserves further research in melanoma.Citation64 EZH2 mediated H3K27 hypermethylation may also silence important immune-protective genes allowing melanoma cells to evade immune surveillance.Citation63 EZH2 inhibitors are already entering clinical trials in lymphoma and sarcoma, thus future studies should look into combining an EZH2 inhibitor with a checkpoint inhibitor in melanoma tumor models to overcome a range of resistant mechanisms and enhance the clinical efficacy of immunotherapy.Citation63
Lack of T cell infiltration
IHC-based studies also support the notion that CD8+, CD4+, PD-1+, and programmed death-ligand 1–positive (PD-L1+) cell densities in pretreatment biopsies can predict response to therapy.Citation14,41,65 The absolute numbers of CD8+ T-cells infused during adoptive cell therapy for patients with metastatic melanoma strongly correlates with response to treatment, which suggests an important role for MHC class I restricted CTL-mediated tumor killing.Citation66,67 Oncogenic and epigenetic changes, as discussed earlier, may alter the function of certain immune-protective genes, such as with defective chemokine production, leading to a decreased homing of T cells to the tumor microenvironment and a decreased response to immunotherapy.Citation68 Down-regulation of adhesion molecules in melanoma may also inhibit homing of activated T cells to the melanoma microenvironment leading to continued tumor progression despite appropriate activation of T cells.Citation43,69
Intralesional injection of the oncolytic Newcastle disease virus (NDV) resulted in increased T cell infiltration locally at the injection site and distally at distant sites in a murine B16 melanoma model. NDV enhanced the efficacy of cytotoxic T lymphocyte 4 (CTLA4) inhibitor, perhaps due to the increased presence of tumor infiltrating lymphocytes, and resulted in rejection of pre-established distant tumors. These observations, though limited to a single model, provide a rationale for exploring the potential of oncolytic viruses such as NDV to enhance T-cell homing to the microenvironment, potentially in combination with immune checkpoint antibodies.Citation70
Factors that interfere with the function of activated T cells
The checkpoints
T cells possess inhibitory molecules, namely CTLA4 and the programmed T cell death 1 (PD1) receptor that, when coupled with their ligand, negatively regulate T cell activation and help prevent autoimmunity.Citation71 Normally, CTLA4 expressed on T cells competes with the CD28 receptor on the T cell, for binding to B7 molecules on APCs, causing T cell anergy by interruption of the co-stimulatory signal.Citation72 CTLA4 expression on T cells is upregulated in melanoma; hence the T cells are primed but turned off. Inhibiting CTLA-4 by targeted ipilimumab and thereby interrupting the negative inhibitory signal helps restore T cell function against melanoma. In addition, CTLA4 is expressed on regulatory T-cells (Tregs), and the immune effects observed with anti-CTLA4 antibodies may be a combination of enhancing effector T-cell function while blocking Tregs activity.Citation73 CTLA4 is also expressed on tumor cell linesCitation74 and in human melanoma.Citation75 Blockade of CTLA4 in vitro induces apoptosis of melanoma cells, indicating that CTLA4 might have additional, non-immune functions as well. CTLA-4 blockade enhances T cell responses, including production of IFN-y.Citation76 Patient identified as non-responders to CTLA-4 blockade have tumors with genomic defects in IFN-y pathway genes. Mice bearing melanoma tumors with knockdown of IFN-y receptor (IFNGR1) have impaired tumor rejection upon anti-CTLA-4 therapy. Loss of the IFN-y signaling pathway is associated with primary resistance to anti CTLA-4. Loss of IFNGR1, IFNGR2, Jak2, and IRF1 genes would impair IFN-y signaling pathway.Citation59,76 Other genomic losses in these non-responders include downstream IFN-y pathway genes such as the family of IFIT1, IFIT2, and IFIT3.Citation76
Melanoma cells can also evade immune surveillance via upregulation of the programmed death-ligand 1 (PDL1) which binds to PD1 receptors on T cells, thereby suppressing their function.Citation1 Anti PD1 agents that block the interaction between the PD1 receptors on T cells and the PDL1 ligand on tumor cells can restore T cell function against melanoma as evidenced by the success of the targeted drugs pembroluzimab and nivolumab.Citation1
Likewise, after chronic exposure to tumor antigens, CD8+ T cells begin to further express immune inhibitory receptors beyond PD-1 such as lymphocyte-activation gene-3 (LAG3), T-cell immunoglobulin mucin-3 (TIM3), and B- and T-lymphocyte attenuator (BTLA); the exact mechanisms and roles of these receptors in melanoma are yet to be determined.Citation77
Another mechanism of melanoma immune evasion is the production of IFN-γ by the effector T cells. This results in induction of PD-L1 expression on tumor cells that, in turn, deactivate T cell function by binding to the PD1 receptors.Citation79 However, IFNγ has been shown to enhance the MHC-I antigen processing and presentation pathway.Citation59 Buferne et al. have showed that CD8+ T cells specific for tumor antigen/MHC-I were efficient at inducing regression of the MHC-I-deficient melanoma, provided that the T cells were endowed with properties permitting their migration into the tumor and their efficient production of IFNγ.Citation79 The amount of IFNγ produced ex vivo from T cells correlated with an increase in surface expression of MHC-I molecules by the melanoma cells. These CD8+ T cells expressed PD-1 and upregulated its PDL-1 ligand on melanoma cells within the tumor. Despite upregulation of this immunosuppressive pathway, efficient IFNγ production in the melanoma microenvironment was actually found to be associated with resistance of the CD8+ T cells to inhibition both by PD-1/PDL-1 engagement and by TGFβ1, 2 main immune regulatory mechanisms hampering the efficiency of immunotherapy in patients.Citation79
An adaptive immune resistant process occurs in response to interferon gamma (IFN-γ) signaling in which melanoma cells would reactively overexpress PDL1 that binds to PD1 receptors on cytotoxic T cells causing its deactivation and allowing melanoma cells to escape immune attack.Citation58 Lack of INF y signaling due to loss-of-function mutations in Jak1/2 has been linked to an acquired resistant to PD1 inhibitors in patients with metastatic melanoma.Citation58,80
Gene expression profiling (GEP), in longitudinal tumor biopsies, predicted response in patients with metastatic melanoma treated with sequential CTLA4 and PD-1 blockade.Citation41 While no significant GEP differences were found at pretreatment CTLA4 blockade, on-treatment CTLA4 blockade, and pretreatment PD-1 blockade, between responders and nonresponders; there was an early on treatment difference in samples of patients on anti–PD-1 therapy.Citation41 The latter showed 411 significantly differentially expressed genes (DEG) in responders (P < 0.05), mostly upregulated as compared with nonresponders, including cytolytic markers, HLA molecules, IFNγ pathway effectors, chemokines and select adhesion molecules.Citation41 Notably, vascular endothelial growth factor (VEGFA), was lower in responders than in nonresponders on PD-1 blockadeCitation41 suggesting a possible role of angiogenesis in resistant to immunotherapy and could potentially be a target of therapy.Citation81-83
The secretion of inhibitory molecules
Many types of molecules and cells in the melanoma microenvironment have been shown to suppress immune responses via cell-cell interactions and/or the production of immune suppressing molecules. Transforming growth factor β (TGF-β), interleukin-10 (IL-10) and enzyme indoleamine-2, 3-dioxygenase (IDO) may have a direct negative effect on the function of T-cells in the tumor microenvironment,Citation84-86 or an indirect one via the recruitment of different immunosuppressive cells that could be used by the tumor cells to evade immune surveillance. Tolerogenic immature DCs, myeloid derived-suppressor cells (MDSCs), immunosuppressive macrophages, regulatory B cells, or regulatory CD4+ T-cells are all examples of immunosuppressive cells that can be deregulated by melanoma to facilitate its progression and evasion of the immune surveillance.Citation87-89 Murine models have demonstrated that CD4+, CD25+, FOXP3+ Tregs hamper the efficacy of CTLs. Therefore, selective depletion of Tregs could be beneficial.Citation25 A recent study found that the T cell subset most induced by IL-2 treatment in melanoma patients is upregulation of Tregs expressing CD4, CD25, Foxp3 and the inducible T cell co-stimulator (ICOS). High levels of ICOS-expressing peripheral Tregs were a strong predictor of the lack of response to IL-2.Citation90 MDSCs have been implicated as an important component in suppression of immune responses in melanoma. A higher numbers of these cells in serum samples of patients with advanced melanoma is associated with worse prognoses.Citation91,92 Strategies targeting these cells are being developed to improve the efficiency of adoptive cell transfer therapy in melanoma.Citation93
The infiltrating T cells in the tumor microenvironment are highly regulated by the amino acid tryptophan levels. The enzyme indoleamine-2,3-dioxygenase (IDO), upregulated by CD8+ cells in the tumor microenvironment, converts tryptophan into the immunosuppressive molecule kynurenine.Citation94-96 Inhibitors of IDO may thus provide a targetable strategy for blocking this innate immunosuppression. INCB024360 and 1-methyl-D-tryptophan are in several clinical trials for different malignancies including metastatic melanoma.Citation61
TGF-β is a cytokine that affects proliferation, activation, and differentiation of cells of innate and adaptive immunity and thus inhibits the anti-tumor immune response. The vascular endothelial growth factor (VEGF)Citation97 inhibits the differentiation of progenitors into DCs. Prostaglandins,Citation98 interleukin-10,Citation99 and soluble tumor gangliosidesCitation100 are all immunosuppressive factors that may contribute to melanoma immune escape. Although the etiology is unclear, a subset of CD4+ T cells seems to suppress the T cell responses against melanoma.Citation101
Factors that interfere with the ability of activated T cells to identify melanoma cells
Defects in the melanoma-antigen processing machinery
In order for them to be recognized by the activated T cells, melanoma-specific antigens need to be loaded onto the MHC-I molecules. Defects in this antigen-processing machinery can thus lead to immune escape.Citation22,23 The transporter associated with antigen processing (TAP) and tapasin are 2 proteins involved in loading antigenic peptides onto MHC-I molecules.Citation102,103 By forming physical links between class I heavy chains and TAP molecules, tapasin plays an important role in the assembly of MHC class I molecules with peptides in the endoplasmic reticulum.Citation104 Tapasin downregulation has been significantly associated with melanoma progression, perhaps due to immune escape.Citation104 Chang et al. reported an irreversible tapasin mutation that was associated with loss of the HLA haplotype and selective epigenetic silencing of HLA-A3 gene responsiveness to IFN-y in a melanoma cell line obtained from an axillary lymph node metastasis removed from a patient with a progressive disease despite surgery, vaccination with C-Vax/BCG, and chemotherapy combined with high-dose IL2.Citation105
Total loss of MHC-I by melanoma cells results in their complete resistance to recognition and destruction by CTLs.Citation106-108 The mechanism underlying this abnormal tumor phenotype has frequently been the loss of the β2-microglobulin (β2m), which is required for the formation of HLA class I H chain-β2m-peptide complexes and their transport to the cell surface.Citation109 Two genetic events – namely, mutations of one copy of the β2m gene and loss of the other copy [i.e., loss of heterozygosity (LOH)] – have been found to underlie β2m loss in malignant cells, although the chronological sequence of the 2 events is not known.Citation22,102 It has been demonstrated that in melanoma patients undergoing immunotherapy, a lack of response to immunotherapy and generation of progressing metastases appears to be associated with immune selection of MHC-I negative melanoma cell variants.Citation110 Porgador et al described a very high prevalence (5 in 13 cases) of irreversible, complete loss of MHC-I expression in patients with metastatic melanoma who were treated with various immunotherapeutic regimens.Citation111 Although the melanoma cells in these 5 patients were very sensitive to autologous natural killer cells in vitro, clinical outcomes were very poor.Citation111 Studies have shown that loss of MHC-I expression may target melanoma cells for NK cell attack; however, the expression of MHC-I surrogates may help melanoma cells resist NK cell-mediated lysis.Citation112,113 Del Campo et al. revealed heterogeneous loss of β2m, caused by either mutation or loss of heterozygosity (LOH), in nests of a patient's primary melanoma; 10 months later the metastasis displayed homogenous loss of β2m- expression.Citation114 Reconstitution of β2m expression has also been shown to restore HLA class I antigen expression in the 5 distinct melanoma cell lines. Immunotherapy imposes a selective pressure, perhaps, leading to an increased survival advantage for melanoma cells lacking the antigen-presenting MHC-I molecule or the tumor-specific antigen that contains the immunogenic peptide epitope (or both).Citation22,23 This selective pressure has the propensity to result in the emergence of immunotherapy resistant clone.Citation105,115 Restoring β2m function and HLA class I expression should be considered therapeutic targets in the development of cancer immunotherapy protocols.Citation114,116,117
Finally, the interferon gamma signaling pathway plays an important role in the function of activated T cells. Upon recognizing their tumor specific antigens on melanoma cell surface, the cytotoxic CD8+ T cells secrets interferon gammaCitation58 which acts via the interferon gamma receptors (the Janus kinases Jak1/2 and the signal transducers and activators of transcription STAT) to stimulate the expression of genes that exhibit anti-tumor effects.Citation58 Interferon gamma increases the antigen presentation machinery by melanoma cells so tumor specific antigens can be easily be recognized by the cytotoxic T cells. Moreover, interferon gamma exhibits direct tumor growth arrest and enhance apoptosis.Citation58 Loss-of-function mutations in Jak1/2 or in β−2-microglobulin resulted in acquired resistance to PD1 blockade in patients with metastatic melanoma.Citation80
Epithelial Mesenchymal Transition
Over time, some melanoma cells switch phenotype from proliferative, or epithelial “E-like,” to more invasive, or mesenchymal “M-like,” state in a process similar to classical epithelial-mesenchymal transition (EMT).Citation118 In a panel of 57 melanoma cell lines derived from patient samples,Citation119 the expression levels of E-cadherin or N-cadherin characterized these cell lines as E-like or mesenchymal-like (M-like), respectively.Citation120 Gene expression profiling enabled a detailed molecular classification of both melanoma cell types.Citation121-123 The proliferative state is characterized by high expression of microphthalmia-associated transcription factor (MITF),Citation124 while the invasive state expresses low or no levels of MITF with increased expression of EMT-related transcription factors.Citation121,122 Expression of Melan-A, tyrosinase, and MITF are significantly downregulated in M-like, compared with E-like, melanoma cells.Citation120 As such, immune responses induced to tumor-associated antigens such as Melan-A and tyrosinase will potentially only be effective against the E-like melanoma cells. EMT like phenotypic switch in melanoma was confirmed as a method of escape from adoptive T cell therapy with T-lymphocytes targeting Melan-A.Citation125 Of interest, there was an upregulation of the SPANX family of cancer testis antigens (CTAgs), which has demonstrated immunogenicityCitation126 in the M-like melanoma cells.Citation118,120 Moreover, high levels of SPANX family antigen expression was found in more aggressive melanoma cell lines.Citation127 Hence, it might be feasible to target the M-type melanoma cells with adoptive cell therapy using T-lymphocytes specific for SPANX family antigens.
Proinflammatory cytokines released in response to therapy-induced tumor tissue injury are critical signals that induce phenotypic plasticity of melanoma and immune cells.Citation128 Proinflammatory signals in the tumor microenvironment may induce EMT and dedifferentiation of melanoma cells. TNF-α resulted in dedifferentiation of melanoma cells, leading to loss of gp 100 (HMB-45) expression and it abrogated immune recognition by gp100-specific adoptive T cell transfer.Citation128 Neutrophil infiltration into the melanoma microenvironment is gaining interest as a therapeutic target because it is a sign of proinflammation that may drive melanoma plasticity toward the dedifferentiated mesenchymal phenotype that easily escapes immune surveillance. Ulceration of primary melanoma promotes neutrophil infiltration and correlates with increased angiotropsim and a higher risk of distant metastasis with poor prognosis.Citation129 Finally, resistance to BRAF inhibitors in both cultured cells and patient samples has been associated with an invasive, M-type phenotype switch with loss of MITF and SOX10 expression (116–118). This change was accompanied by a reduction in CTL infiltrates and an emergence of a CD8+ T cell exhaustion signature in the tumor microenvironment suggesting an acquired lowered state of immune surveillance.
Resistance of melanoma cells to apoptosis
Overexpression of antiapoptotic molecules
Changes in the expression of molecules involved in apoptosis signaling may contribute to melanoma resistance to immunotherapy.Citation10 Several proteins regulate the apoptotic pathway.
FLICE-inhibitory proteins (FLIPs)
FLIPs interfere with the initiation of apoptosis directly at the level of death receptors but do not affect perforin/granzyme induced apoptosis ().Citation130-132 High levels of FLIP expression were reported in tissues from melanoma skin lesions.Citation130,133,134 Overexpression of FLIP by transfection into a Fas- and TRAIL-sensitive human melanoma cell line rendered this cell line more resistant to death mediated by both TRAIL and FasL.Citation135 Selective expression of FLIP by human melanomas may represent a mechanism by which melanoma cells escape immune destruction.Citation135
BCL2 family of antiapoptotic proteins
The Bcl−2 family has both pro- and anti-apoptotic proteins. Pro-apoptotic Bcl−2 proteins include Bak and Bax which oligomerize in the mitochondrial membrane to allow release of cytochrome c and other apoptotic effectors into the cytoplasm (). Bak and Bax activity are facilitated by other pro-apoptotis Bcl2 family proteins such as Bim, Bid, Bad, Noxa, and Puma. The anti-apoptotic Bcl−2 proteins include Bcl−2, Bcl−xL, Mcl−1, Bcl−w and A1.Citation136 Overexpression of the antiapoptotic molecules of the Bcl−2 family, which regulate apoptosis at the mitochondrial level, is associated with poor outcomes in melanoma.Citation10,32,131,137 Bcl−2 expression, both in in vitro and in vivo models, confers resistance to CD95L and other apoptosis stimuli.Citation61 Activating ras mutations, seen in 15% of melanomas, are associated with more progressive disease. N-ras mutation in particular causes resistant to apoptosis due to increased levels of Bcl−2 in melanoma.Citation138
The BH3 mimetic ABT-737 (inhibiting both Bcl2 and Bcl−xL) sensitizes human melanoma cells to marked BIM (Bcl−2-interacting mediator of cell death) -mediated apoptosis to BRAF inhibitors or dacarbazine in vitro.Citation61,139 ABT-737 deserves further study in combination with immunotherapy to enhance efficacy and overcome resistance.Citation140 Attempts to target BCL2 with antisense RNA (in effect inhibiting its action) in melanoma patients have not yet been successful. An international randomized controlled trial of 771 melanoma patients comparing DTIC and oblimersen (an antisense agent that targets Bcl−2) with DTIC alone resulted in an increased median PFS, but no significant difference in OS.Citation141 It was never adequately established that this agent modulated Bcl−2 sufficiently to render cells more susceptible to cytotoxicity.Citation142 A phase I-II clinical study investigated the combination of antisense oligonucleotides (ASO) targeted against BCL2 and dacarbazine in 14 patients with advanced malignant melanoma expressing BCL2.Citation142 40% exhibited decreased Bcl−2 expression, increased tumor cell apoptosis, and increased antitumor responses.Citation142 Obatoclax, a drug inhibiting interaction of BCL2 with the pro-apoptotic proteins Bax and Bak, is being investigated in clinical trials for other malignancies.Citation61
Mcl−1, a structurally distinct member of the anti-apoptotic Bcl−2 family, is strongly expressed at all stages of melanoma,Citation143 and is highly selective for BAK inhibition.Citation144 The combination of DTIC plus antisense oligonucleotide against Mcl−1-sensitized melanomas to DTIC in a mouse model.Citation145 Preclinical data suggest that taxane resistance may be due to increased MEK signaling resulting in anti-apoptotic changes.Citation146 MEK inhibitors achieve much of their apoptotic effect through Mcl−1 suppression.Citation147 These data suggest that dual MEK/Mcl−1 inhibition could be an effective means of improving clinical response.
The inhibitors of apoptosis proteins (IAP)
The IAPs bind to and inhibit caspases ().Citation134,148,149 In melanoma, 2 members of the IAP family (survivin and ML-IAP/Livin) and FLIP have been associated with melanomagenesis, as they are strongly expressed in melanocytic nevi and further overexpressed in invasive and metastatic melanomas.Citation150,151
A key antiapoptotic function of survivin relates to inhibition of mitochondrial and apoptosis inducing factor-dependent apoptotic pathways. Its expression in melanoma likely protects against both caspase-independent and -dependent apoptosis.Citation152 Blocking the expression of survivin triggers spontaneous apoptosis in melanoma cell lines.Citation153 Livin selectively binds to SMAC (IAP antagonist) and caspase-3, caspase-7, and caspase-9, resulting in blockade of both the death receptor and mitochondria-based apoptotic pathways.Citation154,155 Smac/DIABLO () released from the mitochondria during apoptosis promotes caspase activation by binding to IAPs.Citation134,148 In the cytoplasm, SMAC binds and inhibits IAPs and thereby acts as a proapoptotic protein.Citation156 Livin binds and sequesters SMAC, thereby permitting other IAPs, such as XIAP, to function uninhibited.Citation157 Hence livin is considered a sink for SMAC.Citation157
Given its role as a downstream inhibitor of apoptosis and due to its limited expression in normal adult tissues, ML-IAP/Livin has been identified as a potential selective target for immune-mediated tumor destruction in melanoma vaccine trials.Citation149,158 The presence of anti-livin antibodies in the serum of patients with GI, breast, and lung malignancies support anti-livin based immunotherapy as a therapeutic approach.Citation159-161 Spontaneous CTL responses to ML-IAP/Livin epitopes have been found in melanoma patients and a limited number of healthy controls.Citation162 Spontaneous immune responses to ML-IAP/Livin in melanoma patients demonstrate both specific cellular and humoral activity as well as correlations with favorable clinical outcomes.Citation149 Therefore, inhibitors of livin could be useful adjuncts in the treatment of melanoma.Citation163
Expression of the proteinase inhibitor 9 PI-9
The caspase 8 and caspase-10 dependent Fas Ligand pathway becomes activated when tumor specific antigens, displayed on the surface of melanoma cells, are engaged with the cytotoxic T lymphocyte receptors on the immune cells. Activation of the Fas receptor pathway leads to, a cascade of caspase activation ending with apoptotic death of melanoma cells. Granzyme proteases are released by this process. Citation33 After entry into melanoma cells, granzyme B cleaves the precursor forms of several caspases and induces apoptosis as well.Citation166 In humans, Proteinase inhibitor 9 (PtdIns-9) is the only intracellular inhibitor of granzyme B. Expression of PtdIns-9 in metastatic melanoma cells is associated with unfavorable overall and disease-free survival.Citation33 Expression of PtdIns-9 could be an important immune escape mechanism, and its inhibition may enhance the efficacy of immunotherapy.Citation164,165
Loss of Apoptotic protease activating factor 1 (Apaf-1)
p53 is a tumor suppresser gene that promotes apoptosis and cell cycle arrest in response to DNA damage due to radiation or chemotherapy. Mutation of the p53, though rare in melanoma, has been associated with chemoresistance (3). Apaf-1, a critical downstream mediator of p53-dependent apoptosis, and an integral part of the apoptosome, is frequently lost in metastatic melanoma.Citation167 A high rate of allelic loss of the Apaf-1 locus is seen, with the remaining allele transcriptionally inactivated by gene methylation. Apaf-1-negative melanomas are invariably resistant to treatment and are unable to execute a p53 induced apoptosis. Restoring physiologic levels of Apaf-1 through gene transfer or 5aza2dC treatment markedly enhances chemosensitivity and rescues the apoptotic defects associated with Apaf-1 loss.Citation168 Restoring Apaf-1 regulation to some melanomas may have therapeutic benefit, though; it is unclear if restoring Apaf-1 could enhance the effects of immunotherapy in melanoma.
Downregulation of FAS/CD95 signaling
While the Fas-apoptotic pathway is active in early stages of melanomas, it is often impaired in metastatic advanced-phase melanomas.Citation169,170 Functional impairment of the Fas/CD95 receptor-ligand interaction is associated with the development and progression of malignancies.Citation171 One possible cause might be the expression of soluble receptors such as soluble CD95 (sCD95) and decoy receptor 3 (DcR3) that act as decoys for the death ligands, thus interfering with apoptosis via competitive inhibition of Fas/CD95 signaling.Citation171 Indeed, increased soluble CD95 serum levels correlate with poor prognoses in melanoma patients.Citation171-173 Elevated sFas/CD95 serum levels were associated with poor overall (P < 0.005) and low progression-free (P < 0.0005) survival.Citation171 Multivariate analysis revealed sFas/CD95 serum concentration as an independent predictive factor for progression-free survival (P = 0.011), but not overall survival (P = 0.078), in patients with melanoma.Citation171
Downregulation of Fas signaling can also be attributed to changes at the genomic, transcriptional and post-translational levels.Citation174 Mutations in the death domain of Fas have been reported to take place in 7% of metastatic melanomas.Citation175 These mutated forms of CD95 may interfere in a dominant-negative way with apoptosis induction via CD95. Hypermethylation of Fas receptor promoter may represent a more common event in melanoma, resulting in transcriptional downregulation of Fas expression.Citation174
High levels of FasL expression have been implicated in suppression of the immune response in metastatic melanoma.Citation176,177 An inverse correlation between expression of FasL and that of Fas has been found during melanoma progression.Citation178,179 Transfection and overexpression of exogenous Fas in melanoma cells with high levels of endogenous FasL induce efficient cell death.Citation180 Similarly, expression of exogenous FasL in tumor cells that express endogenous Fas receptor has often been tested as a possible anticancer treatment.Citation181
Downregulation of TRAIL signaling
TNF-related apoptosis-inducing ligand (TRAIL/Apo2L) is another major cytotoxic mechanism used by CTLs and NK cells to eradicate melanoma cells. TRAIL/Apo2L interacts with its cognate receptors (DR4, DR5, DcR1, DcR2, and osteoprotegerin) located on the melanoma cell surface, leading to melanoma cell death by apoptosis. Downregulation or malfunction of DR4 and the initiator caspase-8,Citation184 decreased release of Smac/DIABLO and thereby insufficient caspase-3 activation,Citation183 NF-κB mediated survival,Citation185 high expression of cFLIP and anti-apoptotic molecules such as Bcl−2, Bcl−XL, Mcl−1 and inhibitors of apoptosis (IAPs) as well as loss of Bax and Bak function have been reported to be of importance in TRAIL and apoptosis resistance in melanoma.Citation186
Cultured melanoma cells that express high levels of TRAIL receptors are highly sensitive to TRAIL-mediated apoptosis.Citation133 Decreased expression or function of TRAIL in melanoma cells resulted in attenuation of TRAIL-dependent apoptosis.Citation182,183 Moreover, some melanoma cells have completely lost expression of TRAIL receptors, presumably as a result of chromosome 8p22–21 lossCitation182,183 or through an epigenetic silencing mechanism.Citation174 Furthermore, at least 2 decoy receptors bind TRAIL without the ability to induce apoptosis, thereby pointing to an additional layer of regulation for the TRAIL apoptosis cascade.Citation187
Due to its apoptotic activity in a wide range of cancer cell types and low toxicity in most normal cells, TRAIL/Apo2L has become a promising anticancer agent.Citation188 TRAIL induces apoptosis in melanoma cell lines resistant to TNF-α, FASL and CD40L.Citation189 Agonistic monoclonal antibodies directed against TRAIL/Apo2L death-inducing receptors have become an attractive immunological therapeutic tool in clinical oncology due to their selective killing of tumors and lack of affinity toward healthy cells. Downregulation of Bcl−2, FLIP, or IAPs (XIAP and survivin) by siRNAs sensitizes resistant melanoma cells to Apo2L/TRAIL-induced apoptosis.Citation190 Full-length TRAIL, delivered from an adenoviral vector (Ad-hTRAIL), was studied in combination with DTIC or the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) in human melanoma cell lines.Citation191 Administration of Ad-hTRAIL in combination with DTIC or SAHA enhances apoptosis in human melanoma cell lines, and suggests that the therapeutic potential of such treatment strategies should be further evaluated for possible clinical useCitation191,192 HDACIs, inhibitors of histone deacetylation; a process essential in the regulation of gene expression, have proved to induce apoptosis, differentiation and cell cycle arrest in a variety of tumor cell lines, and to have anticancer potential in malignant melanoma.Citation193
The tumor counterattack
Melanomas with low T cell infiltration have poor prognoses and do not respond well to immunotherapy. The acquisition of death receptor ligand expression (FasL or CD95L) enables melanoma cells to deliver death signals to activate Fas (CD95)-positive CTLs leading to the death of CTLs. This mechanism is called the “tumor counterattack.”Citation10,187,194-197 Immune-privileged sites such as the eye, the brain, and the testis have a high level of CD95L expression in the cells native to these sites, which explains why corneal transplants are accepted without tissue matching or immunosuppressive therapy in humans.Citation10,133,197-199 Moreover, this may explain why immunotherapy may not be effective in melanoma involving the brain or the eye.
Melanoma cells not only can acquire resistance to Fas-induced apoptosis, which preserves them from apoptosis-mediated killing by cytotoxic T cells, but also can overexpress FasL which they use to deliver death signals and counterattack cytotoxic T lymphocytes in vitro.Citation10,176,200 The growth of a subcutaneously injected CD95L-positive murine melanoma cell line was slightly faster in wild-type compared with mutated CD95 receptor mice, suggesting impaired cell death in the faster growing tumor.Citation176 Another study hypothesized that CD95L is expressed intracellularly in melanoma cells. Melanosome preparations purified from melanoma cells induced dose-dependent Fas-mediated apoptosis of a cell line susceptible to Fas-dependent apoptosis.Citation200,201 This data suggested a new mechanism whereby FasL may be secreted by FasL-bearing vesicles inside the melanoma cells to mediate Fas-dependent apoptosis in the tumor microenvironment that does not necessarily imply cell-to-cell contact.Citation200,201 Contrary to the studies that support the counterattack phenomenon, others have shown that CD95L expression has a proinflammatory function leading to rejection of CD95L-expressing tumors by neutrophils.Citation10,181,202 To date, it is still unclear whether CD95L expression by tumor cells will provide them with an immune escape mechanism or will make them prone to rejection by the CD95L induced neutrophilic infiltration in vivo.Citation10
Similar to CD95L, the death ligand TRAIL has been suggested to suppress tumor-specific T cells.Citation203 Moreover, the membrane protein RCAS1Citation204 not only inhibits proliferation, but also induces apoptosis in T cells in vitro. Chemokine production by tumor cells may also sensitize T cells to apoptosis.Citation205 Additionally, the combination with treatments aimed at down-modulating FasL expression or at reducing microvesicle release by tumor cells may represent a strategy for improving clinical efficacy of active or adoptive immunotherapy in cancer patients.Citation200
Conclusions
Understanding the mechanisms of resistance to immunotherapy will help us find new therapies and candidate targets in melanoma to prevent or overcome resistance. Mutational load is important for T lymphocyte activation, and we have learned that immunotherapy works best in tumors with a high, as opposed to low, number of mutations. We have also learned that activated T lymphocytes must need to successfully return home to the melanoma microenvironment for optimal activity. An immune cell gene signature for T cell homing may include expression of adhesion molecules, retention integrins, and the chemokine ligands for CCR5, CCR9, and CXCR3. Defects in expression of these proteins, through genetic mutations or epigenetic silencing, may interfere with homing of activated T cells and contribute to melanoma-immune escape. The acquisition of new mutations in melanoma cells may give rise to new and/or the loss of original tumor-specific antigens to which the T cells were activated. Epigenetic silencing or genetic mutations may contribute to downregulation of death ligands and/or their receptors leading to failure of melanoma cells to undergo apoptosis. Future studies should look into combining an EZH2 inhibitor with a checkpoint inhibitor in melanoma tumor models to overcome a variety of resistance mechanisms.
The Intralesional injections of oncolytic viruses, such as the FDA approved agent Talimogen Laherparepvec or the NDV (currently being studied as an investigational new drug) resulted in increased T cell infiltration in the tumor microenvironment. Future studies should look into combining these viruses with anti PD1 agents to enhance the clinical efficacy of immunotherapy. The production of IFN-γ by the effector T cells induce PD-L1 expression on tumor cells that in turn deactivate T cell function by binding to the PD1. In that regard, IFN-γ may enhance the clinical efficacy of anti PD1 therapies. Moreover, IFN-γ enhances the MHC-I antigen processing and presentation pathway. Hence, the combination of IFN-γ with anti PD1 inhibitors may be an alternative approach and deserve further investigation.
The recruitment of different immunosuppressive cells can help melanoma cells evade immune surveillance; hence, strategies targeting these cells may be of benefit. Likewise, finding new therapies that target immunosuppressive molecules in the melanoma microenvironment such as IDO, TGF-B, IL-10 may enhance the efficacy of immunotherapy. Inhibitors of IDO are in several clinical trials for different malignancies, including a randomized phase I-II trial combining ipilimumab with INCB024360 (NCT01604889).Citation61 It appears that treatment with BRAF inhibitors provides a great landscape for the checkpoints inhibitors to work. BRAF or BRAF+MEK inhibitors enhance melanoma- antigen expression, increase CD8+ T lymphocyte infiltration, and decrease the immunosuppressive cytokines IL-6 and IL-8. Hence, it makes sense to start patients on BRAF inhibitors first to enhance the efficacy and benefit of checkpoint inhibitors later. However, the sequencing of current FDA approved agents is an area of active research and ongoing trials. Interestingly, inhibitory PD-1 was increased on T cells and its immunosuppressive ligand PDL1 was also increased with BRAF inhibition. This supports the hypothesis that targeting inhibitory immune interactions may be critical in augmenting responses to BRAF-targeted therapy in patients with melanoma.Citation206
Even if we succeed in all the above, resistance to immunotherapy may still emerge if the apoptotic machinery is dysfunctional. Targeting apoptotic inhibitors such as survivin and ML-IAP/Livin and using AKT inhibitors to reduce the anti-apoptotic Bcl2 family proteins and Mcl−1 are all attractive therapeutic strategies to enhance apoptosis. These strategies deserve further investigation combined with the current available anti PD1 and CTLA4 inhibitors to overcome resistance. p53 is a tumor suppresser gene that promotes apoptosis and cell cycle arrest in response to DNA damage due to radiation or chemotherapy. Hence, combining immunotherapy with checkpoint inhibitors with either stereotactic radiation therapy or chemotherapy may enhance an effective immune response by providing more local inflammation that attracts inflammatory cells, and more tumor specific antigens to the cytotoxic T cells, in addition to their role in enhancing p53-induced apoptosis. These areas of translational research are urgently needed as there currently are no available treatments of patients with metastatic melanoma who have failed checkpoint inhibitors, other than adoptive cell transfer which is done only in major academic centers. The future advances in immunotherapy in the treatment of melanoma will likely involve combinatorial therapy to that allows different therapeutic agents to synergistically work together to optimize tumor antigenicity and the host immune anti-tumor response.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgment
Dr. Mahmoud wishes to acknowledge Dorothy Graves for her critical review of this manuscript.
References
- Atrash S, Makhoul I, Mizell JS, Hutchins L, Mahmoud F. Response of metastatic mucosal melanoma to immunotherapy: It can get worse before it gets better. J Oncol Pharm Pract 2017 Apr; 23(3):215-219; PMID:26811403; https://doi.org/10.1177/1078155215627503
- Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363(8):711-23; PMID:20525992; https://doi.org/10.1056/NEJMoa1003466
- Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, Weber JS, Joshua AM, Hwu WJ, Gangadhar TC, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: A randomised dose-comparison cohort of a phase 1 trial. Lancet 2014; 384(9948):1109-17; PMID:25034862; https://doi.org/10.1016/S0140-6736(14)60958-2
- Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, Patt D, Chen TT, Berman DM, Wolchok JD. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol 2015; 33(17):1889-94; PMID:25667295; https://doi.org/10.1200/JCO.2014.56.2736
- Weber JS, D'Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, Hoeller C, Khushalani NI, Miller WH Jr, Lao CD, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): A randomised, controlled, open-label, phase 3 trial. Lancet Oncol 2015; 16(4):375-84; PMID:25795410; https://doi.org/10.1016/S1470-2045(15)70076-8
- Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015; 373(1):23-34; PMID:26027431; https://doi.org/10.1056/NEJMoa1504030
- Restifo NP, Smyth MJ, Snyder A. Acquired resistance to immunotherapy and future challenges. Nat Rev Cancer 2016; 16(2):121-6; PMID:26822578; https://doi.org/10.1038/nrc.2016.2
- O'Donnell JS, Long GV, Scolyer RA, Teng MW, Smyth MJ. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat Rev 2017; 52:71-81; PMID:27951441; https://doi.org/10.1016/j.ctrv.2016.11.007
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012; 366(26):2443-54; PMID:22658127; https://doi.org/10.1056/NEJMoa1200690
- Igney FH, Krammer PH. Immune escape of tumors: Apoptosis resistance and tumor counterattack. J Leukoc Biol 2002; 71(6):907-20; PMID:12050175
- Schwarz BA, Bhandoola A. Trafficking from the bone marrow to the thymus: A prerequisite for thymopoiesis. Immunol Rev 2006; 209:47-57; PMID:16448533; https://doi.org/10.1111/j.0105-2896.2006.00350.x
- Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer 2012; 12(4):265-77; PMID:22437871; https://doi.org/10.1038/nrc3258
- Cui J, Chen Y, Wang HY, Wang RF. Mechanisms and pathways of innate immune activation and regulation in health and cancer. Hum Vaccin Immunother 2014; 10(11):3270-85; PMID:25625930; https://doi.org/10.4161/21645515.2014.979640
- Erdag G, Schaefer JT, Smolkin ME, Deacon DH, Shea SM, Dengel LT, Patterson JW, Slingluff CL Jr. Immunotype and immunohistologic characteristics of tumor-infiltrating immune cells are associated with clinical outcome in metastatic melanoma. Cancer Res 2012; 72(5):1070-80; PMID:22266112; https://doi.org/10.1158/0008-5472.CAN-11-3218
- Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, McKee M, Gajewski TF. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res 2009; 69(7):3077-85; PMID:19293190; https://doi.org/10.1158/0008-5472.CAN-08-2281
- Kelderman S, Schumacher TN, Haanen JB. Acquired and intrinsic resistance in cancer immunotherapy. Mol Oncol 2014; 8(6):1132-9; PMID:25106088; https://doi.org/10.1016/j.molonc.2014.07.011
- Ferguson AR, Engelhard VH. CD8 T cells activated in distinct lymphoid organs differentially express adhesion proteins and coexpress multiple chemokine receptors. J Immunol 2010; 184(8):4079-86; PMID:20212096; https://doi.org/10.4049/jimmunol.0901903
- Sheasley-O'Neill SL, Brinkman CC, Ferguson AR, Dispenza MC, Engelhard VH. Dendritic cell immunization route determines integrin expression and lymphoid and nonlymphoid tissue distribution of CD8 T cells. J Immunol 2007; 178(3):1512-22; PMID:17237400; https://doi.org/10.4049/jimmunol.178.3.1512
- Salerno EP, Olson WC, McSkimming C, Shea S, Slingluff CL Jr. T cells in the human metastatic melanoma microenvironment express site-specific homing receptors and retention integrins. Int J Cancer 2014; 134(3):563-74; PMID:23873187; https://doi.org/10.1002/ijc.28391
- Gajewski TF, Louahed J, Brichard VG. Gene signature in melanoma associated with clinical activity: A potential clue to unlock cancer immunotherapy. Cancer J 2010; 16(4):399-403; PMID:20693853; https://doi.org/10.1097/PPO.0b013e3181eacbd8
- Weiss GR, Grosh WW, Chianese-Bullock KA, Zhao Y, Liu H, Slingluff CL Jr, Marincola FM, Wang E. Molecular insights on the peripheral and intratumoral effects of systemic high-dose rIL-2 (aldesleukin) administration for the treatment of metastatic melanoma. Clin Cancer Res 2011; 17(23):7440-50; PMID:21976537; https://doi.org/10.1158/1078-0432.CCR-11-1650
- Garrido F, Cabrera T, Aptsiauri N. “Hard” and “soft” lesions underlying the HLA class I alterations in cancer cells: Implications for immunotherapy. Int J Cancer 2010; 127(2):249-56; PMID:20178101; https://doi.org/10.1002/ijc.25270
- Fruci D, Benevolo M, Cifaldi L, Lorenzi S, Lo Monaco E, Tremante E, Giacomini P. Major histocompatibility complex class i and tumour immuno-evasion: How to fool T cells and natural killer cells at one time. Curr Oncol 2012; 19(1):39-41; PMID:22328841; https://doi.org/10.3747/co.19.945
- Shresta S, Pham CT, Thomas DA, Graubert TA, Ley TJ. How do cytotoxic lymphocytes kill their targets? Curr Opin Immunol 1998; 10(5):581-7; PMID:9794837; https://doi.org/10.1016/S0952-7915(98)80227-6
- Jazirehi AR, Arle D. Epigenetic regulation of the TRAIL/Apo2L apoptotic pathway by histone deacetylase inhibitors: An attractive approach to bypass melanoma immunotherapy resistance. Am J Clin Exp Immunol 2013; 2(1):55-74; PMID:23885325
- Schmitz I, Kirchhoff S, Krammer PH. Regulation of death receptor-mediated apoptosis pathways. Int J Biochem Cell Biol 2000; 32(11-12):1123-36; PMID:11137452; https://doi.org/10.1016/S1357-2725(00)00048-0
- Kischkel FC, Lawrence DA, Chuntharapai A, Schow P, Kim KJ, Ashkenazi A. Apo2L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptors 4 and 5. Immunity 2000; 12(6):611-20; PMID:10894161; https://doi.org/10.1016/S1074-7613(00)80212-5
- Muzio M, Chinnaiyan AM, Kischkel FC, O'Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz JD, Zhang M, Gentz R, et al. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (fas/APO-1) death–inducing signaling complex. Cell 1996; 85(6):817-27; PMID:8681377; https://doi.org/10.1016/S0092-8674(00)81266-0
- Sprick MR, Weigand MA, Rieser E, Rauch CT, Juo P, Blenis J, Krammer PH, Walczak H. FADD/MORT1 and caspase-8 are recruited to TRAIL receptors 1 and 2 and are essential for apoptosis mediated by TRAIL receptor 2. Immunity 2000; 12(6):599-609; PMID:10894160; https://doi.org/10.1016/S1074-7613(00)80211-3
- Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli KJ, Debatin KM, Krammer PH, Peter ME. Two CD95 (APO-1/fas) signaling pathways. EMBO J 1998; 17(6):1675-87; PMID:9501089; https://doi.org/10.1093/emboj/17.6.1675
- Martinou JC, Green DR. Breaking the mitochondrial barrier. Nat Rev Mol Cell Biol 2001; 2(1):63-7; PMID:11413467; https://doi.org/10.1038/35048069
- Zamzami N, Kroemer G. The mitochondrion in apoptosis: How pandora's box opens. Nat Rev Mol Cell Biol 2001; 2(1):67-71; PMID:11413468; https://doi.org/10.1038/35048073
- van den Broek ME, Kagi D, Ossendorp F, Toes R, Vamvakas S, Lutz WK, Melief CJ, Zinkernagel RM, Hengartner H. Decreased tumor surveillance in perforin-deficient mice. J Exp Med 1996; 184(5):1781-90; PMID:8920866; https://doi.org/10.1084/jem.184.5.1781
- Kagi D, Ledermann B, Burki K, Seiler P, Odermatt B, Olsen KJ, Podack ER, Zinkernagel RM, Hengartner H. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature 1994; 369(6475):31-7; PMID:8164737; https://doi.org/10.1038/369031a0
- Heusel JW, Wesselschmidt RL, Shresta S, Russell JH, Ley TJ. Cytotoxic lymphocytes require granzyme B for the rapid induction of DNA fragmentation and apoptosis in allogeneic target cells. Cell 1994; 76(6):977-87; PMID:8137431; https://doi.org/10.1016/0092-8674(94)90376-X
- Medema JP, Toes RE, Scaffidi C, Zheng TS, Flavell RA, Melief CJ, Peter ME, Offringa R, Krammer PH. Cleavage of FLICE (caspase-8) by granzyme B during cytotoxic T lymphocyte-induced apoptosis. Eur J Immunol 1997; 27(12):3492-8; PMID:9464839; https://doi.org/10.1002/eji.1830271250
- Heibein JA, Goping IS, Barry M, Pinkoski MJ, Shore GC, Green DR, Bleackley RC. Granzyme B-mediated cytochrome c release is regulated by the bcl-2 family members bid and bax. J Exp Med 2000; 192(10):1391-402; PMID:11085742; https://doi.org/10.1084/jem.192.10.1391
- Kubica AW, Brewer JD. Melanoma in immunosuppressed patients. Mayo Clin Proc 2012; 87(10):991-1003; PMID:23036673; https://doi.org/10.1016/j.mayocp.2012.04.018
- Akbar AN, Fletcher JM. Memory T cell homeostasis and senescence during aging. Curr Opin Immunol 2005; 17(5):480-5; PMID:16098721; https://doi.org/10.1016/j.coi.2005.07.019
- Messaoudi I, Lemaoult J, Guevara-Patino JA, Metzner BM, Nikolich-Zugich J. Age-related CD8 T cell clonal expansions constrict CD8 T cell repertoire and have the potential to impair immune defense. J Exp Med 2004; 200(10):1347-58; PMID:15545358; https://doi.org/10.1084/jem.20040437
- Chen PL, Roh W, Reuben A, Cooper ZA, Spencer CN, Prieto PA, Miller JP, Bassett RL, Gopalakrishnan V, Wani K, et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov 2016; 6(8):827-37; PMID:27301722; https://doi.org/10.1158/2159-8290.CD-15-1545
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013; 499(7457):214-8; PMID:23770567; https://doi.org/10.1038/nature12213
- Rosenberg SA. Progress in human tumour immunology and immunotherapy. Nature 2001; 411(6835):380-4; PMID:11357146; https://doi.org/10.1038/35077246
- Houghton AN, Gold JS, Blachere NE. Immunity against cancer: Lessons learned from melanoma. Curr Opin Immunol 2001; 13(2):134-40; PMID:11228404; https://doi.org/10.1016/S0952-7915(00)00195-3
- Lee PP, Yee C, Savage PA, Fong L, Brockstedt D, Weber JS, Johnson D, Swetter S, Thompson J, Greenberg PD, et al. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat Med 1999; 5(6):677-85; PMID:10371507; https://doi.org/10.1038/9525
- Brown SD, Warren RL, Gibb EA, Martin SD, Spinelli JJ, Nelson BH, Holt RA. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res 2014; 24(5):743-50; PMID:24782321; https://doi.org/10.1101/gr.165985.113
- Champiat S, Ferte C, Lebel-Binay S, Eggermont A, Soria JC. Exomics and immunogenics: Bridging mutational load and immune checkpoints efficacy. Oncoimmunology 2014; 3(1):e27817; PMID:24605269; https://doi.org/10.4161/onci.27817
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, et al. Signatures of mutational processes in human cancer. Nature 2013; 500(7463):415-21; PMID:23945592; https://doi.org/10.1038/nature12477
- Aris M, Barrio MM, Mordoh J. Lessons from cancer immunoediting in cutaneous melanoma. Clin Dev Immunol 2012; 2012:192719; PMID:22924051; https://doi.org/10.1155/2012/192719
- Kim R, Emi M, Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunology 2007; 121(1):1-14; PMID:17386080; https://doi.org/10.1111/j.1365-2567.2007.02587.x
- Vesely MD, Schreiber RD. Cancer immunoediting: Antigens, mechanisms, and implications to cancer immunotherapy. Ann N Y Acad Sci 2013; 1284:1-5; PMID:23651186; https://doi.org/10.1111/nyas.12105
- Maeurer MJ, Gollin SM, Martin D, Swaney W, Bryant J, Castelli C, Robbins P, Parmiani G, Storkus WJ, Lotze MT. Tumor escape from immune recognition: Lethal recurrent melanoma in a patient associated with downregulation of the peptide transporter protein TAP-1 and loss of expression of the immunodominant MART-1/melan-A antigen. J Clin Invest 1996; 98(7):1633-41; PMID:8833913; https://doi.org/10.1172/JCI118958
- Stackpole CW, Cremona P, Leonard C, Stremmel P. Antigenic modulation as a mechanism for tumor escape from immune destruction: Identification of modulation-positive and modulation-negative mouse lymphomas with xenoantisera to murine leukemia virus gp70. J Immunol 1980; 125(4):1715-23; PMID:6251136
- Ellis LM, Hicklin DJ. VEGF-targeted therapy: Mechanisms of anti-tumour activity. Nat Rev Cancer 2008; 8(8):579-91; PMID:18596824; https://doi.org/10.1038/nrc2403
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012; 12(4):252-64; PMID:22437870; https://doi.org/10.1038/nrc3239
- Yaguchi T, Goto Y, Kido K, Mochimaru H, Sakurai T, Tsukamoto N, Kudo-Saito C, Fujita T, Sumimoto H, Kawakami Y. Immune suppression and resistance mediated by constitutive activation of wnt/beta-catenin signaling in human melanoma cells. J Immunol 2012; 189(5):2110-7; PMID:22815287; https://doi.org/10.4049/jimmunol.1102282
- Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015; 523(7559):231-5; PMID:25970248; https://doi.org/10.1038/nature14404
- Shin DS, Zaretsky JM, Escuin-Ordinas H, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov 2017; 7(2):188-201; PMID:27903500; https://doi.org/10.1158/2159-8290.CD-16-1223
- Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol 2006; 6(11):836-48; PMID:17063185; https://doi.org/10.1038/nri1961
- Dunn GP, Ikeda H, Bruce AT, Koebel C, Uppaluri R, Bui J, Chan R, Diamond M, White JM, Sheehan KC, et al. Interferon-gamma and cancer immunoediting. Immunol Res 2005; 32(1-3):231-45; PMID:16106075; https://doi.org/10.1385/IR:32:1-3:231
- Shtivelman E, Davies MQ, Hwu P, Yang J, Lotem M, Oren M, Flaherty KT, Fisher DE. Pathways and therapeutic targets in melanoma. Oncotarget 2014; 5(7):1701-52; PMID:24743024; https://doi.org/10.18632/oncotarget.1892
- Sengupta D, Byrum SD, Avaritt NL, Davis L, Shields B, Mahmoud F, Reynolds M, Orr LM, Mackintosh SG, Shalin SC, et al. Quantitative histone mass spectrometry identifies elevated histone H3 lysine 27 (Lys27) trimethylation in melanoma. Mol Cell Proteomics 2016; 15(3):765-75; PMID:26621846; https://doi.org/10.1074/mcp.M115.053363
- Mahmoud F, Shields B, Makhoul I, Hutchins LF, Shalin SC, Tackett AJ. Role of EZH2 histone methyltrasferase in melanoma progression and metastasis. Cancer Biol Ther 2016; 2:17(6):579-91; https://doi.org/10.1080/15384047.2016.1167291
- Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, Sun Y, Zhao E, Vatan L, Szeliga W, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015; 527(7577):249-53; PMID:26503055; https://doi.org/10.1038/nature15520
- Clemente CG, Mihm MC Jr, Bufalino R, Zurrida S, Collini P, Cascinelli N. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer 1996; 77(7):1303-10; PMID:8608507; https://doi.org/10.1002/(SICI)1097-0142(19960401)77:7%3c1303::AID-CNCR12%3e3.3.CO;2-0 10.1002/(SICI)1097-0142(19960401)77:7%3c1303::AID-CNCR12%3e3.0.CO;2-5
- Besser MJ, Shapira-Frommer R, Itzhaki O, Treves AJ, Zippel DB, Levy D, Kubi A, Shoshani N, Zikich D, Ohayon Y, et al. Adoptive transfer of tumor-infiltrating lymphocytes in patients with metastatic melanoma: Intent-to-treat analysis and efficacy after failure to prior immunotherapies. Clin Cancer Res 2013; 19(17):4792-800; PMID:23690483; https://doi.org/10.1158/1078-0432.CCR-13-0380
- Besser MJ. Is there a future for adoptive cell transfer in melanoma patients? Oncoimmunology 2013; 2(10):e26098; PMID:24353909; https://doi.org/10.4161/onci.26098
- Ochsenbein AF, Sierro S, Odermatt B, Pericin M, Karrer U, Hermans J, Hemmi S, Hengartner H, Zinkernagel RM. Roles of tumour localization, second signals and cross priming in cytotoxic T-cell induction. Nature 2001; 411(6841):1058-64; PMID:11429607; https://doi.org/10.1038/35082583
- Wick M, Dubey P, Koeppen H, Siegel CT, Fields PE, Chen L, Bluestone JA, Schreiber H. Antigenic cancer cells grow progressively in immune hosts without evidence for T cell exhaustion or systemic anergy. J Exp Med 1997; 186(2):229-38; PMID:9221752; https://doi.org/10.1084/jem.186.2.229
- Zamarin D, Holmgaard RB, Subudhi SK, Park JS, Mansour M, Palese P, Merghoub T, Wolchok JD, Allison JP. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med 2014; 6(226):226ra32; PMID:24598590; https://doi.org/10.1126/scitranslmed.3008095
- Yun S, Vincelette ND, Green MR, Wahner Hendrickson AE, Abraham I. Targeting immune checkpoints in unresectable metastatic cutaneous melanoma: A systematic review and meta-analysis of anti-CTLA-4 and anti-PD-1 agents trials. Cancer Med 2016; 5(7):1481-91; PMID:27167347; https://doi.org/10.1002/cam4.732
- Yao S, Zhu Y, Zhu G, Augustine M, Zheng L, Goode DJ, Broadwater M, Ruff W, Flies S, Xu H, et al. B7-h2 is a costimulatory ligand for CD28 in human. Immunity 2011; 34(5):729-40; PMID:21530327; https://doi.org/10.1016/j.immuni.2011.03.014
- Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med 2009; 206(8):1717-25; PMID:19581407; https://doi.org/10.1084/jem.20082492
- Contardi E, Palmisano GL, Tazzari PL, Martelli AM, Falà F, Fabbi M, Kato T, Lucarelli E, Donati D, Polito L, et al. CTLA-4 is constitutively expressed on tumor cells and can trigger apoptosis upon ligand interaction. Int J Cancer 2005; 117(4):538-50; PMID:15912538; https://doi.org/10.1002/ijc.21155
- Shah KV, Chien AJ, Yee C, Moon RT. CTLA-4 is a direct target of wnt/beta-catenin signaling and is expressed in human melanoma tumors. J Invest Dermatol 2008; 128(12):2870-9; PMID:18563180; https://doi.org/10.1038/jid.2008.170
- Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, Chen T, Roszik J, Bernatchez C, Woodman SE, et al. Loss of IFN-gamma pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell 2016; 167(2):397-404.e9; PMID:27667683; https://doi.org/10.1016/j.cell.2016.08.069
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012; 12(4):252-64; PMID:22437870; https://doi.org/10.1038/nrc3239
- Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, Cucolo L, et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016; 167(6):1540-1554.e12; https://doi.org/10.1016/j.cell.2016.11.022
- Buferne M, Chasson L, Grange M, Mas A, Arnoux F, Bertuzzi M, Naquet P, Leserman L, Schmitt-Verhulst AM, Auphan-Anezin N. IFNgamma producing CD8 T cells modified to resist major immune checkpoints induce regression of MHC class I-deficient melanomas. Oncoimmunology 2015; 4(2):e974959; PMID:25949872; https://doi.org/10.4161/2162402X.2014.974959
- Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med 2016; 375(9):819-29; PMID:27433843; https://doi.org/10.1056/NEJMoa1604958
- Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature 2005; 438(7070):967-74; PMID:16355214; https://doi.org/10.1038/nature04483
- Ott PA, Hodi FS, Buchbinder EI. Inhibition of immune checkpoints and vascular endothelial growth factor as combination therapy for metastatic melanoma: An overview of rationale, preclinical evidence, and initial clinical data. Front Oncol 2015; 5:202; PMID:26442214; https://doi.org/10.3389/fonc.2015.00202
- Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL, Latreche S, Bergaya S, Benhamouda N, Tanchot C, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med 2015; 212(2):139-48; PMID:25601652; https://doi.org/10.1084/jem.20140559
- Geissmann F, Revy P, Regnault A, Lepelletier Y, Dy M, Brousse N, Amigorena S, Hermine O, Durandy A. TGF-beta 1 prevents the noncognate maturation of human dendritic langerhans cells. J Immunol 1999; 162(8):4567-75; PMID:10201996
- Pickup MW, Laklai H, Acerbi I, Owens P, Gorska AE, Chytil A, Aakre M, Weaver VM, Moses HL. Stromally derived lysyl oxidase promotes metastasis of transforming growth factor-beta-deficient mouse mammary carcinomas. Cancer Res 2013; 73(17):5336-46; PMID:23856251; https://doi.org/10.1158/0008-5472.CAN-13-0012
- Steinbrink K, Jonuleit H, Muller G, Schuler G, Knop J, Enk AH. Interleukin-10-treated human dendritic cells induce a melanoma-antigen-specific anergy in CD8(+) T cells resulting in a failure to lyse tumor cells. Blood 1999; 93(5):1634-42; PMID:10029592
- Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: Expect the unexpected. J Clin Invest 2015; 125(9):3356-64; PMID:26168215; https://doi.org/10.1172/JCI80005
- Ortiz ML, Kumar V, Martner A, Mony S, Donthireddy L, Condamine T, Seykora J, Knight SC, Malietzis G, Lee GH, et al. Immature myeloid cells directly contribute to skin tumor development by recruiting IL-17-producing CD4+ T cells. J Exp Med 2015; 212(3):351-67; PMID:25667306; https://doi.org/10.1084/jem.20140835
- Vukmanovic-Stejic M, Zhang Y, Cook JE, Fletcher JM, McQuaid A, Masters JE, Rustin MH, Taams LS, Beverley PC, Macallan DC, et al. Human CD4+ CD25hi Foxp3+ regulatory T cells are derived by rapid turnover of memory populations in vivo. J Clin Invest 2006; 116(9):2423-33; PMID:16955142; https://doi.org/10.1172/JCI28941
- Sim GC, Martin-Orozco N, Jin L, Yang Y, Wu S, Washington E, Sanders D, Lacey C, Wang Y, Vence L, et al. IL-2 therapy promotes suppressive ICOS+ treg expansion in melanoma patients. J Clin Invest 2014; 124(1):99-110; PMID:24292706; https://doi.org/10.1172/JCI46266
- Jordan KR, Amaria RN, Ramirez O, Callihan EB, Gao D, Borakove M, Manthey E, Borges VF, McCarter MD. Myeloid-derived suppressor cells are associated with disease progression and decreased overall survival in advanced-stage melanoma patients. Cancer Immunol Immunother 2013; 62(11):1711-22; PMID:24072401; https://doi.org/10.1007/s00262-013-1475-x
- Weide B, Martens A, Zelba H, Stutz C, Derhovanessian E, Di Giacomo AM, Maio M, Sucker A, Schilling B, Schadendorf D, et al. Myeloid-derived suppressor cells predict survival of patients with advanced melanoma: Comparison with regulatory T cells and NY-ESO-1- or melan-A-specific T cells. Clin Cancer Res 2014; 20(6):1601-9; PMID:24323899; https://doi.org/10.1158/1078-0432.CCR-13-2508
- Mok S, Koya RC, Tsui C, Xu J, Robert L, Wu L, Graeber TG, West BL, Bollag G, Ribas A. Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res 2014; 74(1):153-61; PMID:24247719; https://doi.org/10.1158/0008-5472.CAN-13-1816
- Chouaib S, Asselin-Paturel C, Mami-Chouaib F, Caignard A, Blay JY. The host-tumor immune conflict: From immunosuppression to resistance and destruction. Immunol Today 1997; 18(10):493-7; PMID:9357142; https://doi.org/10.1016/S0167-5699(97)01115-8
- Munn DH. Blocking IDO activity to enhance anti-tumor immunity. Front Biosci (Elite Ed) 2012; 4:734-45; PMID:22201909; https://doi.org/10.2741/e414
- Munn DH. Indoleamine 2,3-dioxygenase, tregs and cancer. Curr Med Chem 2011; 18(15):2240-6; PMID:21517755; https://doi.org/10.2174/092986711795656045
- Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, Nadaf S, Kavanaugh D, Carbone DP. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med 1996; 2(10):1096-103; PMID:8837607; https://doi.org/10.1038/nm1096-1096
- Plescia OJ, Grinwich K, Plescia AM. Subversive activity of syngeneic tumor cells as an escape mechanism from immune surveillance and the role of prostaglandins. Ann N Y Acad Sci 1976; 276:455-65; PMID:1071971; https://doi.org/10.1111/j.1749-6632.1976.tb41670.x
- Matsuda M, Salazar F, Petersson M, Masucci G, Hansson J, Pisa P, Zhang QJ, Masucci MG, Kiessling R. Interleukin 10 pretreatment protects target cells from tumor- and allo-specific cytotoxic T cells and downregulates HLA class I expression. J Exp Med 1994; 180(6):2371-6; PMID:7964510; https://doi.org/10.1084/jem.180.6.2371
- McKallip R, Li R, Ladisch S. Tumor gangliosides inhibit the tumor-specific immune response. J Immunol 1999; 163(7):3718-26; PMID:10490967
- Takahashi K, Ono K, Hirabayashi Y, Taniguchi M. Escape mechanisms of melanoma from immune system by soluble melanoma antigen. J Immunol 1988; 140(9):3244-8; PMID:2452202
- Restifo NP, Marincola FM, Kawakami Y, Taubenberger J, Yannelli JR, Rosenberg SA. Loss of functional beta 2-microglobulin in metastatic melanomas from five patients receiving immunotherapy. J Natl Cancer Inst 1996; 88(2):100-8; PMID:8537970; https://doi.org/10.1093/jnci/88.2.100
- Rotem-Yehudar R, Groettrup M, Soza A, Kloetzel PM, Ehrlich R. LMP-associated proteolytic activities and TAP-dependent peptide transport for class 1 MHC molecules are suppressed in cell lines transformed by the highly oncogenic adenovirus 12. J Exp Med 1996; 183(2):499-514; PMID:8627162; https://doi.org/10.1084/jem.183.2.499
- Dissemond J, Kothen T, Mors J, Weimann TK, Lindeke A, Goos M, Wagner SN. Downregulation of tapasin expression in progressive human malignant melanoma. Arch Dermatol Res 2003; 295(2):43-9; PMID:12682852; https://doi.org/10.1007/s00403-003-0393-8
- Chang CC, Pirozzi G, Wen SH, Chung IH, Chiu BL, Errico S, Luongo M, Lombardi ML. Ferrone S multiple structural and epigenetic defects in the human leukocyte antigen class I antigen presentation pathway in a recurrent metastatic melanoma following immunotherapy. J Biol Chem 2015; 290(44):26562-75; PMID:26381407; https://doi.org/10.1074/jbc.M115.676130
- Hicklin DJ, Marincola FM, Ferrone S. HLA class I antigen downregulation in human cancers: T-cell immunotherapy revives an old story. Mol Med Today 1999; 5(4):178-86; PMID:10203751; https://doi.org/10.1016/S1357-4310(99)01451-3
- Hicklin DJ, Wang Z, Arienti F, Rivoltini L, Parmiani G, Ferrone S. Beta2-microglobulin mutations, HLA class I antigen loss, and tumor progression in melanoma. J Clin Invest 1998; 101(12):2720-9; PMID:9637706; https://doi.org/10.1172/JCI498
- Seung S, Urban JL, Schreiber H. A tumor escape variant that has lost one major histocompatibility complex class I restriction element induces specific CD8+ T cells to an antigen that no longer serves as a target. J Exp Med 1993; 178(3):933-40; PMID:8394406; https://doi.org/10.1084/jem.178.3.933
- Arce-Gomez B, Jones EA, Barnstable CJ, Solomon E, Bodmer WF. The genetic control of HLA-A and B antigens in somatic cell hybrids: Requirement for beta2 microglobulin. Tissue Antigens 1978; 11(2):96-112; PMID:77067; https://doi.org/10.1111/j.1399-0039.1978.tb01233.x
- Aptsiauri N, Carretero R, Garcia-Lora A, Real LM, Cabrera T, Garrido F. Regressing and progressing metastatic lesions: Resistance to immunotherapy is predetermined by irreversible HLA class I antigen alterations. Cancer Immunol Immunother 2008; 57(11):1727-33; PMID:18491093; https://doi.org/10.1007/s00262-008-0532-3
- Porgador A, Mandelboim O, Restifo NP, Strominger JL. Natural killer cell lines kill autologous beta2-microglobulin-deficient melanoma cells: Implications for cancer immunotherapy. Proc Natl Acad Sci U S A 1997; 94(24):13140-5; PMID:9371813; https://doi.org/10.1073/pnas.94.24.13140
- Farrell HE, Vally H, Lynch DM, Fleming P, Shellam GR, Scalzo AA, Davis-Poynter NJ. Inhibition of natural killer cells by a cytomegalovirus MHC class I homologue in vivo. Nature 1997; 386(6624):510-4; PMID:9087412; https://doi.org/10.1038/386510a0
- Cretney E, Degli-Esposti MA, Densley EH, Farrell HE, Davis-Poynter NJ, Smyth MJ. m144, a murine cytomegalovirus (MCMV)-encoded major histocompatibility complex class I homologue, confers tumor resistance to natural killer cell-mediated rejection. J Exp Med 1999; 190(3):435-44; PMID:10430631; https://doi.org/10.1084/jem.190.3.435
- del Campo AB, Kyte JA, Carretero J, Zinchencko S, Méndez R, González-Aseguinolaza G, Ruiz-Cabello F, Aamdal S, Gaudernack G, Garrido F, et al. Immune escape of cancer cells with beta2-microglobulin loss over the course of metastatic melanoma. Int J Cancer 2014; 134(1):102-13; PMID:23784959; https://doi.org/10.1002/ijc.28338
- Chang CC, Campoli M, Restifo NP, Wang X, Ferrone S. Immune selection of hot-spot beta 2-microglobulin gene mutations, HLA-A2 allospecificity loss, and antigen-processing machinery component down-regulation in melanoma cells derived from recurrent metastases following immunotherapy. J Immunol 2005; 174(3):1462-71; PMID:15661905; https://doi.org/10.4049/jimmunol.174.3.1462
- Bernal M, Ruiz-Cabello F, Concha A, Paschen A, Garrido F. Implication of the beta2-microglobulin gene in the generation of tumor escape phenotypes. Cancer Immunol Immunother 2012; 61(9):1359-71; PMID:22833104; https://doi.org/10.1007/s00262-012-1321-6
- Maleno I, Aptsiauri N, Cabrera T, Gallego A, Paschen A, López-Nevot MA, Garrido F. Frequent loss of heterozygosity in the beta2-microglobulin region of chromosome 15 in primary human tumors. Immunogenetics 2011; 63(2):65-71; PMID:21086121; https://doi.org/10.1007/s00251-010-0494-4
- Woods K, Pasam A, Jayachandran A, Andrews MC, Cebon J. Effects of epithelial to mesenchymal transition on T cell targeting of melanoma cells. Front Oncol 2014; 4:367; PMID:25566505; https://doi.org/10.3389/fonc.2014.00367
- Behren A, Anaka M, Lo PH, Vella LJ, Davis ID, Catimel J, Cardwell T, Gedye C, Hudson C, Stan R, et al. The ludwig institute for cancer research melbourne melanoma cell line panel. Pigment Cell Melanoma Res 2013; 26(4):597-600; PMID:23527996; https://doi.org/10.1111/pcmr.12097
- Jayachandran A, Anaka M, Prithviraj P, Hudson C, McKeown SJ, Lo PH, Vella LJ, Goding CR, Cebon J, Behren A. Thrombospondin 1 promotes an aggressive phenotype through epithelial-to-mesenchymal transition in human melanoma. Oncotarget 2014; 5(14):5782-97; PMID:25051363; https://doi.org/10.18632/oncotarget.2164
- Hoek KS, Eichhoff OM, Schlegel NC, Döbbeling U, Kobert N, Schaerer L, Hemmi S, Dummer R. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res 2008; 68(3):650-6; PMID:18245463; https://doi.org/10.1158/0008-5472.CAN-07-2491
- Hoek KS, Schlegel NC, Brafford P, Sucker A, Ugurel S, Kumar R, Weber BL, Nathanson KL, Phillips DJ, Herlyn M, et al. Metastatic potential of melanomas defined by specific gene expression profiles with no BRAF signature. Pigment Cell Res 2006; 19(4):290-302; PMID:16827748; https://doi.org/10.1111/j.1600-0749.2006.00322.x
- Verfaillie A, Imrichova H, Atak ZK, Dewaele M, Rambow F, Hulselmans G, Christiaens V, Svetlichnyy D, Luciani F, Van den Mooter L, et al. Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat Commun 2015; 6:6683; PMID:25865119; https://doi.org/10.1038/ncomms7683
- Hsiao JJ, Fisher DE. The roles of microphthalmia-associated transcription factor and pigmentation in melanoma. Arch Biochem Biophys 2014; 563:28-34; PMID:25111671; https://doi.org/10.1016/j.abb.2014.07.019
- Landsberg J, Kohlmeyer J, Renn M, Bald T, Rogava M, Cron M, Fatho M, Lennerz V, Wölfel T, Hölzel M, et al. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature 2012; 490(7420):412-6; PMID:23051752; https://doi.org/10.1038/nature11538
- Almanzar G, Olkhanud PB, Bodogai M, Dell'agnola C, Baatar D, Hewitt SM, Ghimenton C, Tummala MK, Weeraratna AT, Hoek KS, et al. Sperm-derived SPANX-B is a clinically relevant tumor antigen that is expressed in human tumors and readily recognized by human CD4+ and CD8+ T cells. Clin Cancer Res 2009; 15(6):1954-63; PMID:19276289; https://doi.org/10.1158/1078-0432.CCR-08-1290
- Westbrook VA, Schoppee PD, Diekman AB, Klotz KL, Allietta M, Hogan KT, Slingluff CL, Patterson JW, Frierson HF, Irvin WP Jr, et al. Genomic organization, incidence, and localization of the SPAN-x family of cancer-testis antigens in melanoma tumors and cell lines. Clin Cancer Res 2004; 10(1 Pt 1):101-12; PMID:14734458; https://doi.org/10.1158/1078-0432.CCR-0647-3
- Kohlmeyer J, Cron M, Landsberg J, Bald T, Renn M, Mikus S, Bondong S, Wikasari D, Gaffal E, Hartmann G, et al. Complete regression of advanced primary and metastatic mouse melanomas following combination chemoimmunotherapy. Cancer Res 2009; 69(15):6265-74; PMID:19622767; https://doi.org/10.1158/0008-5472.CAN-09-0579
- Eggermont AM, Suciu S, Testori A, Kruit WH, Marsden J, Punt CJ, Santinami M, Salès F, Schadendorf D, Patel P, et al. Ulceration and stage are predictive of interferon efficacy in melanoma: Results of the phase III adjuvant trials EORTC 18952 and EORTC 18991. Eur J Cancer 2012; 48(2):218-25; PMID:22056637; https://doi.org/10.1016/j.ejca.2011.09.028
- Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schröter M, Burns K, Mattmann C, et al. Inhibition of death receptor signals by cellular FLIP. Nature 1997; 388(6638):190-5; PMID:9217161; https://doi.org/10.1038/40657
- Kataoka T, Schroter M, Hahne M, Schneider P, Irmler M, Thome M, Froelich CJ, Tschopp J. FLIP prevents apoptosis induced by death receptors but not by perforin/granzyme B, chemotherapeutic drugs, and gamma irradiation. J Immunol 1998; 161(8):3936-42; PMID:9780161
- Krueger A, Baumann S, Krammer PH, Kirchhoff S. FLICE-inhibitory proteins: Regulators of death receptor-mediated apoptosis. Mol Cell Biol 2001; 21(24):8247-54; PMID:11713262; https://doi.org/10.1128/MCB.21.24.8247-8254.2001
- Griffith TS, Chin WA, Jackson GC, Lynch DH, Kubin MZ. Intracellular regulation of TRAIL-induced apoptosis in human melanoma cells. J Immunol 1998; 161(6):2833-40; PMID:9743343
- Deveraux QL, Reed JC. IAP family proteins–suppressors of apoptosis. Genes Dev 1999; 13(3):239-52; PMID:9990849; https://doi.org/10.1101/gad.13.3.239
- Bullani RR, Huard B, Viard-Leveugle I, Byers HR, Irmler M, Saurat JH, Tschopp J, French LE. Selective expression of FLIP in malignant melanocytic skin lesions. J Invest Dermatol 2001; 117(2):360-4; PMID:11511316; https://doi.org/10.1046/j.0022-202x.2001.01418.x
- Keuling AM, Felton KE, Parker AA, Akbari M, Andrew SE, Tron VA. RNA silencing of mcl-1 enhances ABT-737-mediated apoptosis in melanoma: Role for a caspase-8-dependent pathway. PLoS One 2009; 4(8):e6651; PMID:19684859; https://doi.org/10.1371/journal.pone.0006651
- Senft D, Berking C, Graf SA, Kammerbauer C, Ruzicka T, Besch R. Selective induction of cell death in melanoma cell lines through targeting of mcl-1 and A1. PLoS One 2012; 7(1):e30821; PMID:22292048; https://doi.org/10.1371/journal.pone.0030821
- Jansen B, Schlagbauer-Wadl H, Eichler HG, Wolff K, van Elsas A, Schrier PI, Pehamberger H. Activated N-ras contributes to the chemoresistance of human melanoma in severe combined immunodeficiency (SCID) mice by blocking apoptosis. Cancer Res 1997; 57(3):362-5; PMID:9012455
- Wroblewski D, Mijatov B, Mohana-Kumaran N, Lai F, Gallagher SJ, Haass NK, Zhang XD, Hersey P. The BH3-mimetic ABT-737 sensitizes human melanoma cells to apoptosis induced by selective BRAF inhibitors but does not reverse acquired resistance. Carcinogenesis 2013; 34(2):237-47; PMID:23087082; https://doi.org/10.1093/carcin/bgs330
- Anvekar RA, Asciolla JJ, Lopez-Rivera E, Floros KV, Izadmehr S, Elkholi R, Belbin G, Sikora AG, Chipuk JE. Sensitization to the mitochondrial pathway of apoptosis augments melanoma tumor cell responses to conventional chemotherapeutic regimens. Cell Death Dis 2012; 3:e420; PMID:23152056; https://doi.org/10.1038/cddis.2012.161
- Bedikian AY, Millward M, Pehamberger H, Conry R, Gore M, Trefzer U, Pavlick AC, DeConti R, Hersh EM, Hersey P, et al. Bcl-2 antisense (oblimersen sodium) plus dacarbazine in patients with advanced melanoma: The oblimersen melanoma study group. J Clin Oncol 2006; 24(29):4738-45; PMID:16966688; https://doi.org/10.1200/JCO.2006.06.0483
- Jansen B, Wacheck V, Heere-Ress E, Schlagbauer-Wadl H, Hoeller C, Lucas T, Hoermann M, Hollenstein U, Wolff K, Pehamberger H, et al. Chemosensitisation of malignant melanoma by BCL2 antisense therapy. Lancet 2000; 356(9243):1728-33; PMID:11095261; https://doi.org/10.1016/S0140-6736(00)03207-4
- Tang L, Tron VA, Reed JC, Mah KJ, Krajewska M, Li G, Zhou X, Ho VC, Trotter MJ. Expression of apoptosis regulators in cutaneous malignant melanoma. Clin Cancer Res 1998; 4(8):1865-71; PMID:9717813
- Zhai D, Jin C, Huang Z, Satterthwait AC, Reed JC. Differential regulation of bax and bak by anti-apoptotic bcl-2 family proteins bcl-B and mcl-1. J Biol Chem 2008; 283(15):9580-6; PMID:18178565; https://doi.org/10.1074/jbc.M708426200
- Thallinger C, Wolschek MF, Wacheck V, Maierhofer H, Günsberg P, Polterauer P, Pehamberger H, Monia BP, Selzer E, Wolff K, et al. Mcl-1 antisense therapy chemosensitizes human melanoma in a SCID mouse xenotransplantation model. J Invest Dermatol 2003; 120(6):1081-6; PMID:12787138; https://doi.org/10.1046/j.1523-1747.2003.12252.x
- Haass NK, Sproesser K, Nguyen TK, Contractor R, Medina CA, Nathanson KL, Herlyn M, Smalley KS. The mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitor AZD6244 (ARRY-142886) induces growth arrest in melanoma cells and tumor regression when combined with docetaxel. Clin Cancer Res 2008; 14(1):230-9; PMID:18172275; https://doi.org/10.1158/1078-0432.CCR-07-1440
- Wang YF, Jiang CC, Kiejda KA, Gillespie S, Zhang XD, Hersey P. Apoptosis induction in human melanoma cells by inhibition of MEK is caspase-independent and mediated by the bcl-2 family members PUMA, bim, and mcl-1. Clin Cancer Res 2007; 13(16):4934-42; PMID:17652623; https://doi.org/10.1158/1078-0432.CCR-07-0665
- Deveraux QL, Stennicke HR, Salvesen GS, Reed JC. Endogenous inhibitors of caspases. J Clin Immunol 1999; 19(6):388-98; PMID:10634212; https://doi.org/10.1023/A:1020502800208
- Zhou J, Yuen NK, Zhan Q, Velazquez EF, Murphy GF, Giobbie-Hurder A, Hodi FS. Immunity to the melanoma inhibitor of apoptosis protein (ML-IAP; livin) in patients with malignant melanoma. Cancer Immunol Immunother 2012; 61(5):655-65; PMID:22033581; https://doi.org/10.1007/s00262-011-1124-1
- Gong J, Chen N, Zhou Q, Yang B, Wang Y, Wang X. Melanoma inhibitor of apoptosis protein is expressed differentially in melanoma and melanocytic naevus, but similarly in primary and metastatic melanomas. J Clin Pathol 2005; 58(10):1081-5; PMID:16189155; https://doi.org/10.1136/jcp.2005.025817
- Schimmer AD. Inhibitor of apoptosis proteins: Translating basic knowledge into clinical practice. Cancer Res 2004; 64(20):7183-90; PMID:15492230; https://doi.org/10.1158/0008-5472.CAN-04-1918
- Liu T, Brouha B, Grossman D. Rapid induction of mitochondrial events and caspase-independent apoptosis in survivin-targeted melanoma cells. Oncogene 2004; 23(1):39-48; PMID:14712209; https://doi.org/10.1038/sj.onc.1206978
- Grossman D, Kim PJ, Schechner JS, Altieri DC. Inhibition of melanoma tumor growth in vivo by survivin targeting. Proc Natl Acad Sci U S A 2001; 98(2):635-40; PMID:11149963; https://doi.org/10.1073/pnas.98.2.635
- Vucic D, Deshayes K, Ackerly H, Pisabarro MT, Kadkhodayan S, Fairbrother WJ, Dixit VM. SMAC negatively regulates the anti-apoptotic activity of melanoma inhibitor of apoptosis (ML-IAP). J Biol Chem 2002; 277(14):12275-9; PMID:11801603; https://doi.org/10.1074/jbc.M112045200
- Vucic D, Fairbrother WJ. The inhibitor of apoptosis proteins as therapeutic targets in cancer. Clin Cancer Res 2007; 13(20):5995-6000; PMID:17947460; https://doi.org/10.1158/1078-0432.CCR-07-0729
- Huang Y, Rich RL, Myszka DG, Wu H. Requirement of both the second and third BIR domains for the relief of X-linked inhibitor of apoptosis protein (XIAP)-mediated caspase inhibition by smac. J Biol Chem 2003; 278(49):49517-22; PMID:14512414; https://doi.org/10.1074/jbc.M310061200
- Vucic D, Franklin MC, Wallweber HJ, Das K, Eckelman BP, Shin H, Elliott LO, Kadkhodayan S, Deshayes K, Salvesen GS, et al. Engineering ML-IAP to produce an extraordinarily potent caspase 9 inhibitor: Implications for smac-dependent anti-apoptotic activity of ML-IAP. Biochem J 2005; 385(Pt 1):11-20; PMID:15485396; https://doi.org/10.1042/BJ20041108
- Schmollinger JC, Vonderheide RH, Hoar KM, Maecker B, Schultze JL, Hodi FS, Soiffer RJ, Jung K, Kuroda MJ, Letvin NL, et al. Melanoma inhibitor of apoptosis protein (ML-IAP) is a target for immune-mediated tumor destruction. Proc Natl Acad Sci U S A 2003; 100(6):3398-403; PMID:12626761; https://doi.org/10.1073/pnas.0530311100
- Yagihashi A, Asanuma K, Tsuji N, Torigoe T, Sato N, Hirata K, Watanabe N. Detection of anti-livin antibody in gastrointestinal cancer patients. Clin Chem 2003; 49(7):1206-8; PMID:12816927; https://doi.org/10.1373/49.7.1206
- Yagihashi A, Asanuma K, Kobayashi D, Tsuji N, Shijubo Y, Abe S, Hirohashi Y, Torigoe T, Sato N, Watanabe N. Detection of autoantibodies to livin and survivin in sera from lung cancer patients. Lung Cancer 2005; 48(2):217-21; PMID:15829321; https://doi.org/10.1016/j.lungcan.2004.11.002
- Yagihashi A, Ohmura T, Asanuma K, Kobayashi D, Tsuji N, Torigoe T, Sato N, Hirata K, Watanabe N. Detection of autoantibodies to survivin and livin in sera from patients with breast cancer. Clin Chim Acta 2005; 362(1-2):125-30; PMID:16026775; https://doi.org/10.1016/j.cccn.2005.06.009
- Andersen MH, Reker S, Becker JC, thor Straten P. The melanoma inhibitor of apoptosis protein: A target for spontaneous cytotoxic T cell responses. J Invest Dermatol 2004; 122(2):392-9; PMID:15009721; https://doi.org/10.1046/j.0022-202X.2004.22242.x
- Chang H, Schimmer AD. Livin/melanoma inhibitor of apoptosis protein as a potential therapeutic target for the treatment of malignancy. Mol Cancer Ther 2007; 6(1):24-30; PMID:17237263; https://doi.org/10.1158/1535-7163.MCT-06-0443
- Bird CH, Sutton VR, Sun J, Hirst CE, Novak A, Kumar S, Trapani JA, Bird PI. Selective regulation of apoptosis: The cytotoxic lymphocyte serpin proteinase inhibitor 9 protects against granzyme B-mediated apoptosis without perturbing the fas cell death pathway. Mol Cell Biol 1998; 18(11):6387-98; PMID:9774654; https://doi.org/10.1128/MCB.18.11.6387
- Medema JP, de Jong J, Peltenburg LT, Verdegaal EM, Gorter A, Bres SA, Franken KL, Hahne M, Albar JP, Melief CJ, et al. Blockade of the granzyme B/perforin pathway through overexpression of the serine protease inhibitor PI-9/SPI-6 constitutes a mechanism for immune escape by tumors. Proc Natl Acad Sci U S A 2001; 98(20):11515-20; PMID:11562487; https://doi.org/10.1073/pnas.201398198
- Cunningham TD, Jiang X, Shapiro DJ. Expression of high levels of human proteinase inhibitor 9 blocks both perforin/granzyme and fas/fas ligand-mediated cytotoxicity. Cell Immunol 2007; 245(1):32-41; PMID:17490628; https://doi.org/10.1016/j.cellimm.2007.03.004
- Soengas MS, Capodieci P, Polsky D, Mora J, Esteller M, Opitz-Araya X, McCombie R, Herman JG, Gerald WL, Lazebnik YA, et al. Inactivation of the apoptosis effector apaf-1 in malignant melanoma. Nature 2001; 409(6817):207-11; PMID:11196646; https://doi.org/10.1038/35051606
- Soengas MS, Capodieci P, Polsky D, Mora J, Esteller M, Opitz-Araya X, McCombie R, Herman JG, Gerald WL, Lazebnik YA, et al. Inactivation of the apoptosis effector apaf-1 in malignant melanoma. Nature 2001; 409(6817):207-11; PMID:11196646; https://doi.org/10.1038/35051606
- Chan H, Bartos DP, Owen-Schaub LB. Activation-dependent transcriptional regulation of the human fas promoter requires NF-kappaB p50-p65 recruitment. Mol Cell Biol 1999; 19(3):2098-108; PMID:10022897; https://doi.org/10.1128/MCB.19.3.2098
- Owen-Schaub LB, Zhang W, Cusack JC, Angelo LS, Santee SM, Fujiwara T, Roth JA, Deisseroth AB, Zhang WW, Kruzel E, et al. Wild-type human p53 and a temperature-sensitive mutant induce fas/APO-1 expression. Mol Cell Biol 1995; 15(6):3032-40; PMID:7539102; https://doi.org/10.1128/MCB.15.6.3032
- Ugurel S, Rappl G, Tilgen W, Reinhold U. Increased soluble CD95 (sFas/CD95) serum level correlates with poor prognosis in melanoma patients. Clin Cancer Res 2001; 7(5):1282-6; PMID:11350895
- Cheng J, Zhou T, Liu C, Shapiro JP, Brauer MJ, Kiefer MC, Barr PJ, Mountz JD. Protection from fas-mediated apoptosis by a soluble form of the fas molecule. Science 1994; 263(5154):1759-62; PMID:7510905; https://doi.org/10.1126/science.7510905
- Mouawad R, Khayat D, Soubrane C. Plasma fas ligand, an inducer of apoptosis, and plasma soluble fas, an inhibitor of apoptosis, in advanced melanoma. Melanoma Res 2000; 10(5):461-7; PMID:11095407; https://doi.org/10.1097/00008390-200010000-00008
- Ivanov VN, Bhoumik A, Ronai Z. Death receptors and melanoma resistance to apoptosis. Oncogene 2003; 22(20):3152-61; PMID:12789291; https://doi.org/10.1038/sj.onc.1206456
- Shin MS, Park WS, Kim SY, Kim HS, Kang SJ, Song KY, Park JY, Dong SM, Pi JH, Oh RR, et al. Alterations of fas (apo-1/CD95) gene in cutaneous malignant melanoma. Am J Pathol 1999; 154(6):1785-91; PMID:10362803; https://doi.org/10.1016/S0002-9440(10)65434-X
- Hahne M, Rimoldi D, Schroter M, Romero P, Schreier M, French LE, Schneider P, Bornand T, Fontana A, Lienard D, et al. Melanoma cell expression of fas(apo-1/CD95) ligand: Implications for tumor immune escape. Science 1996; 274(5291):1363-6; PMID:8910274; https://doi.org/10.1126/science.274.5291.1363
- O'Connell J, Bennett MW, Nally K, O'Sullivan GC, Collins JK, Shanahan F. Interferon-gamma sensitizes colonic epithelial cell lines to physiological and therapeutic inducers of colonocyte apoptosis. J Cell Physiol 2000; 185(3):331-8; PMID:11056003; https://doi.org/10.1002/1097-4652(200012)185:3%3c331::AID-JCP3%3e3.0.CO;2-V
- Ekmekcioglu S, Okcu MF, Colome-Grimmer MI, Owen-Schaub L, Buzaid AC, Grimm EA. Differential increase of fas ligand expression on metastatic and thin or thick primary melanoma cells compared with interleukin-10. Melanoma Res 1999; 9(3):261-72; PMID:10465582; https://doi.org/10.1097/00008390-199906000-00008
- Soubrane C, Mouawad R, Antoine EC, Verola O, Gil-Delgado M, Khayat D. A comparative study of fas and fas-ligand expression during melanoma progression. Br J Dermatol 2000; 143(2):307-12; PMID:10951137; https://doi.org/10.1046/j.1365-2133.2000.03655.x
- Aragane Y, Maeda A, Cui CY, Tezuka T, Kaneda Y, Schwarz T. Inhibition of growth of melanoma cells by CD95 (fas/APO-1) gene transfer in vivo. J Invest Dermatol 2000; 115(6):1008-14; PMID:11121134; https://doi.org/10.1046/j.1523-1747.2000.00164.x
- Arai H, Gordon D, Nabel EG, Nabel GJ. Gene transfer of fas ligand induces tumor regression in vivo. Proc Natl Acad Sci U S A 1997; 94(25):13862-7; PMID:9391118; https://doi.org/10.1073/pnas.94.25.13862
- Zhang XY, Zhang XD, Borrow JM, Nguyen T, Hersey P. Translational control of tumor necrosis factor-related apoptosis-inducing ligand death receptor expression in melanoma cells. J Biol Chem 2004; 279(11):10606-14; PMID:14688276; https://doi.org/10.1074/jbc.M308211200
- Zhang XD, Zhang XY, Gray CP, Nguyen T, Hersey P. Tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis of human melanoma is regulated by smac/DIABLO release from mitochondria. Cancer Res 2001; 61(19):7339-48; PMID:11585775
- Kurbanov BM, Fecker LF, Geilen CC, Sterry W, Eberle J. Resistance of melanoma cells to TRAIL does not result from upregulation of antiapoptotic proteins by NF-kappaB but is related to downregulation of initiator caspases and DR4. Oncogene 2007; 26(23):3364-77; PMID:17160022; https://doi.org/10.1038/sj.onc.1210134
- Franco AV, Zhang XD, Van Berkel E, Sanders JE, Zhang XY, Thomas WD, Nguyen T, Hersey P. The role of NF-kappa B in TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis of melanoma cells. J Immunol 2001; 166(9):5337-45; PMID:11313369; https://doi.org/10.4049/jimmunol.166.9.5337
- Griffith TS, Chin WA, Jackson GC, Lynch DH, Kubin MZ. Intracellular regulation of TRAIL-induced apoptosis in human melanoma cells. J Immunol 1998; 161(6):2833-40; PMID:9743343
- Krammer PH. CD95s deadly mission in the immune system. Nature 2000; 407(6805):789-95; PMID:11048730; https://doi.org/10.1038/35037728
- Ashkenazi A. Targeting death and decoy receptors of the tumour-necrosis factor superfamily. Nat Rev Cancer 2002; 2(6):420-30; PMID:12189384; https://doi.org/10.1038/nrc821
- Thomas WD, Hersey P. TNF-related apoptosis-inducing ligand (TRAIL) induces apoptosis in fas ligand-resistant melanoma cells and mediates CD4 T cell killing of target cells. J Immunol 1998; 161(5):2195-200; PMID:9725211
- Chawla-Sarkar M, Bae SI, Reu FJ, Jacobs BS, Lindner DJ, Borden EC. Downregulation of bcl-2, FLIP or IAPs (XIAP and survivin) by siRNAs sensitizes resistant melanoma cells to Apo2L/TRAIL-induced apoptosis. Cell Death Differ 2004; 11(8):915-23; PMID:15118763; https://doi.org/10.1038/sj.cdd.4401416
- Lillehammer T, Engesaeter BO, Prasmickaite L, Maelandsmo GM, Fodstad O, Engebraaten O. Combined treatment with ad-hTRAIL and DTIC or SAHA is associated with increased mitochondrial-mediated apoptosis in human melanoma cell lines. J Gene Med 2007; 9(6):440-51; PMID:17410615; https://doi.org/10.1002/jgm.1036
- Lee J, Hampl M, Albert P, Fine HA. Antitumor activity and prolonged expression from a TRAIL-expressing adenoviral vector. Neoplasia 2002; 4(4):312-23; PMID:12082547; https://doi.org/10.1038/sj.neo.7900245
- Boyle GM, Martyn AC, Parsons PG. Histone deacetylase inhibitors and malignant melanoma. Pigment Cell Res 2005; 18(3):160-6; PMID:15892712; https://doi.org/10.1111/j.1600-0749.2005.00228.x
- Krammer PH. The tumor strikes back: New data on expression of the CD95(APO-1/fas) receptor/ligand system may cause paradigm changes in our view on drug treatment and tumor immunology. Cell Death Differ 1997; 4(5):362-4; PMID:16465254; https://doi.org/10.1038/sj.cdd.4400252
- Igney FH, Krammer PH. Tumor counterattack: Fact or fiction? Cancer Immunol Immunother 2005; 54(11):1127-36; PMID:15889255; https://doi.org/10.1007/s00262-005-0680-7
- Igney FH, Behrens CK, Krammer PH. Tumor counterattack–concept and reality. Eur J Immunol 2000; 30(3):725-31; PMID:10741386; https://doi.org/10.1002/1521-4141(200003)30:3%3c725::AID-IMMU725%3e3.0.CO;2-D 10.1002/1521-4141(200003)30:3%3c725::AID-IMMU725%3e3.3.CO;2-4
- French LE, Wilson A, Hahne M, Viard I, Tschopp J, MacDonald HR. Fas ligand expression is restricted to nonlymphoid thymic components in situ. J Immunol 1997; 159(5):2196-202; PMID:9278307
- Griffith TS, Brunner T, Fletcher SM, Green DR, Ferguson TA. Fas ligand-induced apoptosis as a mechanism of immune privilege. Science 1995; 270(5239):1189-92; PMID:7502042; https://doi.org/10.1126/science.270.5239.1189
- Stuart PM, Griffith TS, Usui N, Pepose J, Yu X, Ferguson TA. CD95 ligand (FasL)-induced apoptosis is necessary for corneal allograft survival. J Clin Invest 1997; 99(3):396-402; PMID:9022072; https://doi.org/10.1172/JCI119173
- Rivoltini L, Carrabba M, Huber V, Castelli C, Novellino L, Dalerba P, Mortarini R, Arancia G, Anichini A, Fais S, et al. Immunity to cancer: Attack and escape in T lymphocyte-tumor cell interaction. Immunol Rev 2002; 188:97-113; PMID:12445284; https://doi.org/10.1034/j.1600-065X.2002.18809.x
- Andreola G, Rivoltini L, Castelli C, Huber V, Perego P, Deho P, Squarcina P, Accornero P, Lozupone F, Lugini L, et al. Induction of lymphocyte apoptosis by tumor cell secretion of FasL-bearing microvesicles. J Exp Med 2002; 195(10):1303-16; PMID:12021310; https://doi.org/10.1084/jem.20011624
- Seino K, Ogino T, Fukunaga K, Taniguchi H, Takada Y, Yuzawa K, Otsuka M, Yagita H, Okumura K, Fukao K. Attempts to reveal the mechanism of CD95-ligand-mediated inflammation. Transplant Proc 1999; 31(5):1942-3; PMID:10455926; https://doi.org/10.1016/S0041-1345(99)00219-5
- Giovarelli M, Musiani P, Garotta G, Ebner R, Di Carlo E, Kim Y, Cappello P, Rigamonti L, Bernabei P, Novelli F, et al. A “stealth effect:” Adenocarcinoma cells engineered to express TRAIL elude tumor-specific and allogeneic T cell reactions. J Immunol 1999; 163(9):4886-93; PMID:10528190
- Nakashima M, Sonoda K, Watanabe T. Inhibition of cell growth and induction of apoptotic cell death by the human tumor-associated antigen RCAS1. Nat Med 1999; 5(8):938-42; PMID:10426319; https://doi.org/10.1038/11383
- Mellado M, de Ana AM, Moreno MC, Martinez C, Rodriguez-Frade JM. A potential immune escape mechanism by melanoma cells through the activation of chemokine-induced T cell death. Curr Biol 2001; 11(9):691-6; PMID:11369232; https://doi.org/10.1016/S0960-9822(01)00199-3
- Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, Mitra D, Boni A, Newton LP, Liu C, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res 2013; 19(5):1225-31; PMID:23307859; https://doi.org/10.1158/1078-0432.CCR-12-1630