
ABSTRACT
Nutlin-3a is a small molecule MDM2 antagonist and potent activator of wild-type p53. Nutlin-3a disrupts MDM2 binding to p53, thus increasing p53 levels and allowing p53 to inhibit proliferation or induce cell death. Factors that control sensitivity to Nutlin-3a-induced apoptosis are incompletely understood. In this study we isolated cisplatin-resistant clones from MHM cells, an MDM2-amplified and p53 wild-type osteosarcoma cell line. Cisplatin resistance in these clones resulted in part from heightened activation of the IGF-1R/AKT pathway. Interestingly, these cisplatin resistant clones showed hyper-sensitivity to Nutlin-3a induced apoptosis. Increased Nutlin-3a sensitivity was associated with reduced authophagy flux and a greater increase in p53 levels in response to Nutlin-3a treatment. IGF-1R and AKT inhibitors further increased apoptosis by Nutlin-3a in parental MHM cells and the cisplatin-resistant clones, confirming IGF-1R/AKT signaling promotes apoptosis resistance. However, IGF-1R and AKT inhibitors also reduced p53 accumulation in Nutlin-3a treated cells and increased autophagy flux, which we showed can promote apoptosis resistance. We conclude the IGF-1R/AKT pathway has opposing effects on Nutlin-3a-induced apoptosis. First, it can inhibit apoptosis, consistent with its well-established role as a survival-signaling pathway. Second, it can enhance Nutlin-3a induced apoptosis through a combination of maintaining p53 levels and inhibiting pro-survival autophagy.
Introduction
P53 is a stress-responsive transcription factor and potent tumor suppressor. P53 levels are low in most cells because of MDM2, an E3 ubiquitin-ligase that binds p53 and promotes its degradation.Citation1,2 However, DNA damage and other stresses induce post-translational modifications in p53 and MDM2 that disrupt their binding and cause p53 protein levels to increase.Citation3 Increased levels of p53 then activate expression of downstream target genes whose protein products can cause apoptosis or cell cycle arrest.Citation4 In recent years small molecule MDM2 antagonists have been developed as potential therapeutic agents. These compounds occupy the p53 binding site in MDM2, thus blocking p53-MDM2 binding and unleashing p53 to induce cell cycle arrest or apoptosis. Nutlin-3a (Nutlin) is the prototype MDM2 antagonist first described in 2004.Citation5 Nutlin has been shown to inhibit proliferation and induce apoptosis in p53 wild-type cancer cell lines and block the growth of p53 wild-type human tumors grown in mice.Citation6,7 Second generation Nutlin derivatives have entered clinical trials against various solid and hematologic cancers.
Not all p53 wild-type cancer cells respond to MDM2 antagonist treatment in the same way. For example, most hematologic cancer cell lines undergo apoptosis as their primary response to Nutlin, whereas most but not all non-hematologic cancer cell lines undergo cell cycle arrest.Citation7,8 Tovar et al reported that SJSA-1 and MHM, 2 osteosarcoma cell lines with amplification of the MDM2 gene, were highly sensitive to Nutlin-induced apoptosis whereas HCT116 (colon), A549 (lung), and H460 (lung), which contain only one MDM2 gene, were least sensitive.Citation7 This suggested MDM2 gene amplification may predispose to Nutlin-induced apoptosis. In contrast, in the study by Kitagawa et al it was found Nutlin treatment did not induce abundant apoptosis in the choriocarcinoma cell line JAR, which is known to have MDM2 gene amplification.Citation9 This would suggest MDM2 amplification is not a perfect predictor of Nutlin sensitivity. We and others found that the cell cycle arrest induced by Nutlin is reversible and, in some cases, can give rise to tetraploid cells that are resistant to radiation and chemotherapy induced apoptosis.Citation10-12 Thus, being able to target Nutlin treated cells down the more desirable apoptotic pathway could, conceivably, increase its therapeutic potential. It is therefore important to identify factors that regulate whether cells undergo apoptosis or arrest in response to Nutlin treatment.
The IGF-1R/AKT/mTORC1 pathway is activated in multiple cancers and is associated with chemotherapy resistance and poor patient outcome.Citation13 In this pathway, ligands IGF-1 and-2 bind the receptor IGF-1R, stimulating its auto-phosphorylation on tyrosines. This leads to recruitment and activation of PI3-K. The kinase AKT is subsequently activated by phosphorylation at 2 sites: S473 is phosphorylated by mTORC2 and T308 is phosphorylated by PDK1. Activated AKT can promote survival by phosphorylating and inhibiting/activating various pro/anti-apoptotic factors.Citation14-16 mTORC1 is activated downstream of AKT and promotes protein synthesis and cell growth by phosphorylating its substrates (e.g. S6K).Citation17,18 Importantly, activated mTORC1 also inhibits autophagy,Citation19 the self-eating process in which cells degrade damaged organelles and proteins to maintain nutrient and energy levels and survive.
There is abundant crosstalk between p53 and the IGF-1R/AKT/mTORC1 pathway that could potentially influence cancer cell sensitivity to Nutlin or other MDM2 antagonists. For example, Zhu et al reported that leukemia cells with basal or elevated PTEN expression, and thus low PI3K/AKT signaling, were more susceptible to Nutlin induced apoptosis than cells without PTEN expression.Citation20 More recently, Saiki et al reported that AKT and mTORC1 inhibitors could synergize with the MDM2 antagonist C-25 to reduce viability in a subset of p53 wild-type cancer cell lines.Citation21 Together, these findings support the idea that AKT/mTORC1 signaling can reduce apoptosis sensitivity in response to MDM2 antagonists like Nutlin. In contrast, we and others found that the IGF-1R/AKT/mTORC1 signaling promotes p53 protein synthesis and maintains p53 expression levels in stressed cells.Citation22-25 These findings raise the possibility that heightened IGF-1R/AKT/mTORC1 activation could potentially increase cancer cell sensitivity to Nutlin by maintaining high p53 protein levels. Finally, we recently found the autophagy inhibitors bafilomycin A1 and chloroquine could increase apoptosis sensitivity in Nutlin treated cells, indicating that autophagy promotes apoptosis resistance.Citation26 Given that AKT/mTORC1 signaling inhibits autophagy, the results suggest heightened AKT/mTORC1 activation could increase apoptosis in Nutlin treated cells by blocking or reducing pro-survival autophagy.
In the current report we isolated cisplatin (CP) resistant clones from the MHM osteosarcoma cell line. The clones showed heightened basal IGF-1R/AKT activation that contributes to their CP resistance. Interestingly, the clones displayed hypersensitivity to apoptosis by Nutlin. IGF-1R and AKT inhibitors increased apoptosis in response to Nutlin, demonstrating IGF-1R/AKT activation can promote Nutlin resistance in these cells. Notably, however, p53 was induced to a higher level and AKT was more activated in response to Nutlin in the CP-resistant clones compared with parental MHM cells. IGF-1R and AKT inhibitors reduced p53 levels in response to Nutlin, demonstrating IGF-1R/AKT activation also contributes to the accumulation of p53. Finally, the CP-resistant and Nutlin hypersensitive clones showed reduced autophagy flux that was AKT-dependent, and autophagy inhibitors increased Nutlin sensitivity in parental MHM cells. Our findings suggest that in addition to reducing apoptosis in response to Nutlin, IGF-1R/AKT pathway activation can also increase apoptosis sensitivity through a combination of maintaining p53 protein levels and inhibiting pro-survival autophagy.
Results
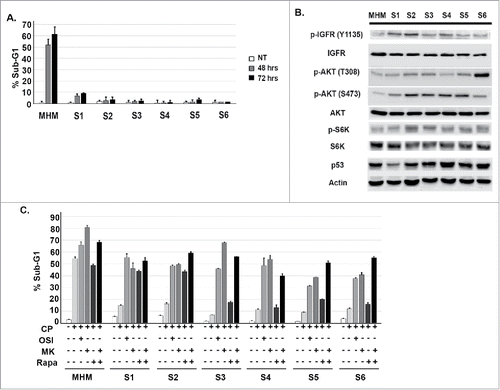
Osteosarcoma is an aggressive bone cancer that primarily affects children and adolescents.Citation27 Standard OS treatment includes pre and post- surgical chemotherapy and surgical resection.Citation28 Nonetheless, ∼30% of patients fail treatment and die due to tumor relapse at metastatic sites.Citation29 The primary reason for treatment failure is tumor therapy resistance. Cisplatin is a standard chemotherapy agent for OS. MHM is an OS cell line that has MDM2 gene amplification and expresses wild-type p53. We treated MHM with increasing doses of CP and expanded surviving clones (the clones are termed S1-S6). In our previous study we reported clone S4 is resistant to CP-induced apoptosis compared with MHM.Citation23 In the current study we compared parental MHM cells and clones S1-S6 in a side-by-side experiment to determine apoptosis sensitivity in response to CP. For this purpose we monitored the percentage of cells with sub-G1 DNA content as an indicator of apoptosis. As shown in , the S1-S6 clones were resistant to CP-induced apoptosis compared with MHM. We previously reported that the IGF-1R/AKT/mTORC1 pathway can contribute to CP resistance in primary OS cells and OS cell lines.Citation23 We therefore assessed activation of this pathway by monitoring phosphorylated (activated) levels of IGF-1R, AKT, and S6K in MHM cells and the CP-resistant clones. As shown in , the clones S1-S6 expressed basally higher levels of pIGF-1R, pAKT, and pS6K (indicative of mTORC1 activity) when compared with parental MHM cells. Finally, we monitored apoptosis by determining the percentage of cells with sub-G1 DNA content after treatment with CP alone or in combination with an IGF-1R inhibitor (OSI-906), AKT inhibitor (MK2206), mTORC1 inhibitor (rapamycin), or combination rapamycin plus MK2206. As shown in , co-treatment with OSI-906 and MK2206 increased apoptosis in CP-treated MHM cells and each of the CP resistant clones, supporting the idea that IGF-1R and AKT contribute to apoptosis resistance. Interestingly, rapamycin also increased CP-induced apoptosis and this effect of rapamycin was especially evident in clones S1 and S2. This suggests S1 and S2 are more dependent on mTORC1 for apoptosis resistance than either MHM or clones S3-S6. Rapamycin has been reported to cause feedback activation of AKT,Citation30 and we therefore tested the combined effect of rapamycin plus the AKT inhibitor MK2206. We found rapamycin plus MK2206 increased apoptosis in S3-S6 clones above the level seen with MK2206 or rapamycin alone (). The results suggest the relative lack of apoptosis sensitivity in the rapamycin treated S3-S6 clones could result from feedback activation of AKT.
Figure 1. Cisplatin resistant cells have heightened activation of the IGF-1R/AKT/mTORC1 pathway. (A) MHM cells and clones S1-S6 were treated with 10 μM CP for 48 and 72 hours. The percentage of cells with sub-G1 DNA was determined. Shown are the mean results of 3 separate experiments. (B) Parental and CP resistant clones were immunoblotted for the indicated proteins under untreated conditions. (C) Parental MHM and CP resistant clones were untreated or treated with CP (10 μM) alone or in combination with OSI906 (10 μM), MK2206 (10 μM), rapamycin (0.5 μM), or rapamycin plus MK2206 for 72 hours. Apoptosis was determined by percentage of cells with sub-G1 DNA content.
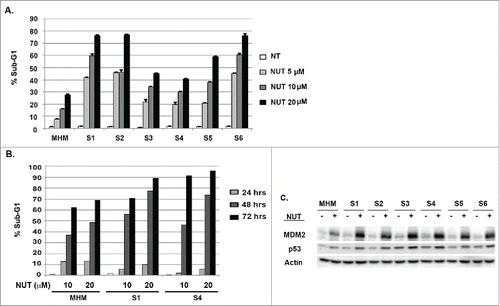
Because MHM cells express wild-type p53, we considered that the CP-resistant MHM clones (S1-S6) might be sensitive to MDM2 antagonists that stabilize and activate p53. To test this possibility, we first treated MHM and clones S1-S6 with increasing doses of the MDM2 antagonist Nutlin-3a (Nutlin) for 72 hrs. We then monitored apoptosis by determining the percentage of cells with sub-G1 DNA content (). In these studies, MHM cells showed about 30% apoptosis in response to the highest (20 μM) Nutlin dose tested. In contrast, clones S1-S6 were hyper-sensitive to Nutlin-induced apoptosis, in some cases showing 75–80% apoptotic cells in response to 20 μM Nutlin (). Next, we performed a time-course experiment by treating MHM and 2 of the CP-resistant clones (S1 and S4) with Nutlin (10 or 20 μM) and monitoring apoptosis between 24 and 72 hrs after treatment. As shown in , MHM, S1, and S4 cells had comparable and relatively low levels of apoptosis when Nutlin-treated for 24 hrs. However, S1 and S4 cells displayed much higher levels of apoptosis than MHM when treated with Nutlin for 48 and 72 hrs, again indicating that at these later time points the CP-resistant S cells were hyper-sensitive to apoptosis by Nutlin. Finally, we performed immunoblotting to ask if increased apoptosis sensitivity coincided with higher levels of induced p53 (). The data revealed that p53 was induced to a higher level upon Nutlin treatment in the S1-S6 clones compared with parental MHM cells, and MDM2 was also induced to a higher level, supporting the idea that higher levels of p53 led to increased p53 activity (). In summary, the S1-S6 clones are resistant to CP-induced apoptosis but are hypersensitive to apoptosis by Nutlin, and the Nutlin hypersensitivity is associated with a greater induction of p53.
Figure 2. CP-resistant cells are hyper-sensitive to Nutlin induced apoptosis. (A) Parental MHM and CP resistant S1-S6 cells were untreated or treated with Nutlin (5, 10 and 20 μM) for 48 hours and percentage of cells with sub-G1 DNA content was determined. (B) Parental MHM and CP-resistant S1 and S4 cells were untreated or treated with Nutlin (10 μM or 20 μM) for 24, 48 and 72 hours and percentage of cells with sub-G1 DNA content was determined. (C) MHM and CP resistant S1-S6 cells were untreated or treated with Nutlin (10 μM) for 24 hours and p53, MDM2, and Actin levels were determined by immunoblotting.
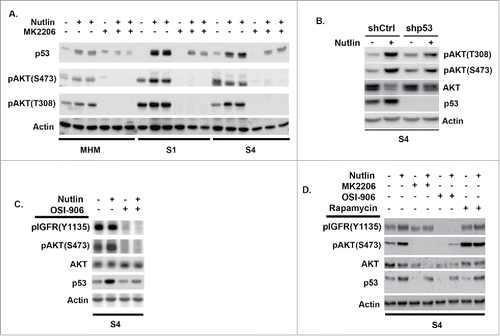
Next, we wished to examine the relationship between IGF-1R/AKT signaling and p53 induction by Nutlin in MHM cells and the CP resistant clones. To this end, we treated MHM cells and 2 of the CP resistant clones (S1 and S4) for 24 hrs with Nutlin alone or Nutlin plus the allosteric AKT inhibitor MK2206. As shown in , p53 was again induced to a higher level by Nutlin in S1 and S4 cells compared with MHM. Interestingly, levels of activated AKT (phopshorylated at S473 and T308) were increased in the Nutlin-treated cells, and to a higher level in S1 and S4 than in MHM (). pAKT was less induced in S4 cells in which p53 was depleted by shRNA, confirming the increase in pAKT is largely p53-dependent (). As expected, pAKT was not detected in cells co-treated with Nutlin and the allosteric AKT inhibitor MK2206. Importantly, p53 was less induced in cells co-treated with Nutlin plus MK2206 compared with cells treated with Nutlin alone, indicating that AKT activation contributes to the accumulation of p53 in Nutlin treated cells. To test if IGF-1R contributes to the accumulation of p53, we monitored p53 levels in S4 cells treated with Nutlin alone or Nutlin plus the IGF-1R inhibitor OSI-906. As shown in , co-treatment with OSI-906 reduced p53 accumulation in Nutlin treated S4 cells, indicating IGF-1R contributes to the accumulation of p53. Levels of activated (S473 phosphorylated) AKT were completely absent in cells treated with OSI-906, indicating AKT activation in these cells is IGF-1R dependent (). Finally, to ask if mTORC1 activity is required for p53 accumulation, we co-treated S4 cells with Nutlin and the mTORC1 inhibitor rapamycin. As shown in , OSI-906 and MK2206 again reduced p53 levels in Nutlin treated S4 cells, indicating IGF-1R and AKT contribute to p53 accumulation in Nutlin-treated cells. However, rapamycin did not reduce p53 accumulation in Nutlin-treated S4 cells (), suggesting mTORC1 activity is not required for the accumulation of p53. mTORC1 inhibition can cause feedback activation of AKT.Citation30 Consistent with this, pAKT(S473) levels were increased in S4 cells treated with either rapamycin alone or Nutlin plus rapamycin (). In total, the results of indicate that p53 can activate AKT in Nutlin treated cells in a manner that is IGF-1R-dependent, that S1 and S4 cells express higher levels of activated AKT in response to Nutlin compared with MHM cells, and that IGF-1R and AKT contribute to the Nutlin-induced accumulation of p53.
Figure 3. The IGF-1R/AKT/mTORC1 pathway is more highly activated in Nutlin treated S1 and S4 cells and contributes to apoptosis resistance. (A) MHM, S1, and S4 cells were treated with Nutlin (10 mM) for 24 hrs. Levels of p53, AKT phosphorylated at S473, AKT phosphorylated at T308, and Actin were determined by immunoblotting. (B) S4 cells stably infected with virus encoding control shRNA (shCtrl) or p53 shRNA (shp53) wer treated with Nutlin (10 μM) for 24 hrs and immunoblotted for the indicated proteins. (C) S4 cells were treated with Nutlin (10 μM) and OSI-906 (10 μM) for 24 hrs and immunoblotted for the indicated proteins. (D) S4 cells were treated for 24 hrs with Nutlin (10 μM), OSI-906 (10 μM), MK2206 (10 μM), and rapamycin (0.5 μM) as indicated, and immunoblotted for the indicated proteins.
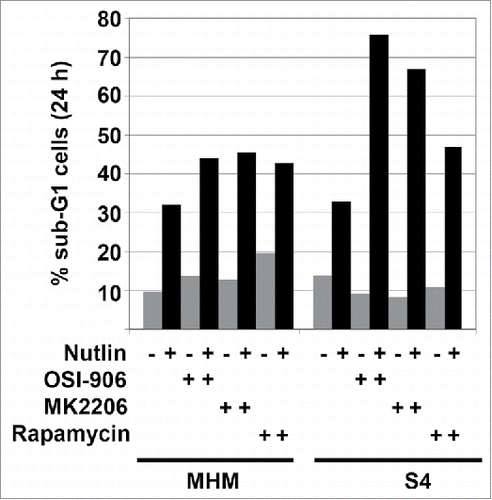
Higher levels of p53 usually lead to increased apoptosis. The fact that IGF-1R/AKT activity contributes to p53 accumulation in Nutlin-treated cells suggests IGF-1R and AKT could increase p53-dependent apoptosis. However, it is well established that IGF-1R/AKT signaling can inhibit apoptosis and increase survival by altering the activity of various apoptosis regulators. To examine the effect of IGF-1R and AKT and mTORC1 on apoptosis in Nutlin-treated cells, we treated MHM and S4 cells with Nutlin alone or in combination with OSI-906, MK2206, or rapamycin. Apoptosis was determined by the percentage of cells with sub-G1 DNA content. As shown in , about 30% of MHM cells treated for 24 hrs with 10 uM Nutlin had sub-G1 DNA content (apoptosis). Co-treatment with OSI-906, MK2206, or rapamycin increased the percentage of apoptotic, sub-G1 MHM cells to 40–50% (). About 30% of S4 cells also had sub-G1 DNA content (apoptosis) when treated for 24 hrs with 10 uM Nutlin. Co-treatment of S4 cells with Nutlin and either OSI-906 or MK2206 increased the percentage of apoptotic, sub-G1 cells to ∼65–75%, and co-treatment with rapamycin increased the percentage of apoptotic, sub-G1 cells to ∼45% (). The results indicate IGF-1R/AKT/mTORC1 signaling promotes apoptosis resistance in Nutlin-treated MHM and S4 cells. IGF-1R/AKT activation appears to contribute more to apoptosis resistance in S4 cells than in MHM cells.
Figure 4. The effect of IGF-1R/AKT inhibitors on Nutlin-induced apoptosis. MHM, S1, and S4 cells were treated with Nutlin alone (10 μM) or in combination with inhibitors of IGF-1R (OSI-906 10 μM), AKT (MK2206, 10 μM), rapamycin (0.5 μM) or rapamycin plus MK2206 for 24 or 48 hrs. The percentage of cells with sub-G1 DNA content was determined.
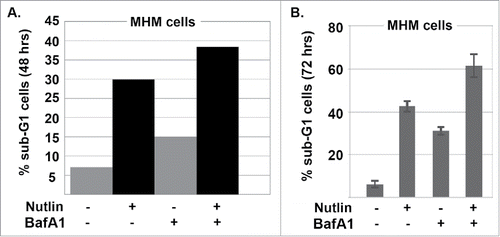
Autophagy is a process of “self-eating” in which damaged organelles, mis-folded proteins, and other components are broken down and degraded in autophagolysosomes. This degradation allows cells to maintain nutrient and energy levels critical for survival.Citation31,32 mTORC1 is activated downstream of AKT and normally inhibits autophagy by phosphorylating and inhibiting ULK1 and ULK2, which are components of the autophagy-initiating complex.Citation31,32 We previously reported that autophagy inhibits apoptosis and promotes survival in Nutlin-treated cells.Citation26 We therefore wished to ask if AKT regulates autophagy in MHM, S1, and S4 cells and if this affects apoptosis sensitivity in response to Nutlin. P62 is an autophagy protein that is degraded in autophagolysosomes and that facilitates recruitment of misfolded, ubiquitinated proteins and damaged organelles to autophagolysosomes for degradation. Bafilomycin A1 disrupts autophagosomes and inhibits autophagic protein degradation, including degradation of p62. Thus, the extent to which p62 increases in response to bafilomycin A1 reflects the rate with which autophagic degradation is occurring, or “autophagy flux.” We compared autophagy flux in MHM, S1, and S4 cells by treating the cells with Bafilomycin A1 for 8 or 24 hrs and monitoring p62 levels by immunoblotting (). p62 levels increased in MHM cells treated with bafilomycin A1 alone, indicating autophagic degradation was occurring. We quantified the extent to which p62 levels increased in bafilomycin A1 treated cells as an indication of autophagy flux, using actin as a normalization control. The results showed that p62 levels increased approximately 8–15-fold in MHM cells in response to 8 or 24 hrs bafilomycin A1 treatment. Basal p62 levels were strikingly higher in S1 and S4 cells compared with MHM and, moreover, p62 increased to a lesser extent (only 1.5 to 2-fold) in S1 and S4 cells treated with bafilomycin A1 for 8 or 24 hrs. These results indicate that S1 and S4 cells have reduced autophagic flux compared with MHM. Next, we wished to ask if AKT regulates autophagic flux in these cells, and its effect on Nutlin sensitivity. For this, S4 cells were treated with bafilomycin A1 and the AKT inhibitor MK2206 either alone or in combination, and p62 levels determined. As shown in , p62 increased by 1.8-fold in S4 cells treated with bafilomycin A1 alone, but increased by 2.6-fold in cells co-treated with bafilomycin A1 and MK2206. Thus, AKT inhibition by MK2206 increased autophagy flux in these cells, indicating AKT activation normally inhibits autophagy. We speculated that reduced autophagy in S1 and S4 cells may contribute to or lead to their increased sensitivity to apoptosis by Nutlin. If this is true, then autophagy inhibition should increase apoptosis in Nutlin treated MHM cells. To test this, we treated MHM cells with Nutlin alone or in combination with bafilomycin A1 for 48–72 hrs, and then monitored apoptosis by the percentage of cells with sub-G1 DNA content. As shown in and , the combination of Nutlin plus bafilomycin A1 caused a greater amount of apoptosis in MHM cells than did either agent alone, indicating autophagy inhibition can increase Nutlin-induced apoptosis. We conclude reduced autophagy flux, that is dependent on AKT activity, contributes to greater apoptosis sensitivity in Nutlin-treated S1 and S4 cells.
Figure 5. Autophagy flux is different in MHM, S1, and S4 cells. (A) MHM, S1, and S4 cells were treated with bafilomycin A1 (10 nM) for 8 and 24 hrs. p62 levels were determined by immunoblotting. p62 and actin levels were quantified using Image-J software. The relative p62 level is indicated after normalization to actin levels. (B) p62 levels were determined in S4 cells treated with bafilomycin A1 (10 nM) alone or in combination with MK2206 (10 μM) for 16 hrs. The relative p62 level is indicated after normalization to actin levels. P62 increased to a greater extent in cells co-treated with bafilomycin A1 plus MK2206 compared with bafilomycin A1 alone, indicated that MK2206 increased autophagy flux.
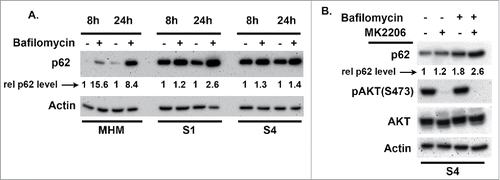
Figure 6. Autophagy inhibition increases Nutlin-induced apoptosis. (A) and (B) MHM cells were treated Nutlin alone (10 μM) or in combination with bafilomycin A1 (10 nM) as indicated. The percentage of cells with sub-G1 DNA content was determined 48 hrs (A) and 72 hrs (B) after treatment.
Discussion
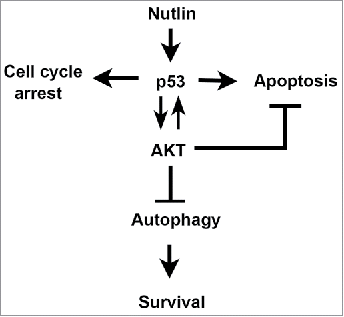
In recent years MDM2 antagonists (e.g., Nutlin) have been developed as potential therapeutics in cancers with wild-type p53. Nutlin occupies the p53-binding pocket in MDM2, thus blocking p53-MDM2 binding and unleashing p53 to inhibit cancer cell proliferation or induce apoptosis. Notably, some p53 wild-type cancer cells are resistant to apoptosis in response to Nutlin and can survive and resume proliferation upon Nutlin removal. Thus, an important goal is to identify factors that control apoptosis in MDM2 antagonist (Nutlin) treated cells, as these factors could be potential targets to enhance the therapeutic response. MHM is an MDM2-amplified osteosarcoma cell line that is relatively sensitive to Nutlin-induced apoptosis. We isolated cisplatin (CP)-resistant MHM cell clones after repeated exposure to increasing CP doses. These clones (termed S1-S6) had heightened IGF-1R/AKT signaling that contributes to CP resistance. Surprisingly, these CP-resistant clones expressed elevated p53 levels in response to Nutlin compared with the parental MHM cells and were hyper-sensitive to Nutlin-induced apoptosis. IGF-1R and AKT inhibitors increased apoptosis in response to Nutlin, confirming IGF-1R/AKT activation can promote apoptosis resistance. However, IGF-1R/AKT signaling also contributed to p53 accumulation in Nutlin-treated cells and reduced autophagy, which we showed can protect cells against apoptosis. Based on these findings we propose IGF-1R/AKT pathway signaling has dual and opposing effects on Nutlin sensitivity (). First, it can inhibit apoptosis, consistent with its well-established role as a survival-signaling pathway. Second, it can enhance Nutlin sensitivity through a combination of maintaining p53 levels and inhibiting pro-survival autophagy.
Figure 7. Model for how IGF-1R/AKT activation affects apoptosis sensitivity in response to Nutlin. P53 induced by Nutlin promotes AKT activation dependent on IGF-1R. IGF-1R/AKT activation also promotes the accumulation of p53. Activated AKT can reduce apoptosis and promote survival by regulating the activity of pro- and anti-apoptotic factors that localize to the mitochondria, such as bcl-2, bax, and bim. AKT also inhibits autophagy, which can promote survival.
The survival kinase AKT is activated downstream of IGF-1R. AKT can promote survival by phosphorylating and inhibiting the activity of pro-apoptotic bcl-2 family members such as Bad and Bax, while also phosphorylating and inhibiting the activity of Foxo-3a, a transcription factor that promotes apoptosis by activating expression of BH3 domain-containing proteins like PUMA and Bim.Citation14-16 In the current study, IGF-1R and AKT inhibitors increased apoptosis by Nutlin, and it is likely that IGF-1R/AKT signaling promotes survival, at least in part, through AKT-dependent phosphorylation of one or more bcl-2 family members and/or Foxo-3a. In addition to promoting survival, our results suggest IGF-1R/AKT signaling can increase apoptosis through a combination of maintaining p53 protein levels and inhibiting autophagy. Evidence that IGF-1R and AKT maintain p53 protein levels comes from the finding that IGF-1R/AKT inhibitors reduced p53 accumulation in Nutlin-treated cells. Increasing Nutlin doses cause a progressive increase in p53 levels and a corresponding increase in apoptosis. The finding that IGF-1R/AKT inhibitors reduced p53 suggests IGF-1R and AKT could contribute to apoptosis in Nutlin-treated cells by maintaining/promoting high p53 protein levels. mTORC1 can be activated downstream of AKT, and previous studies have suggested mTORC1 can promote p53 protein synthesis.Citation23,25 However, in our studies the mTORC1 inhibitor rapamycin did not reduce p53 accumulation in Nutlin treated cells (). This suggests the mechanism by which AKT maintains p53 protein levels is not through mTORC1-mediated p53 synthesis. In continuing studies in other cells we have found that AKT inhibition reduces p53 protein levels in Nutlin-treated cells without reducing levels of p53 mRNA. This suggests IGF-1R and AKT affect p53 levels at a post-transcriptional level. Blattner and colleagues reported that AKT is required for efficient stabilization of p53 in response to ionizing radiation.Citation24 The model proposed in these previous studies was that AKT promotes p53 stabilization by phosphorylating and inhibiting GSK-3b, which normally functions with MDM2 to promote p53 degradation. It is possible that, in the current study, AKT maintained p53 protein levels in Nutlin-treated cells through a similar mechanism.
Most studies suggest the IGF-1R/AKT pathway and p53 have opposing effects on cancer cell survival. IGF-1R/AKT signaling promotes cancer cell survival, while p53 inhibits cancer cell proliferation and promotes apoptosis. It was therefore interesting in the current study that Nutlin-induced p53 caused activation of AKT. This raises the question of why p53 would activate AKT. While p53 can promote cancer cell killing in response to stress, it can also promote survival. The choice between p53-mediated cell killing and survival is dependent on multiple factors including the level of stress.Citation3 Thus, low levels of DNA damaging stress trigger p53-dependent cell cycle arrests, which allow DNA repair and survival, whereas high levels of DNA damage can trigger p53-dependent apoptosis.Citation3 It is possible that the ability of p53 to activate AKT constitutes part of the mechanism by which p53 promotes survival. There are several possible ways p53 induced by Nutlin in the current study could activate AKT. The finding that OSI-906 blocked AKT activation by Nutlin indicates it is IGF-1R dependent. Thus, one possibility is that p53 in some way increases IGF-1R activity, leading to AKT activation. Against this idea are reports that p53 can repress expression of both IGF-1R and its activating ligand IGF-1.Citation33,34 However, we note that in some but not all experiments we observed increased activation of IGF-1R (phosphorylation at Y1135) in Nutlin-treated cells (e.g., compare and ), suggesting increased p53 may lead to IGF-1R activation. A second possibility is that p53 activates AKT via the AMPK-TSC2-mTORC2 pathway. P53 can activate AMPK, which can then activate TSC2.Citation35 TSC2, in turn, can activate mTORC2, which can directly phosphorylate AKT at S473.Citation36 Thus, Nutlin-induced p53 could potentially increase AKT by activating AMPK, TSC2, and mTORC2. P53 has been reported to inhibit mTORC1,Citation37 and inhibition of mTORC1 can cause feedback activation of AKT by relieving feedback inhibition of PI3K-AKT signaling mediated by S6K.Citation30 Thus, a third possibility is that p53 could activate AKT by inhibiting mTORC1. Finally, Manfe et al (2012) reported p53 can increase expression of miR-122, which can then trigger AKT activation.Citation38 Thus, a fourth possibility is that p53 activates AKT by increasing miR-122. These possibilities are not mutually exclusive. Notably, AKT was activated to a greater extent by Nutlin in S1 and S4 cells than in MHM. Thus, whatever the mechanisms are for AKT activation by Nutlin they appears to be more active in S1 and S4 cells than in MHM.
In summary, our findings indicate AKT, activated downstream of IGF-1R, can inhibit apoptosis in Nutlin treated cells, but can also increase apoptosis through maintaining p53 protein levels and inhibiting autophagy. These findings have potential clinical implications. For example, several IGF-1R and AKT inhibitors have been developed for cancer clinical trials. Our results suggest that while these inhibitors may increase cancer cell killing, their effectiveness may be limited by a reduction in p53 protein levels and an increase in pro-survival autophagy. Overcoming these limitations could be an important goal for future drug development.
Materials and methods
Cell lines
MHM are MDM2-amplified osteosarcoma cells and were kindly provided by Dr.Ola Myklebost, Norwegian Radium Hospital. To isolate cisplatin-resistant MHM clones, MHM cells were treated with increasing doses of cisplatin (1, 2.5, 5, and 10 μM) for 48 hrs, and surviving cells expanded between each treatment. Cells expanded after 10 μM cisplatin treatment were plated at single cell density in drug-free medium (minus cisplatin) and colonies that formed after 2 weeks were isolated and expanded. A total of 6 colonies (termed S1-S6) were obtained. MHM and clones S1-S6 were grown in RPMI medium supplemented with 10% fetal bovine serum (FBS), penicillin (100 U/mL) and streptomycin (100 μg/mL).
Drug treatments
Cells were plated 24 hours before being treated. Cisplatin was from Bedford Laboratory. OSI-906, MK2206, and rapamycin were obtained from Selleck Chemicals. Bafilomycin A1 was from Sigma Aldrich. Nutlin-3a was obtained from Cayman Chemical. Cells were treated with these drugs at the following final concentrations: Cisplatin (10 μM), OSI-906 (10 μM), MK2206 (10 μM), rapamycin (0.5 μM), Nutlin-3a (5, 10, 20 μM), bafilomycin A1 (10 nM).
Immunoblotting
Whole cell extracts were prepared by resuspending cell pellets in lysis buffer (150 mM NaCl, 5 mM EDTA, 0.5% Nonidet P-40, 50 mM Tris, pH 7.5), resolved by SDS-PAGE, and transferred to polyvinylidene difluoride membranes (NEN Life Science Products). The following antibodies were used at a 1:1000 dilution and were obtained from Cell Signaling: pIGF-1R (Y1135), IGF-1R, pAKT (S473), pAKT (T308), AKT (C67E7), p-p70S6K (T389), p70S6K (49D7), and p62 (5114). Antibodies to β-Actin (C4) and p53 (Ab-6) were from Santa Cruz Biotechnology and were also used at a 1:1000 dilution. Primary antibodies were detected with goat anti-mouse (Pierce) or goat anti-rabbit (Life Technologies) secondary antibodies conjugated to horseradish peroxidase, using Clarity chemiluminescence (Bio-Rad). Both secondary antibodies were used at a 1:10,000 dilution.
Flow cytometry analysis
For apoptosis (% sub-G1 cells) and cell cycle analysis, cells were harvested and fixed in 25% ethanol overnight. The cells were then stained with propidium iodide (25 μg/ml; Calbiochem). Flow cytometry analysis was performed on Gallios™ flow cytometer (Beckman Coulter) and analyzed with FlowJo 8.7 (Treestar, Inc.). For each sample, 10,000 events were collected.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by National Cancer Institute grant 1 R21 CA185036–01A1 (to C.G.M) and by a grant from the Swim Across America Foundation (to C.G.M).
References
- Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature 1997; 387(6630):296-9; PMID:9153395; https://doi.org/10.1038/387296a0
- Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature 1997; 387(6630):299-303; PMID:9153396; https://doi.org/10.1038/387299a0
- Vousden KH, Prives C. Blinded by the light: The growing complexity of p53. Cell 2009; 137(3):413-31; PMID:19410540; https://doi.org/10.1016/j.cell.2009.04.037
- Laptenko O, Prives C. Transcriptional regulation by p53: One protein, many possibilities. Cell Death Differ 2006; 13(6):951-61; PMID:16575405; https://doi.org/10.1038/sj.cdd.4401916
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. In vivo activation of the p53 pathway by small-moleculeantagonists of MDM2. Science 2004; 303(5659):844-8; PMID:14704432; https://doi.org/10.1126/science.1092472
- Tovar C, Graves B, Packman K, Filipovic Z, Higgins B, Xia M, Tardell C, Garrido R, Lee E, Kolinsky K, et al. MDM2 small-molecule antagonist RG7112 activates p53signaling and regresses human tumors in preclinical cancer models. Cancer Res 2013; 73(8):2587-97; PMID:23400593; https://doi.org/10.1158/0008-5472.CAN-12-2807
- Tovar C, Rosinski J, Filipovic Z, Higgins B, Kolinsky K, Hilton H, Zhao X, Vu BT, Qing W, Packman K, et al. Small-molecule MDM2 antagonists reveal aberrant p53 signalingin cancer: Implications for therapy. Proc Natl Acad Sci U S A 2006; 103(6):1888-93; PMID:16443686; https://doi.org/10.1073/pnas.0507493103
- Hasegawa H, Yamada Y, Iha H, Tsukasaki K, Nagai K, Atogami S, Sugahara K, Tsuruda K, Ishizaki A, Kamihira S. Activation of p53 by Nutlin-3a, an antagonist of MDM2, inducesapoptosis and cellular senescence in adult T-cell leukemia cells. Leukemia 2009; 23(11):2090-101; PMID:19710698; https://doi.org/10.1038/leu.2009.171
- Kitagawa M, Aonuma M, Lee SH, Fukutake S, McCormick F. E2F-1 transcriptional activity is a criticaldeterminant of Mdm2 antagonist-induced apoptosis in human tumor cell lines. Oncogene 2008; 27(40):5303-14; PMID:18521084; https://doi.org/10.1038/onc.2008.164
- Shen H, Maki CG. Persistent p21 expression after Nutlin-3a removal is associated with senescence-likearrest in 4N cells. J Biol Chem 2010; 285(30):23105-14; PMID:20489208; https://doi.org/10.1074/jbc.M110.124990
- Shen H, Moran DM, Maki CG. Transient nutlin-3a treatment promotes endoreduplication and thegeneration of therapy-resistant tetraploid cells. Cancer Res 2008; 68(20):8260-8; PMID:18922897; https://doi.org/10.1158/0008-5472.CAN-08-1901
- Huang B, Deo D, Xia M, Vassilev LT. Pharmacologic p53 activation blocks cell cycle progression butfails to induce senescence in epithelial cancer cells. Mol Cancer Res 2009; 7(9):1497-509; PMID:19737973; https://doi.org/10.1158/1541-7786.MCR-09-0144
- Denduluri SK, Idowu O, Wang Z, Liao Z, Yan Z, Mohammed MK, Ye J, Wei Q, Wang J, Zhao L, et al. Insulin-like growth factor (IGF) signaling in tumorigenesis andthe development of cancer drug resistance. Genes Dis 2015; 2(1):13-25; PMID:25984556; https://doi.org/10.1016/j.gendis.2014.10.004
- Abedini MR, Muller EJ, Bergeron R, Gray DA, Tsang BK. Akt promotes chemoresistance in humanovarian cancer cells by modulating cisplatin-induced, p53-dependent ubiquitination of FLICE-likeinhibitory protein. Oncogene 2009; 29(1):11-25; PMID:19802016; https://doi.org/10.1038/onc.2009.300
- Parcellier A, Tintignac LA, Zhuravleva E, Hemmings BA. PKB and the mitochondria: AKTing onapoptosis. Cell Signal 2008; 20(1):21-30; PMID:17716864; https://doi.org/10.1016/j.cellsig.2007.07.010
- Zhang X, Tang N, Hadden TJ, Rishi AK. Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta 2011; 1813(11):1978-86; PMID:21440011; https://doi.org/10.1016/j.bbamcr.2011.03.010
- Pene F, Claessens YE, Muller O, Viguié F, Mayeux P, Dreyfus F, Lacombe C, Bouscary D. Role of the phosphatidylinositol 3-kinase/Akt and mTOR/P70S6-kinase pathways in the proliferation and apoptosis in multiple myeloma. Oncogene 2002; 21(43):6587-97; PMID:12242656; https://doi.org/10.1038/sj.onc.1205923
- Rashmi R, DeSelm C, Helms C, Bowcock A, Rogers BE, Rader JL, Grigsby PW, Schwarz JK. AKT inhibitors promote cell death in cervical cancer throughdisruption of mTOR signaling and glucose uptake. PloS One 2014; 9(4):e92948; PMID:24705275; https://doi.org/10.1371/journal.pone.0092948
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through directphosphorylation of Ulk1. Nat Cell Biol 2011; 13(2):132-41; PMID:21258367; https://doi.org/10.1038/ncb2152
- Zhu N, Gu L, Li F, Zhou M. Inhibition of the Akt/survivin pathway synergizes the antileukemia effect ofnutlin-3 in acute lymphoblastic leukemia cells. Mol Cancer Ther 2008; 7(5):1101-9; PMID:18483299; https://doi.org/10.1158/1535-7163.MCT-08-0179
- Saiki AY, Caenepeel S, Yu D, Lofgren JA, Osgood T, Robertson R, Canon J, Su C, Jones A, Zhao X, et al. MDM2 antagonists synergize broadly and robustly with compoundstargeting fundamental oncogenic signaling pathways. Oncotarget 2014; 5(8):2030-43; PMID:24810962; https://doi.org/10.18632/oncotarget.1918
- Xiong L, Kou F, Yang Y, Wu J. A novel role for IGF-1R in p53-mediated apoptosis through translationalmodulation of the p53-Mdm2 feedback loop. J Cell Biol 2007; 178(6):995-1007; PMID:17846171; https://doi.org/10.1083/jcb.200703044
- Davaadelger B, Duan L, Perez RE, Gitelis S, Maki CG. Crosstalk between the IGF-1R/AKT/mTORC1pathway and the tumor suppressors p53 and p27 determines cisplatin sensitivity and limits theeffectiveness of an IGF-1R pathway inhibitor. Oncotarget 2016; 7(19):27511-26; PMID:27050276; https://doi.org/10.18632/oncotarget.8484
- Boehme KA, Kulikov R, Blattner C. p53 stabilization in response to DNA damage requires Akt/PKB andDNA-PK. Proc Natl Acad Sci (USA) 2008; 105(22):7785-90; PMID:18505846; https://doi.org/10.1073/pnas.0703423105
- Vadysirisack DD, Baenke F, Ory B, Lei K, Ellisen LW. Feedback control of p53 translation by REDD1and mTORC1 limits the p53-dependent DNA damage response. Mol Cell Biol 2011; 31(21):4356-65; PMID:21896779; https://doi.org/10.1128/MCB.05541-11
- Duan L, Perez RE, Davaadelger B, Dedkova EN, Blatter LA, Maki CG. p53-regulated autophagy iscontrolled by glycolysis and determines cell fate. Oncotarget 2015; 6(27):23135-56; PMID:26337205; https://doi.org/10.18632/oncotarget.5218
- Ottaviani G, Jaffe N. The etiology of osteosarcoma. Cancer Treat Res 2009; 152:15-32; PMID:20213384; https://doi.org/10.1007/978-1-4419-0284-9_2
- O'Day K, Gorlick R. Novel therapeutic agents for osteosarcoma. Expert Rev Anticancer Ther 2009; 9(4):511-23; PMID:18505846; https://doi.org/10.1586/era.09.7
- Kempf-Bielack B, Bielack SS, Jurgens H, Branscheid D, Berdel WE, Exner GU, Göbel U, Helmke K, Jundt G, Kabisch H, et al. Osteosarcoma relapse after combined modality therapy: An analysis of unselected patients in the Cooperative Osteosarcoma Study Group (COSS). J Clin Oncol 2005; 23(3):559-68; PMID:15659502; https://doi.org/10.1200/JCO.2005.04.063
- O'Reilly KE, Rojo, F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling andactivates AKT. Cancer Res 2006; 66(3)1500-8; PMID:16452206; https://doi.org/10.1158/0008-5472.CAN-05-2925
- Russell RC, Yuan H-X, Guan K-L. Autophagy regulation by nutrient sensing. Cell Res 2014; 24:42-57; PMID:24343578; https://doi.org/10.1038/cr.2013.166
- Kim YC, Guan K-L. mTOR: A pharmacologic target for autophagy regulation. J Clin Invest 2015; 125(1) 25-32; PMID:25654547; https://doi.org/10.1172/JCI73939
- Werner H, Karnieli E, Rauscher FJ, LeRoith D. Wild-type and mutant p53 differentially regulatetranscription of the insulin-like growth factor I receptor gene. Proc Natl Acad Sci U S A 1996; 93:8318-23; PMID:8710868; https://doi.org/10.1073/pnas.93.16.8318
- Goetz EM, Shankar B, Zou Y, Morales JC, Luo X, Araki S, Bachoo R, Mayo LD, Boothman DA. ATM-dependent IGF-1 induction regulates secretory clusterin expression after DNA damage and in geneticinstability. Oncogene 2011; 30:3745-54; PMID:21460853; https://doi.org/10.1038/onc.2011.92
- Ryan KM, Vousden KH. p53 and metabolism. Nat Rev Cancer 2009; 9:691-700; PMID:19759539; https://doi.org/10.1038/nrc2715
- Huang J, Dibble CC, Matsuzaki M, Manning BD. The TSC1-TSC2 complex is required for properactivation of mTOR complex 2. Mol Cell Biol 2008; 28(12) 4104-15; PMID:18411301; https://doi.org/10.1128/MCB.00289-08
- Hay N. p53 strikes mTORC1 by employing sestrins. Cell Metab 2008; 3(3) 184-5; PMID:18762019; https://doi.org/10.1016/j.cmet.2008.08.010
- Manfe V, Biskup E, Rosbjerg A, Kamstrup M, Guldhammer S, Lerche CM, Lauenborg BT, Odum N, Gniadecki R. miR-122 regulates p53/AKT signaling and the chemotherapy-induced apoptosis incutaneous T-cell lymphoma. PLoS One 2012; 7(1): e29541; PMID:22235305; https://doi.org/10.1371/journal.pone.0029541