
ABSTRACT
The oncoprotein c-Myc is often overexpressed in cancer cells, and the stability of this protein has major significance in deciding the fate of a cell. Thus, targeting c-Myc levels is an attractive approach for developing therapeutic agents for cancer treatment. In this study, we report the anti-cancer activity of the macrocyclic peptides [D-Trp]CJ-15,208 (cyclo[Phe-D-Pro-Phe-D-Trp]) and the natural product CJ-15,208 (cyclo[Phe-D-Pro-Phe-Trp]). [D-Trp]CJ-15,208 reduced c-Myc protein levels in prostate cancer cells and decreased cell proliferation with IC50 values ranging from 2.0 to 16 µM in multiple PC cell lines. [D-Trp]CJ-15,208 induced early and late apoptosis in PC-3 cells following 48 hours treatment, and growth arrest in the G2 cell cycle phase following both 24 and 48 hours treatment. Down regulation of c-Myc in PC-3 cells resulted in loss of sensitivity to [D-Trp]CJ-15,208 treatment, while overexpression of c-Myc in HEK-293 cells imparted sensitivity of these cells to [D-Trp]CJ-15,208 treatment. This macrocyclic tetrapeptide also regulated PP2A by reducing the levels of its phosphorylated form which regulates the stability of cellular c-Myc protein. Thus [D-Trp]CJ-15,208 represents a new lead compound for the potential development of an effective treatment of prostate cancer.
Abbreviations
| APC | = | allophycocyanin |
| AR | = | androgen receptor |
| BPH-1 | = | benign prostatic hyperplasia epithelial cells |
| CDK | = | cyclin dependent kinase |
| CR | = | castration resistant |
| DMSO | = | dimethyl sulfoxide |
| HEK | = | human embryonic kidney cells |
| KOR | = | kappa opioid receptor |
| mCRPC | = | metastatic castration resistant prostate cancer |
| PC | = | prostate cancer |
| PI | = | propidium iodide |
| PP2A | = | protein phosphatase 2A |
Introduction
Current therapies, i.e. androgen deprivation therapy or the application of androgen receptor antagonists, are initially effective in treating prostate cancer (PC), but the disease generally recurs with metastasis due to non-responsiveness to these treatments.Citation1-3 These therapies lower the level of the androgens testosterone and dihydrotestosterone or interfere with the binding of androgen ligands to androgen receptors (AR), respectively,Citation4 but the disease typically progresses despite application of these treatments, with the AR typically being expressed in truncated or mutated form.Citation5 Although the mechanism for cancer progression after androgen deprivation/AR antagonist treatment is not fully understood, possible explanations include: i) AR gene amplification, ii) constitutive activation of AR, iii) AR mutation or truncation, iv) activation of AR by growth factors, and/or v) activation of AR by alternative androgen independent signaling pathways.Citation5,Citation6 Thus, identifying compounds that can alter androgen dependent and independent pathways should facilitate drug design and could potentially result in drug development to treat metastatic and castration resistant prostate cancer.
This study focused on the oncoprotein c-MycCitation5,Citation7 in prostate cancer cell growth. c-Myc is a transcription factor that is overexpressed in most cancer types.Citation8-10 It accumulates and is stabilized by phosphorylation as phospho-Ser62-c-Myc.Citation11-13 c-Myc is upregulated in both androgen receptor positive and androgen receptor negative, ligand-independent castration resistant prostate cancers,Citation5,Citation14-16 and recent studies have shown that the androgen receptor directly regulates c-Myc protein levels in an androgen independent manner.Citation5,Citation14,Citation17 Thus, the regulation of c-Myc protein levels in cancer cells holds therapeutic promise to prevent tumor growth and is a potential approach for anti-cancer drug development.
Both linear and cyclic peptidesCitation18,Citation19 have been examined in recent years as potential therapeutic agents for the treatment of several cancers.Citation20 The natural product macrocyclic tetrapeptide CJ-15,208 (cyclo[Phe-D-Pro-Phe-Trp]), which was isolated from the fermentation broth of a fungus,Citation21 was reported to bind to kappa opioid receptors (KOR) and antagonize this receptor in vitro.Citation21 We have synthesized this macrocyclic tetrapeptide CJ-15,208 along with its D-Trp isomer,Citation22,Citation23 and found that both peptides exhibit activity mediated through opioid receptors.Citation23-25 Opioid receptors have been reported on a variety of human tumors cells and cancer cell lines, and evidence suggests that opioids can modulate tumor growth.Citation26 However, there are conflicting reports on the effects of opioids on cancer cell proliferation.Citation26,Citation27 The endogenous KOR peptide dynorphin A (Dyn A) was reported to significantly stimulate the growth of the metastatic prostate cancer cell line DU145.Citation28 In contrast, the selective KOR small molecule agonist U50,488H was reported to decrease the growth of non-small cell lung cancer cells.Citation29
Therefore, we assessed the anti-cancer activity of several KOR peptide ligands, including the 2 lead peptides CJ-15,208 and its D-Trp isomer, in prostate cancer cells. Both of these macrocyclic peptides induced cytotoxicity and lowered c-Myc protein levels in prostate cancer cells. [D-Trp]CJ-15,208 induced early apoptosis and arrested the cell cycle at the G2 phase in PC cells. Moreover, altering c-Myc protein expression levels affected the sensitivity of the cells to [D-Trp]CJ-15,208 treatment. The detailed effects of [D-Trp]CJ-15,208 on the phosphorylation state of several proteins involved in regulating the stability of the c-Myc protein were also evaluated. The results of these studies are described below.
Results
Effect of KOR peptides on c-Myc protein levels in prostate cancer cells
Many cancer cells, including prostate cancer cells, overexpress c-Myc.Citation9,Citation30 We examined the effect of several KOR ligands, including the macrocyclic tetrapeptides CJ-15,208 and its D-Trp isomer ( and ), the endogenous KOR agonist dynorphin A (Dyn A, ),Citation28 and the KOR antagonist zyklophin (),Citation31 on cellular c-Myc protein levels. The cellular levels of this protein decreased 5-fold ( lanes 4 and 5) following treatment with the macrocyclic tetrapeptides CJ-15,208 and its D-Trp isomer, but were not decreased by treatment with dynorphin A or its antagonist analog zyklophin (lanes 2 and 3). A radioligand binding assay for KOR, performed under standard conditions using [3H]diprenorphine (and Dyn A-(1–13) amide to determine nonspecific bindingCitation32), showed no detectable specific KOR binding to PC-3 cells (data not shown). Also treatment of PC-3 cells with 10 µM of the nonselective opioid antagonist naloxone for 48 h had no effect on c-Myc protein levels (lane 3), and did not alter the decrease in c-Myc protein levels caused by [D-Trp]CJ-15,208 (lane 4 vs. lane 2). These results provide strong evidence that the effect of these cyclic tetrapeptides on c-Myc in the prostate cancer cells is not mediated through KOR.
Figure 1. Macrocyclic tetrapeptides CJ-15,208 and [D-Trp]CJ-15,208, but not Dyn A or zyklophin, decrease c-Myc protein levels in PC-3 cells. Structures of (A) CJ-15,208, (B) [D-Trp]CJ-15,208, (C) dynorphin A-(1-13), and (D) zyklophin. (E) Cellular c-Myc protein levels were determined by western blot analysis following treatment with: lane 1: 0.5% DMSO (vehicle), lane 2: 1 µM dynorphin A (Dyn A), lane 3: 10 µM zyklophin, lane 4: 50 µM CJ-15,208, and lane 5: 50 µM [D-Trp]CJ-15,208. A representative immunoblot is shown from duplicate experiments. (F) c-Myc protein levels in PC-3 cells were examined by western blot analysis after treating the cells with naloxone (10 µM) and/or [D-Trp]CJ-15,208 (10 µM). The immunoblot shown is representative of duplicate experiments; lanes 1/2 and 3/4 are from different parts of the same gel.
![Figure 1. Macrocyclic tetrapeptides CJ-15,208 and [D-Trp]CJ-15,208, but not Dyn A or zyklophin, decrease c-Myc protein levels in PC-3 cells. Structures of (A) CJ-15,208, (B) [D-Trp]CJ-15,208, (C) dynorphin A-(1-13), and (D) zyklophin. (E) Cellular c-Myc protein levels were determined by western blot analysis following treatment with: lane 1: 0.5% DMSO (vehicle), lane 2: 1 µM dynorphin A (Dyn A), lane 3: 10 µM zyklophin, lane 4: 50 µM CJ-15,208, and lane 5: 50 µM [D-Trp]CJ-15,208. A representative immunoblot is shown from duplicate experiments. (F) c-Myc protein levels in PC-3 cells were examined by western blot analysis after treating the cells with naloxone (10 µM) and/or [D-Trp]CJ-15,208 (10 µM). The immunoblot shown is representative of duplicate experiments; lanes 1/2 and 3/4 are from different parts of the same gel.](/cms/asset/533627b3-8671-49fc-a891-90956db85d15/kcbt_a_1345384_f0001_b.gif)
Both [D-Trp]CJ-15,208 and CJ-15,208 reduced c-Myc protein levels in PC-3 cells in a concentration dependent manner after 48 h treatment ( and ), causing approximately a 50% reduction at a concentration of 10 µM (lane 3 and lane 4). After 48 h treatment these peptides reduced cell proliferation in PC-3 cells with IC50 values of 10.1 ± 0.9 µM and 16.5 ± 0.7 µM for CJ-15,208 and its D-Trp isomer, respectively (), demonstrating that these cyclic tetrapeptides possess anti-cancer activity in this PC cell line. In contrast, treatment of PC-3 cells with 10 µM of [D-Trp]CJ-15,208 for 0 to 48 h did not affect mRNA expression at any time point (). These results indicate that [D-Trp]CJ-15,208 does not affect c-Myc gene transcription, but decreases c-Myc protein levels in these cancer cells. We further explored the anti-cancer activity of these macrocyclic peptides using [D-Trp]CJ-15,208.
Figure 2. The macrocyclic tetrapeptides [D-Trp]CJ-15,208 and CJ-15,208 reduce c-Myc protein levels and prevent cell growth, but [D-Trp]CJ-15,208 has no effect on c-Myc mRNA expression in PC-3 cells. PC-3 cells were treated with (A) CJ-15,208 and (B) [D-Trp]CJ-15,208 for 48 h at the indicated concentrations, and c-Myc protein levels determined by western blot analysis using c-Myc antibody. The effects of [D-Trp]CJ-15,208 treatment on c-Myc levels from 2 independent experiments are also shown graphically. (C) Cell proliferation was assessed using WST-1 after treating the cells with CJ-15,208 or [D-Trp]CJ-15,208 at the indicated concentrations for 48 h. The data shown are the results from 2 independent experiments, with each data point obtained from triplicate measurements. (D) c-Myc mRNA expression in PC-3 cells was determined following treatment with 10 µM [D-Trp]CJ-15,208 for 0–48 h as described in Materials and Methods.
![Figure 2. The macrocyclic tetrapeptides [D-Trp]CJ-15,208 and CJ-15,208 reduce c-Myc protein levels and prevent cell growth, but [D-Trp]CJ-15,208 has no effect on c-Myc mRNA expression in PC-3 cells. PC-3 cells were treated with (A) CJ-15,208 and (B) [D-Trp]CJ-15,208 for 48 h at the indicated concentrations, and c-Myc protein levels determined by western blot analysis using c-Myc antibody. The effects of [D-Trp]CJ-15,208 treatment on c-Myc levels from 2 independent experiments are also shown graphically. (C) Cell proliferation was assessed using WST-1 after treating the cells with CJ-15,208 or [D-Trp]CJ-15,208 at the indicated concentrations for 48 h. The data shown are the results from 2 independent experiments, with each data point obtained from triplicate measurements. (D) c-Myc mRNA expression in PC-3 cells was determined following treatment with 10 µM [D-Trp]CJ-15,208 for 0–48 h as described in Materials and Methods.](/cms/asset/f66be426-b955-438c-ba33-1146819bf512/kcbt_a_1345384_f0002_b.gif)
Cytotoxicity of [D-Trp]CJ-15,208 in different PC cell lines
We examined the cytotoxicity of this lead compound in several PC cell lines, namely PC-3, LNCaP, DU145, 22Rv1, and C4–2 cells. BPH-1 was used as a normal prostate epithelial cell line and HEK (human embryonic kidney) cells were used as a non-prostate cell line to assess the selectivity of the cytotoxicity for PC cells. Cell proliferation assays () were performed using WST-1 (Roche) as described in the Materials and Methods. [D-Trp]CJ-15,208 inhibited cell growth in most of the PC cell lines (), with IC50 values of 3.0 ± 1.0 µM in LNCaP cells following 72 h treatment, 1.2 ± 0.2 µM in DU145 cells after 48 h treatment, and 8.8 ± 4.0 µM in 22Rv1 cells after 48 h treatment. However, the peptide had no effect on cell proliferation of C4–2 cells, a castration resistant prostate cancer (CRPC) cell line. Also no cytotoxicity of this compound was observed in normal prostate BPH-1 cells or non-prostate HEK cells (). The c-Myc protein levels in the different PC cell lines and normal cells () were consistent with the compound's cytotoxicity. All of the PC cell lines except C4–2 cells expressed high levels of cellular c-Myc protein, while the normal BPH-1 cells and the C4–2 cells had low levels of c-Myc protein. While the HEK-293 cells exhibited higher c-Myc protein levels than expected, overexpression of c-Myc was necessary to make these cells sensitive to [D-Trp]CJ-15,208 (see and related discussion below). The C4–2 cell line was not studied further, and our experiments focused on PC-3, LNCaP, and 22Rv1 cells.
Figure 3. [D-Trp]CJ-15,208 decreases cell proliferation in several PC cell lines, but not in normal prostate epithelial BPH-1 cells or non-prostate HEK cells. Cell proliferation was measured using WST-1 as described in the Materials and Methods. Cells were treated with the compound at the indicated concentrations and time points. The data shown are the results from 2 independent experiments, with each data point obtained from triplicate measurements. (A) PC-3, (B) LNCaP, (C) DU145, (D) 22Rv1, (E) C4–2, (F) BPH-1, and (G) HEK cells. (H) Western blot analysis for c-Myc protein levels in various PC cell lines plus BPH-1 and HEK-293 cells; dashed line indicates separate blots.
![Figure 3. [D-Trp]CJ-15,208 decreases cell proliferation in several PC cell lines, but not in normal prostate epithelial BPH-1 cells or non-prostate HEK cells. Cell proliferation was measured using WST-1 as described in the Materials and Methods. Cells were treated with the compound at the indicated concentrations and time points. The data shown are the results from 2 independent experiments, with each data point obtained from triplicate measurements. (A) PC-3, (B) LNCaP, (C) DU145, (D) 22Rv1, (E) C4–2, (F) BPH-1, and (G) HEK cells. (H) Western blot analysis for c-Myc protein levels in various PC cell lines plus BPH-1 and HEK-293 cells; dashed line indicates separate blots.](/cms/asset/3953f6af-24f8-4b8b-a773-c93f5694c095/kcbt_a_1345384_f0003_b.gif)
Table 1. Effect of [D-Trp]CJ-15,208 on cell growth in various cancer cell lines,Citation46 normal prostate cells,Citation46 and non-prostate cells.
We further confirmed that treatment of PC cells with the peptide resulted in cell death using the Trypan Blue assay as described in the Materials and Methods. Cell viability in the PC cell lines following 48 and 72 h treatment () correlated with the results from the cell proliferation assay. As expected, we did not see any evidence of cell death in BPH-1 cells after treatment with 1–40 µM of the compound for 48 h.
Figure 4. Cytotoxicity of [D-Trp]CJ-15,208 in PC cells as determined by Trypan Blue. The viability of PC cells following treatment with [D-Trp]CJ-15,208 at the indicated concentrations and time points was determined using Trypan Blue as described in the Materials and Methods. Data shown are the results of duplicate experiments, with triplicate measurements for each data point. (A) PC-3, (B) LNCaP, (C) 22RV1, and (D) BPH-1 cells.
![Figure 4. Cytotoxicity of [D-Trp]CJ-15,208 in PC cells as determined by Trypan Blue. The viability of PC cells following treatment with [D-Trp]CJ-15,208 at the indicated concentrations and time points was determined using Trypan Blue as described in the Materials and Methods. Data shown are the results of duplicate experiments, with triplicate measurements for each data point. (A) PC-3, (B) LNCaP, (C) 22RV1, and (D) BPH-1 cells.](/cms/asset/118c88ef-7c97-421c-a708-6461e388556a/kcbt_a_1345384_f0004_b.gif)
[D-Trp]CJ-15,208 regulates cell cycle progression and apoptosis in PC-3 cells
The effect of peptide treatment on cell cycle progression in PC-3 cells was examined by flow cytometry following staining the cells with propidium iodide (PI) as described in the Materials and Methods. Following treatment with 10 µM of the compound for 24 or 48 h, a larger number of cells (at least 8-fold higher) accumulated in the G2 phase compared with vehicle treated control cells () (5 ± 1.5% for control, 42 ± 4% following 24 h, and 58 ± 4% following 48 h treatment). The mean cell population in the G1 phase was 53 ± 6% for control, 32 ± 1% for 24 h, and 31 ± 6% for 48 h treatment. Similarly, the mean cell distribution in S phase was 45 ± 4% for control, 26 ± 4% following 24 h, and 12 ± 1% following 48 h treatment. These results indicate that the peptide [D-Trp]CJ-15,208 significantly induces growth arrest at the G2 phase in cell cycle progression in PC-3 cells. The cell cycle phase distributions are summarized in .
Figure 5. [D-Trp]CJ-15,208 induces G2 cell cycle arrest in PC-3 cells after 24 or 48 h treatment. Following treatment with 10 µM compound for 24 or 48 h, the cells were stained with propidium iodide (PI) before cell cycle analysis using flow cytometry as described in the Materials and Methods. (A) Representative cell cycle phase distribution following 24 and 48 h treatment. (B) Summary of % cell population in different phases. Data shown are the results of duplicate experiments. Statistical analyses were performed as described in the Materials and Methods; * p<0.01, **, p<0.003 and *** p<0.001 compared with vehicle (0.5% DMSO) treated control cells.
![Figure 5. [D-Trp]CJ-15,208 induces G2 cell cycle arrest in PC-3 cells after 24 or 48 h treatment. Following treatment with 10 µM compound for 24 or 48 h, the cells were stained with propidium iodide (PI) before cell cycle analysis using flow cytometry as described in the Materials and Methods. (A) Representative cell cycle phase distribution following 24 and 48 h treatment. (B) Summary of % cell population in different phases. Data shown are the results of duplicate experiments. Statistical analyses were performed as described in the Materials and Methods; * p<0.01, **, p<0.003 and *** p<0.001 compared with vehicle (0.5% DMSO) treated control cells.](/cms/asset/8a9928dc-71bb-461b-942c-1caa07d0e6d5/kcbt_a_1345384_f0005_b.gif)
We further examined whether [D-Trp]CJ-15,208 can induce apoptosis in PC-3 cells. The apoptotic cells were detected using flow cytometry following double staining with annexin V-APC and PI as described in the Materials and Methods. In , the upper left quadrant (Q1) indicates the presence of early apoptotic cells which are positively stained by annexin V but not stained by PI, the upper right quadrant (Q2) indicates the late apoptotic cells which are positively stained by both PI and annexin V, the lower right quadrant (Q3) shows the dead cells that are positively stained only by PI but not by annexin V, and the lower left quadrant (Q4) indicates the viable cells which are not stained by either PI or annexin V. Following 48 h treatment with 10 µM of the peptide, the number of cells in early and late apoptosis increased by 10- and 3-fold, respectively, compared with vehicle treated control cells. No significant change in the number of cells was observed in early or late apoptosis following 24 h treatment with 10 µM of the peptide ().
Figure 6. [D-Trp]CJ-15,208 induces early apoptosis in PC-3 cells following 48 h treatment. Following treatment with 10 µM compound for 24 or 48 h cells were doubly stained with annexin V-APC and PI, and the extent of apoptosis was determined by flow cytometer as described in the Materials and Methods. (A) Q1, the top left quadrant, represents early apoptotic cells which were positively stained with annexin V, but not stained with PI; Q2, the top right quadrant, indicates late apoptotic cells positively stained by both PI and annexin V; the lower right quadrant Q3 were necrotic cells positively stained by PI and not stained by annexin V; and the lower left quadrant Q4 indicates viable cells that were not stained by either PI or annexin V. (B) Summary of flow cytometric analysis showing % cell population in early and late apoptosis. Statistical analyses were performed as described in Materials and Methods; *p<0.02 and ** p<0.01 compared with vehicle treated control cells.
![Figure 6. [D-Trp]CJ-15,208 induces early apoptosis in PC-3 cells following 48 h treatment. Following treatment with 10 µM compound for 24 or 48 h cells were doubly stained with annexin V-APC and PI, and the extent of apoptosis was determined by flow cytometer as described in the Materials and Methods. (A) Q1, the top left quadrant, represents early apoptotic cells which were positively stained with annexin V, but not stained with PI; Q2, the top right quadrant, indicates late apoptotic cells positively stained by both PI and annexin V; the lower right quadrant Q3 were necrotic cells positively stained by PI and not stained by annexin V; and the lower left quadrant Q4 indicates viable cells that were not stained by either PI or annexin V. (B) Summary of flow cytometric analysis showing % cell population in early and late apoptosis. Statistical analyses were performed as described in Materials and Methods; *p<0.02 and ** p<0.01 compared with vehicle treated control cells.](/cms/asset/b9cbec1f-6afc-4052-859d-c88133afee99/kcbt_a_1345384_f0006_b.gif)
Altering c-Myc expression levels affects sensitivity of cells to [D-Trp]CJ-15,208 treatment
To verify the involvement of c-Myc downregulation in the effects of [D-Trp]CJ-15,208, experiments involving both knockdown and overexpression of c-Myc were performed. In the knockdown experiments, PC-3 cells transfected with either siRNA against c-Myc or a nontargeting siRNA were subsequently treated with 10 µM [D-Trp]CJ-15,208 for 48 h, and the cells were analyzed for apoptosis (). Treatment with [D-Trp]CJ-15,208 significantly increased early apoptosis in cells transfected with nontargeting siRNA, but did not significantly affect the percent of cells transfected with the siRNA against c-Myc that underwent early apoptosis (). Western blot analysis confirmed the knockdown of c-Myc in the cells transfected with siRNA against c-Myc ().
Figure 7. Knockdown of c-Myc expression in PC-3 cells results in loss of sensitivity to [D-Trp]CJ-15,208. PC-3 cells were treated with either a nontargeting siRNA or c-Myc siRNA as described in Materials and Methods. (A) Following treatment with 10 µM [D-Trp]CJ-15,208 ([D-Trp]CJ) for 48 h, cells were stained with annexin V-APC and PI and analyzed by flow cytometry. (B) Percent cell population in early and late apoptosis. Statistical analyses were performed as described in Materials and Methods; * p<0.05 compared with cells treated with DMSO containing vehicle. (C) Western blot analysis verified c-Myc knockdown by the c-Myc siRNA.
![Figure 7. Knockdown of c-Myc expression in PC-3 cells results in loss of sensitivity to [D-Trp]CJ-15,208. PC-3 cells were treated with either a nontargeting siRNA or c-Myc siRNA as described in Materials and Methods. (A) Following treatment with 10 µM [D-Trp]CJ-15,208 ([D-Trp]CJ) for 48 h, cells were stained with annexin V-APC and PI and analyzed by flow cytometry. (B) Percent cell population in early and late apoptosis. Statistical analyses were performed as described in Materials and Methods; * p<0.05 compared with cells treated with DMSO containing vehicle. (C) Western blot analysis verified c-Myc knockdown by the c-Myc siRNA.](/cms/asset/8e24bc4f-1270-4fc0-9720-cad888997edd/kcbt_a_1345384_f0007_b.gif)
HEK-293 cells transfected with either a pcDNA containing an HA-tagged human c-Myc insert to overexpress the protein or a vector control were also treated with 10 µM [D-Trp]CJ-15,208 for 48 h, and the cells were analyzed for apoptosis (). While no significant difference was seen in cells transfected with the empty vector, cells that overexpressed c-Myc showed a significant increase in early apotosis when treated with [D-Trp]-CJ-15,208 (), consistent with what was observed in PC-3 cells. Western blot analysis confirmed the overexpression of c-Myc in the cells transfected with the c-Myc containing plasmid ().
Figure 8. Overexpression of c-Myc in HEK-293 cells imparts sensitivity to [D-Trp]CJ-15,208. HEK-293 cells were transfected with either a vector control plasmid or a plasmid containing HA-HA-c-Myc as described in Materials and Methods. (A) Following treatment with 10 µM [D-Trp]CJ-15,208 ([D-Trp]CJ) for 48 h, cells were stained with annexin V-APC and PI and analyzed by flow cytometry. (B) Percent cell population in early and late apoptosis. Statistical analyses were performed as described in Materials and Methods; *p<0.05 compared with cells treated with DMSO containing vehicle. (C) Western blot analysis was used to verify c-Myc transfection in the cells transfected with the plasmid containing HA-HA-c-Myc.
![Figure 8. Overexpression of c-Myc in HEK-293 cells imparts sensitivity to [D-Trp]CJ-15,208. HEK-293 cells were transfected with either a vector control plasmid or a plasmid containing HA-HA-c-Myc as described in Materials and Methods. (A) Following treatment with 10 µM [D-Trp]CJ-15,208 ([D-Trp]CJ) for 48 h, cells were stained with annexin V-APC and PI and analyzed by flow cytometry. (B) Percent cell population in early and late apoptosis. Statistical analyses were performed as described in Materials and Methods; *p<0.05 compared with cells treated with DMSO containing vehicle. (C) Western blot analysis was used to verify c-Myc transfection in the cells transfected with the plasmid containing HA-HA-c-Myc.](/cms/asset/e4fc32f9-ed46-4283-bf6f-e7ae4f777968/kcbt_a_1345384_f0008_b.gif)
[D-Trp]CJ-15,208 regulates the phosphorylation state of PP2A in PC-3 cells which affects c-Myc stability
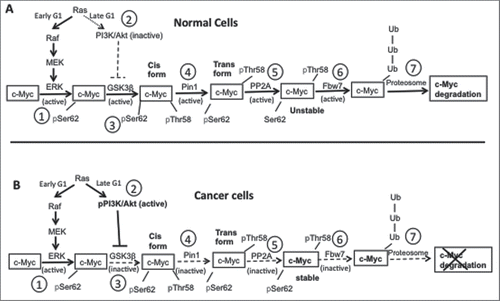
The stability of cellular c-Myc protein is determined by the tight regulation of its phosphorylation state.Citation11,Citation13,Citation33 c-Myc protein degradation is important to maintain normal cellular functions, whereas high levels of c-Myc protein have been found in various cancer cells including prostate cancer.Citation13,Citation16 The schematic diagrams in illustrate the pathway that determines c-Myc stability in normal () vs. cancer cells (). A series of key enzymes, mainly Erk, PI3K/Akt, and PP2A (protein phosphatase 2A), are involved in regulating c-Myc phosphorylation and its degradation via this pathway (see the Discussion section for descriptions of the roles of these enzymes).
Figure 9. A schematic diagram showing c-Myc phosphorylation and degradation in normal vs. cancer cells. (A) c-Myc degradation in normal cells. Step 1: In early G1 phase of the cell cycle, and in response to mitogenic stimulation, Ras is activated and phosphorylates Erk through the Raf/MEK/ERK signaling cascade. Phosphorylation activates Erk, which then phosphorylates c-Myc on Ser62. Step 2: As the cell cycle progresses, during the late G1 phase Ras signaling decreases and does not activate PI3K, which in turn does not inhibit GSK3β. Step 3: Active GSK3β phosphorylates pSer62-c-Myc on Thr58. Step 4: The doubly phosphorylated c-Myc is a substrate for the enzyme PIN1, which causes a conformational change in c-Myc from its cis to trans form. Step 5: The trans form of c-Myc is a substrate for PP2A, which dephosphorylates pSer62 to give Thr58 phosphorylated c-Myc. Step 6: The singly phosphorylated pThr58-c-Myc is a substrate for the ubiquitin ligase Fbw7. Step 7: Ubiquitination of c-Myc targets the protein for degradation through the proteasome. (B) c-Myc degradation in cancer cells. Step 1: Ras is overexpressed, and phosphorylates PI3K simultaneously with Erk phosphorylation and activation (step 2), which causes inhibition of GSK3β by phosphorylated PI3K. As a result, inactive GSK3β fails to phosphorylate c-Myc on Thr58 (step 3), which prevents the rest of the c-Myc degradation pathway (steps 4–7). Thus, c-Myc accumulates in cancer cells, enhancing cell growth. The schematic diagram is adapted from references 8, 11, and 13.
We examined the cellular phosphorylation status and total protein levels of 3 key enzymes, Erk, Akt, and PP2A, following treatment with the peptide at the indicated concentrations for 48 h (). Cellular levels of p-Erk and p-Akt, which are the activated forms of these enzymes, did not change significantly following treatment with increasing concentrations of the compound ( and ). The total Erk protein levels also did not decrease significantly. A significant decrease in total Akt protein levels was observed when cells were treated with the highest concentration (50 µM) of the peptide (), possibly due to the peptide affecting additional target(s) at such a high concentration. PP2A dephosphorylates phospho-Ser62-c-Myc, leading to c-Myc degradation in cells.Citation11,Citation34 Several reports have suggested that phosphorylation of the C-terminal tyrosine 307 of PP2A results in inactivation of its phosphatase activity.Citation16,Citation35,Citation36 The level of pTyr307-PP2A in PC-3 cells was high in vehicle treated cells, but peptide treatment at concentrations ≥10 µM significantly reduced p-PP2A levels in cells (); total PP2A protein levels were not significantly different than in vehicle treated cells.
Figure 10. [D-Trp]CJ-15,208 reduces p-PP2A protein levels in PC-3 cells. PC-3 cells were treated with the peptide at the indicated concentrations for 48 h. Western blot analysis was performed to determine protein levels of (A) p-Erk/total Erk, (B) p-Akt/total Akt, and (C) p-PP2A/total PP2A. Data shown are from 3 experiments. Representative western blots are shown under each graph. Statistical analyses were performed as described in Materials and Methods; * p<0.05,**p<0.01 and **** p<0.0001 compared with vehicle treated control cells. (D) Summary of the results of [D-Trp]CJ-15,208 treatment in PC-3 cells. [D-Trp]CJ-15,208 reduced the phosphorylation of PP2A, which in turn increased c-Myc degradation and decreased cancer cell growth.
![Figure 10. [D-Trp]CJ-15,208 reduces p-PP2A protein levels in PC-3 cells. PC-3 cells were treated with the peptide at the indicated concentrations for 48 h. Western blot analysis was performed to determine protein levels of (A) p-Erk/total Erk, (B) p-Akt/total Akt, and (C) p-PP2A/total PP2A. Data shown are from 3 experiments. Representative western blots are shown under each graph. Statistical analyses were performed as described in Materials and Methods; * p<0.05,**p<0.01 and **** p<0.0001 compared with vehicle treated control cells. (D) Summary of the results of [D-Trp]CJ-15,208 treatment in PC-3 cells. [D-Trp]CJ-15,208 reduced the phosphorylation of PP2A, which in turn increased c-Myc degradation and decreased cancer cell growth.](/cms/asset/91ef51c7-ddfe-43fc-9471-c8bcf507f56d/kcbt_a_1345384_f0010_b.gif)
Taken together, this data suggest that treatment with the peptide [D-Trp]CJ-15,208, which reduces the level of p-PP2A in PC-3 cells, increases c-Myc degradation and thereby reduces cancer cell growth ().
Discussion
We have demonstrated that the macrocyclic tetrapeptides [D-Trp]CJ-15,208 and its isomer the natural product CJ-15,208 exhibit anti-cancer activity against prostate cancer cells. Treatment of several PC cell lines with [D-Trp]CJ-15,208 resulted in decreased cell growth and increased cell death: i) the highly metastatic and androgen independent PC-3 cells, ii) mCRPC 22Rv1 cells, and iii) low metastatic, androgen dependent LNCaP cells, with IC50 values ranging from 2 to 16 µM following 48–72 h treatment (, ). All of these cell lines where [D-Trp]CJ-15,208 decreased cell growth exhibited high c-Myc protein levels regardless of whether they were androgen dependent (LNCaP) or independent metastatic (PC-3)/ castration resistant (22Rv1) prostate cancer cells. Treatment with the peptide for 48 h decreased c-Myc protein levels in a concentration dependent manner in PC cells (). However, treatment with[D-Trp]CJ-15,208 did not prevent cell proliferation in PC cells (C4–2) where c-Myc protein levels were not elevated, nor in normal cells (BPH-1 or HEK cells). Treatment with the peptide also did not alter c-Myc mRNA levels. These results provide strong evidence that [D-Trp]CJ-15,208 inhibits cancer cell growth through its effects on c-Myc protein levels.
[D-Trp]CJ-15,208 treatment induced apoptosis in PC-3 cells in a time-dependent manner and caused cell cycle arrest (). Increased early and late apoptosis were observed after 48 h treatment, but significant apoptosis induction was not found following 24 h treatment with the compound. These results suggesting that c-Myc suppression by [D-Trp]CJ-15,208 caused induction of apoptosis in PC-3 cells are consistent with the findings for other small molecules reported in the literature.Citation37-40 Cell cycle distribution is a complicated process, with c-Myc strictly controlling key cell cycle checkpoint proteins in the G1 to M phases including cyclins, CDKs, p21, and p53.Citation38 Several studies have demonstrated that cell cycle arrest by c-Myc suppression can occur via 2 mechanisms: i) uncontrolled expression of c-Myc-dependent checkpoint genes, such as CDKs or CDKIs (p21), or ii) changes in gene expression in specific cellular metabolic pathways that are directly regulated by c-Myc.Citation41-43 Treatment of PC-3 cells with [D-Trp]CJ-15,208 for 24 or 48 h resulted in cell cycle arrest in the G2 phase, preventing cancer cells from entering mitosis and cell division.
Loss and gain of function experiments also confirmed the involvement of c-Myc in the effects of the peptide on the PC cells. PC-3 cells in which c-Myc was downregulated no longer exhibited an increase in early apoptosis upon treatment with the peptide (). In complimentary gain of function experiments, HEK cells overexpressing c-Myc became sensitive to treatment with [D-Trp]CJ-15,208, exhibiting a significant increase in early apoptosis following treatment with the peptide ().
We also examined the effect of [D-Trp]CJ-15,208 treatment on the phosphorylation state of several proteins involved in the c-Myc degradation pathway. The phosphorylation state of c-Myc plays a major role in determining c-Myc stability and accumulation in cells. Under normal cellular conditions () Ras activation in the early G1 phase of the cell cycle triggers c-Myc phosphorylation and degradation through an orchestrated cascade of enzymatic reactions. In contrast to in normal cells, Ras activates both Erk and Akt by phosphorylation in the late as well as the early G1 phase in malignant cells, enhancing c-Myc stability (). p-Erk phosphorylates c-Myc on Ser62 and p-Akt inhibits GSK-3β phosphorylation of c-Myc on Thr58. These phosphorylation events prevent the rest of the steps in the c-Myc degradation pathway, increasing c-Myc stability in cellsCitation11,Citation13 (). Treatment with [D-Trp]CJ-15,208 did not have significant effects on p-Erk or p-Akt levels, but significantly reduced p-PP2A levels in a concentration dependent manner, leading to c-Myc degradation and reduction in cancer cell growth (). Thus, our data demonstrate that [D-Trp]CJ-15,208 regulates a protein involved in c-Myc degradation, resulting in decreased c-Myc protein levels in prostate cancer cells and decreased cell growth.
While there are several reports of peptides and small molecules that can decrease c-Myc protein levels in cancer cells,Citation5,Citation20,Citation37-40,Citation44,Citation45 the macrocyclic tetrapeptides described here have several advantages as lead compounds with anti-cancer activity. They are active after oral administration and showed no evidence of toxicity in mice at a dose of 60 mg/kg.Citation24,Citation25 An additional advantage is that these macrocyclic tetrapeptides appear to penetrate the blood-brain barrier in mice after systemic administration,Citation24,Citation25 which makes these compounds particularly interesting for prostate cancers that metastasize to the brain.
Taken together, these macrocyclic tetrapeptides show significant potential for development as effective treatments of advanced prostate cancer. Further studies are required to understand the details of how these peptides enhance c-Myc degradation. These studies are currently ongoing in our laboratory.
Materials and methods
Cell lines and cell culture conditions
Human prostate cancer PC-3, DU145, 22Rv1, and LNCaP cell lines were obtained from American Type Culture Collection (ATCC). Normal BPH-1 prostate cells and the C4–2 PC cell line were obtained from Dr. Benyi Li, Department of Urology, University of Kansas Medical Center. PC-3 cells were cultured and maintained in EMEM (ATCC), HEK cells were cultured in DMEM, and DU145, LNCaP, 22Rv1, C4–2, and BPH-1 cells were cultured in RPMI 1640 (ATCC), all containing 10% fetal bovine serum (Atlanta Biologicals) and 1% penicillin/streptomycin (Invitrogen). Cells were grown at 37°C in humidified air with 5% CO2 and passaged every fourth to fifth day following trypsinization with trypsin/EDTA (Invitrogen).Citation16
Cell proliferation assay
PC cells (PC-3, LNCaP, DU145, 22Rv1, and C4–2), normal prostate cells (BPH-1), and non-prostate HEK cells were grown to a density of 2000 cells/well in 96 well plates and treated with [D-Trp]CJ-15,208 or other peptides for 24–72 h in triplicate as indicated. Media containing compound was changed every 24 h. The proliferation of treated cells was determined using the cell proliferation reagent WST-1 (Roche) according to the manufacturer's protocol and compared with vehicle (1% DMSO in media) treated control cells. In brief, after treatment with compound 10 µl of the reagent was added to the well in 100 µl culture media, and the cells incubated at 37°C for 30 min. The absorbance of the media in each well was measured at 410 nm using a plate reader (BioTek). The percentage cell proliferation was normalized to vehicle treated control cells calculated as: (mean absorbance of sample/mean absorbance of control cells) X 100.Citation38
Cell viability assay
The viability of PC cells was determined using Trypan Blue (Sigma) according to the manufacturer's protocol. In brief, cells were grown in 6 well plates to a density of 1 × 105 cells/well. Cells were grown to 75% confluency and treated with compound in 0.5% DMSO at the indicated concentrations for 24–72 h, with 0.5% DMSO as the vehicle control. After treatment, the cell media from each well was collected. The cells were washed with PBS, trypsinized, and collected. All of the cells, combined media, and washes were centrifuged at 250xg for 5 min. The supernatant was discarded, and the cell pellet was resuspended in 500 µl PBS. Trypan Blue (10 µl) was added to 100 µl of resuspended cells, and the solution incubated for 10 min. The cell number in 10 µl was then counted using a hemocytometer. Viable cells (not stained) were distinguished from dead cells (colored blue) and the % viable cells calculated as follows: (number of white cells)/(number of total cells (white+blue))*100, and compared with untreated cells.Citation16
Western blot analysis
Cells were seeded in 6 well plates at a density of 1 × 105 cells/well and grown to 75% confluence. Cells were treated with compound at the indicated concentrations. Cells were then harvested as described above, and the cell pellets lysed in RIPA buffer consisting of 10 mM Tris HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA, 1% Triton-X-100, 0.1% SDS, 1% sodium deoxycholate, and a protease inhibitor cocktail (Sigma, catalog # P8340). Cell lysates were centrifuged at 13000 rpm for 15 min and the supernatants collected. The total protein concentration was measured in cell lysates using the standard Bradford assay. SDS gel electrophoresis was performed using 4–20% polyacrylamide gel (Bio-Rad, catalog # 345–0032). Western blot analysis was performed using the following antibodies: i) c-Myc (rabbit monoclonal, 1:2000 dilution, Cell Signaling Technologies, catalog # 5605); actin (mouse monoclonal, 1:5000 dilution, Sigma, catalog # A2228), PP2A (mouse monoclonal 1:5000 dilution, BD Biosciences, catalog # 610555), p-PP2A (rabbit polyclonal, 1:5000 dilution, Epitomics, catalog # 1155–1), Erk (mouse monoclonal, 1:2000 dilution, Cell Signaling Technologies, catalog # 9107) and p-Erk (rabbit monoclonal, 1:2000, Cell Signaling Technologies, catalog # 4376); Akt (mouse monoclonal, 1:2000 dilution, Cell Signaling Technologies, catalog # 2920), p-Akt (rabbit monoclonal, 1:2000, Cell Signaling Technologies, catalog # 4056). The secondary antibodies were obtained from Jackson ImmunoResearch Laboratory (goat-anti-mouse, catalog # 116–035–003; goat anti-rabbit, catalog # 111–035–003). Signal densities of the protein bands were determined using ImageJ software (NIH, Bethesda) and normalized to the internal loading control (actin).
Cell cycle analysis
PC-3 cells were grown in T-75 flasks to 75% confluency and treated with either 10 µM [D-Trp]CJ-15,208 in 0.5% DMSO or 0.5% DMSO as a vehicle control for 24–48 h. After treatment, the cell media was collected, the cells were washed with ice cold PBS twice, and the washes collected with the media. The cells were then trypsinized and the combined cells and media centrifuged at 1200 rpm for 5 min. The cell pellet was resuspended in PBS, the cells counted using a hemocytometer, and 1 million cells resuspended in 300 µl PBS. The cells were fixed with 700 µl ice cold 100% ethanol overnight and then centrifuged at 1200 rpm. Following removal of the ethanol, the cells were resuspended in 500 µl PBS containing 100 µg/ml RNase and incubated for 30 min at room temperature. Next, propidium iodide (PI) solution (Sigma, 100 µg/ml) was added and the solution then incubated for 30 min at room temperature in the dark. Cell cycle analysisCitation38 was performed using a Beckman-Coulter MoFlo XDP flow cytometer, and the cell cycle phase distribution was analyzed using FlowJo v10.1.
Annexin V-APC/PI assay
The extent of apoptosis was analyzed by detecting surface expression of phosphatidylserine on apoptotic cells using an Annexin V-APC/PI dual staining apoptosis detection kit I (BD Biosciences) according to the manufacturer's protocol. Following treatment of PC-3 cells with 10 µM [D-Trp]CJ-15,208 or vehicle for 24 or 48 h, the cells were collected, washed twice with cold PBS, and resuspended in 1X binding buffer at a concentration of 1 × 106 cells/ml. Cell suspension (100 µl) was transferred to a 5 ml culture tube, and 5 µl of annexin V-APC and 25 µl of PI (50 µg/ml) were added. The cells were incubated for 15 min at room temperature in the dark with gentle vortexing, 400 µl of 1X binding buffer was then added to each sample, and the samples were analyzed using a Beckman-Coulter MoFlo XDP flow cytometer within one hour. The number of viable, apoptotic and necrotic cells were quantified using FlowJo v10.0.7. The apoptotic fraction (%) was calculated as (number of apoptotic cells/the number of total cells observed) x 100%.Citation38
c-Myc mRNA measurement
PC-3 cells were grown in 10 cm petri dishes to 75% confluency, and then treated with [D-Trp]CJ-15,208 (10 µM) for 2–48 h. Cells were harvested and the total RNA from each sample was isolated using the Aurum Total RNA Mini Kit (Bio-Rad, catalog # 732–6820) according to the manufacturer's protocol. The RNA concentration in the sample was determined using a NanoDrop ND-1000 spectrophotometer. Total RNA (1 μg) from each sample was used to synthesize cDNA using the iScript cDNA Synthesis Kit (Bio-Rad). Real-time PCR was performed with a Power-SYBR Green PCR Master Mix (Applied Biosystems) using the following thermal cycles: initial activation at 95°C for 5 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. The fluorescence was determined at the melting temperature of the product for 20 s (ABI PRISM 7000 Sequence Detection System, Applied Biosystems). The following primer sets were used to amplify regions of c-Myc and GAPDH (internal control): c-Myc (forward: 5′-CTG GTG CTC CAT GAG GAG-3′, reverse: 5′-AGG TGA TCC AGA CTC TGA C-3′) and GAPDH (forward: 5′-CCA TGG AGA AGG CTG GGG-3′, reverse: 5′-CAA ATG TGT CAT GGA TGA CC-3′). The levels of c-Myc mRNA were normalized to the GAPDH mRNA levels.Citation37
c-Myc knockdown
PC-3 cells were seeded at a density of 2 × 106 cells in 10 cm plates and grown to 75–85% confluence in EMEM supplemented with 10% FBS and penicillin/streptomycin. The media was then replaced with antibiotic free EMEM containing 10% FBS, and the cells were incubated for a minimum of 12 h. Solutions containing 500 nM siRNA against c-Myc (Dharmacon, catalog # L-003292–02–0005), 500 nM non-targeting siRNA (Dharmacon, catalog # D-001810–10–05), or 5% (v/v) dharmafect 2 (Dharmacon, catalog # T-2002–02) were prepared in optiMEM with Glutamax (Gibco, catalog # 51985–034) to obtain a final volume of 1 ml each. After incubating 5 min at room temperature, the siRNA and dharmafect solutions were combined and incubated for an additional 20 min at room temperature. The transfection complex was then added to the cells in fresh antibiotic free EMEM containing 10% FBS, and the cells were incubated for 24 h. The cells were split into 2 10 cm tissue culture dishes in EMEM supplemented with 10% FBS and penicillin/streptomycin, and allowed to adhere for another 24 h. The cells were treated with 10 µM [D-Trp]CJ-15,208 or 0.5% DMSO vehicle control for a total of 48 h. The cells were then collected and prepared for the annexin V-APC/PI assay as described above. Samples were analyzed using a BD FACSCanto-II cell analyzer in the Interdisciplinary Center for Biotechnology Research at the University of Florida and quantified using FlowJo v10.2.
c-Myc overexpression
Forced expression of c-Myc in HEK-293 cells was performed using X-tremeGENE HP (Sigma, catalog # 6366244001). Cells were cultured in antibiotic free DMEM supplemented with 10% FBS and transfected with pcDNA3-HA-HA-c-Myc (Addgene, # 74164). Empty-vector pcDNA3-EV was used as a control. Transfected cells were selected with 500 μg/ml G418 for 2 weeks. The transfected cells were then seeded onto 10 cm tissue culture plates and allowed to grow to 75% confluence in DMEM supplemented with 10% FBS and penicillin/streptomycin. The cells were treated with 10 µM [D-Trp]CJ-15,208 or 0.5% DMSO vehicle control for a total of 48 h, after which the cells were collected and prepared for the annexin V-APC/PI assay as described above. Samples were analyzed using a BD FACSCanto-II cell analyzer and quantified in FlowJo v10.2.
Statistical Analysis
Each experiment was performed in triplicate unless otherwise stated in the figure legend. GraphPad Prism 5 was used to determine IC50 values. Data represent the mean value +/− SEM. Compound vs. vehicle treated cells in the cell cycle phase and apoptosis experiments () were compared using the student's t-test, with a 2-tailed, unpaired method, using the Prism software. The effects of compound vs. vehicle treatment on protein levels () were evaluated by one-way ANOVA with Dunnett's test for multiple comparisons.
Disclosure of potential conflicts of interest
A patent application has been filed on the anti-cancer activity of these macrocyclic peptides.
Acknowledgments
We would like to thank Robyn Pescatore for her assistance with western blot analysis and flow cytometry, Dr. Yehia Daaka, Chair, Department of Anatomy and Cell Biology at the University of Florida, for facilitating the collaboration with Dr. Hamsa Thayele Purayil, and Dr. Benyi Li, University of Kansas Medical Center, for his kind gift of PC C4–2 cells and normal prostate BPH-1 cells. We would also like to thank Heather Shinogle from the Microscopy and Analytical Imaging Laboratories, the University of Kansas, for helping us with the flow cytometry for cell cycle and apoptosis analysis in PC cells shown in and .
Funding
This work was supported by the University of Kansas J.R. and Inez Jay Fund, the Department of Defense Health Program, through the Prostate Cancer Research Program under Award No. W81XWH-14–1–0330, and the University of Florida College of Pharmacy. Opinions, interpretations, conclusions and recommendations are those of the authors and are not necessarily endorsed by the Department of Defense.
References
- Karanika S, Karantanos T, Kurosaka S, Wang J, Hirayama T, Yang G, Park S, Golstov AA, Tanimoto R, Li L, et al. GLIPR1-DeltaTM synergizes with docetaxel in cell death and suppresses resistance to docetaxel in prostate cancer cells. Mol Cancer 2015; 14:122; PMID:26084402; https://doi.org/10.1186/s12943-015-0395-0
- Amaral TM, Macedo D, Fernandes I, Costa L. Castration-resistant prostate cancer: Mechanisms, targets, and treatment. Prostate Cancer 2012; 2012:327253; PMID:22530130; https://doi.org/10.1155/2012/327253
- Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, de Souza P, Fizazi K, Mainwaring P, Piulats JM, Ng S, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med 2013; 368:138-48; PMID:23228172; https://doi.org/10.1056/NEJMoa1209096
- Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, de Wit R, Mulders P, Chi KN, Shore ND, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med 2012; 367:1187-97; PMID:22894553; https://doi.org/10.1056/NEJMoa1207506
- Gao L, Schwartzman J, Gibbs A, Lisac R, Kleinschmidt R, Wilmot B, Bottomly D, Coleman I, Nelson P, McWeeney S, et al. Androgen receptor promotes ligand-independent prostate cancer progression through c-Myc upregulation. PLoS One 2013; 8:e63563; PMID:23704919; https://doi.org/10.1371/journal.pone.0063563
- Zhang X, Morrissey C, Sun S, Ketchandji M, Nelson PS, True LD, Vakar-Lopez F, Vessella RL, Plymate SR. Androgen receptor variants occur frequently in castration resistant prostate cancer metastases. PloS One 2011; 6:e27970; PMID:22114732; https://doi.org/10.1371/journal.pone.0027970
- Vander Griend DJ, Litvinov IV, Isaacs JT. Conversion of androgen receptor signaling from a growth suppressor in normal prostate epithelial cells to an oncogene in prostate cancer cells involves a gain of function in c-Myc regulation. Int J Biol Sci 2014; 10:627-42; PMID:24948876; https://doi.org/10.7150/ijbs.8756
- Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer 2008; 8:976-90; PMID:19029958; https://doi.org/10.1038/nrc2231
- Miller DM, Thomas SD, Islam A, Muench D, Sedoris K. c-Myc and cancer metabolism. Clin Cancer Res 2012; 18:5546-53; PMID:23071356; https://doi.org/10.1158/1078-0432.CCR-12-0977
- Westermarck J, Hahn WC. Multiple pathways regulated by the tumor suppressor PP2A in transformation. Trends Mol Med 2008; 14:152-60; PMID:18329957; https://doi.org/10.1016/j.molmed.2008.02.001
- Sears RC. The life cycle of C-Myc: From synthesis to degradation. Cell Cycle 2004; 3:1133-7; PMID:15467447; https://doi.org/10.4161/cc.3.9.1145
- Tibbitts DC, Escamilla-Powers JR, Zhang X, Sears RC. Studying c-Myc serine 62 phosphorylation in leukemia cells: Concern over antibody cross-reactivity. Blood 2012; 119:5334-5; PMID:22653959; https://doi.org/10.1182/blood-2012-03-414532
- Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev 2000; 14:2501-14; PMID:11018017; https://doi.org/10.1101/gad.836800
- Bernard D, Pourtier-Manzanedo A, Gil J, Beach DH. Myc confers androgen-independent prostate cancer cell growth. J Clin Invest 2003; 112:1724-31; PMID:14660748; https://doi.org/10.1172/JCI200319035
- Ryan CJ, Molina A, Griffin T. Abiraterone in metastatic prostate cancer. N Engl J Med 2013; 368:1458-9; PMID:23574133; https://doi.org/10.1056/NEJMoa1209096
- Mukhopadhyay A, Tabanor K, Chaguturu R, Aldrich JV. Targeting inhibitor 2 of protein phosphatase 2A as a therapeutic strategy for prostate cancer treatment. Cancer Biol Ther 2013; 14:962-72; PMID:24025258; https://doi.org/10.4161/cbt.25943
- Nadiminty N, Tummala R, Liu C, Lou W, Evans CP, Gao AC. NF-kappaB2/p52:c-Myc:hnRNPA1 pathway regulates expression of androgen receptor splice variants and enzalutamide sensitivity in prostate cancer. Mol Cancer Ther 2015; 14:1884-95; PMID:26056150; https://doi.org/10.1158/1535-7163.MCT-14-1057
- Sivertsen A, Torfoss V, Isaksson J, Ausbacher D, Anderssen T, Brandsdal BO, Havelkova M, Skjorholm AE, Strom MB. Anticancer potency of small linear and cyclic tetrapeptides and pharmacokinetic investigations of peptide binding to human serum albumin. J Peptide Sci 2014; 20:279-91; PMID:24677781; https://doi.org/10.1002/psc.2615
- Murray BC, Peterson MT, Fecik RA. Chemistry and biology of tubulysins: Antimitotic tetrapeptides with activity against drug resistant cancers. Nat Prod Rep 2015; 32:654-62; PMID:25677951; https://doi.org/10.1039/C4NP00036F
- Neviani P, Perrotti D. SETting OP449 into the PP2A-activating drug family. Clin Cancer Res 2014; 20:2026-8; PMID:24634375; https://doi.org/10.1158/1078-0432.CCR-14-0166
- Saito T, Hirai H, Kim YJ, Kojima Y, Matsunaga Y, Nishida H, Sakakibara T, Suga O, Sujaku T, Kojima N. CJ-15,208, a novel kappa opioid receptor antagonist from a fungus, Ctenomyces serratus ATCC15502. J Antibiot (Tokyo) 2002; 55:847-54; PMID:12523817; https://doi.org/10.7164/antibiotics.55.847
- Ross NC, Kulkarni SS, McLaughlin JP, Aldrich JV. Synthesis of CJ-15,208, a novel kappa-opioid receptor antagonist. Tetrahedron Lett 2010; 51:5020-3; PMID:22865937; https://doi.org/10.1016/j.tetlet.2010.07.086
- Ross NC, Reilley KJ, Murray TF, Aldrich JV, McLaughlin JP. Novel opioid cyclic tetrapeptides: Trp isomers of CJ-15,208 exhibit distinct opioid receptor agonism and short-acting kappa opioid receptor antagonism. Br J Pharmacol 2012; 165:1097-108; PMID:21671905; https://doi.org/10.1111/j.1476-5381.2011.01544.x
- Aldrich JV, Senadheera SN, Ross NC, Ganno ML, Eans SO, McLaughlin JP. The macrocyclic peptide natural product CJ-15,208 is orally active and prevents reinstatement of extinguished cocaine-seeking behavior. J Nat Prod 2013; 76:433-8; PMID:23327691; https://doi.org/10.1021/np300697k
- Eans SO, Ganno ML, Reilley KJ, Patkar KA, Senadheera SN, Aldrich JV, McLaughlin JP. The macrocyclic tetrapeptide [D-Trp]CJ-15,208 produces short-acting kappa opioid receptor antagonism in the CNS after oral administration. Br J Pharmacol 2013; 169:426-36; PMID:23425081; https://doi.org/10.1111/bph.12132
- Fichna J, Janecka A. Opioid peptides in cancer. Cancer Metastasis Rev 2004; 23:351-66; PMID:15197335; https://doi.org/10.1023/B:CANC.0000031773.46458.63
- Gach K, Wyrebska A, Fichna J, Janecka A. The role of morphine in regulation of cancer cell growth. Naunyn Schmiedebergs Arch Pharmacol 2011; 384:221-30; PMID:21800094; https://doi.org/10.1007/s00210-011-0672-4
- Moon TD. The effect of opiates upon prostatic carcinoma cell growth. Biochem Biophys Res Commun 1988; 153:722-7; PMID:2898243; https://doi.org/10.1016/S0006-291X(88)81154-9
- Kuzumaki N, Suzuki A, Narita M, Hosoya T, Nagasawa A, Imai S, Yamamizu K, Morita H, Nagase H, Okada Y, et al. Effect of kappa-opioid receptor agonist on the growth of non-small cell lung cancer (NSCLC) cells. Br J Cancer 2012; 106:1148-52; PMID:22343623; https://doi.org/10.1038/bjc.2011.574
- Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, Hahn WC, Stukenberg PT, Shenolikar S, Uchida T, et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol 2004; 6:308-18; PMID:15048125; https://doi.org/10.1038/ncb1110
- Patkar KA, Yan X, Murray TF, Aldrich JV. [Nα-benzylTyr1,cyclo(D-Asp5,Dap8)]-dynorphin A-(1-11)NH2 cyclized in the “address” domain is a novel kappa-opioid receptor antagonist. J Med Chem 2005; 48:4500-3; PMID:15999987; https://doi.org/10.1021/jm050105i
- Arttamangkul S, Ishmael JE, Murray TF, Grandy DK, DeLander GE, Kieffer BL, Aldrich JV. Synthesis and opioid activity of conformationally constrained dynorphin A analogues. 2. Conformational constraint in the “address” sequence. J Med Chem 1997; 40:1211-8; PMID:9111295; https://doi.org/10.1021/jm960753p
- Pelengaris S, Khan M, Evan G. c-MYC: More than just a matter of life and death. Nat Rev Cancer 2002; 2:764-76; PMID:12360279; https://doi.org/10.1038/nrc904
- Oaks JJ, Santhanam R, Walker CJ, Roof S, Harb JG, Ferenchak G, Eisfeld AK, Van Brocklyn JR, Briesewitz R, Saddoughi SA, et al. Antagonistic activities of the immunomodulator and PP2A-activating drug FTY720 (Fingolimod, Gilenya) in Jak2-driven hematologic malignancies. Blood 2013; 122:1923-34; PMID:23926298; https://doi.org/10.1182/blood-2013-03-492181
- Chen J, Martin BL, Brautigan DL. Regulation of protein serine-threonine phosphatase type-2A by tyrosine phosphorylation. Science 1992; 257:1261-4; PMID:1325671; https://doi.org/10.1126/science.1325671
- Neviani P, Santhanam R, Trotta R, Notari M, Blaser BW, Liu S, Mao H, Chang JS, Galietta A, Uttam A, et al. The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein. Cancer Cell 2005; 8:355-68; PMID:16286244; https://doi.org/10.1016/j.ccr.2005.10.015
- Lu JJ, Meng LH, Shankavaram UT, Zhu CH, Tong LJ, Chen G, Lin LP, Weinstein JN, Ding J. Dihydroartemisinin accelerates c-MYC oncoprotein degradation and induces apoptosis in c-MYC-overexpressing tumor cells. Biochem Pharmacol 2010; 80:22-30; PMID:20206143; https://doi.org/10.1016/j.bcp.2010.02.016
- Zhang JY, Tao LY, Liang YJ, Yan YY, Dai CL, Xia XK, She ZG, Lin YC, Fu LW. Secalonic acid D induced leukemia cell apoptosis and cell cycle arrest of G1 with involvement of GSK-3β/β-catenin/c-Myc pathway. Cell Cycle 2009; 8:2444-50; PMID:19571678; https://doi.org/10.4161/cc.8.15.9170
- Thamilselvan V, Menon M, Thamilselvan S. Anticancer efficacy of deguelin in human prostate cancer cells targeting glycogen synthase kinase-3β/β-catenin pathway. Int J Cancer 2011; 129:2916-27; PMID:21472727; https://doi.org/10.1002/ijc.25949
- Huang HL, Weng HY, Wang LQ, Yu CH, Huang QJ, Zhao PP, Wen JZ, Zhou H, Qu LH. Triggering Fbw7-mediated proteasomal degradation of c-Myc by oridonin induces cell growth inhibition and apoptosis. Mol Cancer Ther 2012; 11:1155-65; PMID:22389469; https://doi.org/10.1158/1535-7163.MCT-12-0066
- Zhang Y, Guo Z, Xu L. Tributyltin induces a G2/M cell cycle arrest in human amniotic cells via PP2A inhibition-mediated inactivation of the ERK1/2 cascades. Environ Toxicol Pharmacol 2014; 37:812-8; PMID:24632106; https://doi.org/10.1016/j.etap.2014.02.009
- Song A, Ye J, Zhang K, Sun L, Zhao Y, Yu H. Lentiviral vector-mediated siRNA knockdown of c-MYC: Cell growth inhibition and cell cycle arrest at G2/M phase in Jijoye cells. Biochem Genet 2013; 51:603-17; PMID:23657834; https://doi.org/10.1007/s10528-013-9590-0
- Lin HP, Lin CY, Huo C, Hsiao PH, Su LC, Jiang SS, Chan TM, Chang CH, Chen LT, Kung HJ, et al. Caffeic acid phenethyl ester induced cell cycle arrest and growth inhibition in androgen-independent prostate cancer cells via regulation of Skp2, p53, p21Cip1 and p27Kip1. Oncotarget 2015; 6:6684-707; PMID:25788262; https://doi.org/10.18632/oncotarget.3246
- Jingushi K, Nakamura T, Takahashi-Yanaga F, Matsuzaki E, Watanabe Y, Yoshihara T, Morimoto S, Sasaguri T. Differentiation-inducing factor-1 suppresses the expression of c-Myc in the human cancer cell lines. J Pharmacol Sci 2013; 121:103-9; PMID:23357875; https://doi.org/10.1254/jphs.12204FP
- Akinyeke TO, Stewart LV. Troglitazone suppresses c-Myc levels in human prostate cancer cells via a PPARgamma-independent mechanism. Cancer Biol Ther 2011; 11:1046-58; PMID:21525782; https://doi.org/10.4161/cbt.11.12.15709
- Sampson N, Neuwirt H, Puhr M, Klocker H, Eder IE. In vitro model systems to study androgen receptor signaling in prostate cancer. Endocr Relat Cancer 2013; 20:R49-64; PMID:23447570; https://doi.org/10.1530/ERC-12-0401