
ABSTRACT
Nasopharyngeal carcinoma (NPC) is a common malignant neoplasm of the head and neck which is harmful to human's health. Radiotherapy is commonly used in the treatment of NPC and it induces immediate cell cycle arrest and cell apoptosis. However, the mechanism remains unknown. Evidences suggested the activation of Ataxia telangiectasia mutated (ATM) pathway and Smad pathway are 2 of the important crucial mediators in the function of radiotherapy. In this study, we performed in vitro assays with human nasopharyngeal carcinoma CNE-2 cells and in vivo assays with nude mice to investigate the role of the ATM and Smad pathways in the treatment of nasopharyngeal carcinoma with radiotherapy. The results suggested that radiation induced activation of ATM pathway by inducing expression of p-ATM, p-CHK1, p-CHK2, p15 and inhibiting expression of p-Smad3. In addition, Caspase3 expression was increased while CDC25A was decreased, leading to cell cycle arrest and cell apoptosis. On the other hand, activation of Smad3 can inhibited the ATM pathway and attenuated the efficacy of radiation. In summary, we suggest that both ATM and Smad pathways contribute to the cell cycle arrest and cell apoptosis during nasopharyngeal carcinoma cells treated with radiation.
Introduction
Nasopharyngeal carcinoma (NPC) is one of the most common malignant neoplasm originating from the uppermost region of the pharynx. It has significant demographic variations.Citation1-3 The NPC is endemic in particular such as Southeast Asia, and the annual incidence varies from 30 to 80 per 100,000 people in China.Citation4,5 The etiology of NPC is multifactorial, including the Epstein-Barr virus (EBV) infection, genetic susceptibility, unhealthy lifestyle and environmental hazards.Citation6-9
The regular treatment of NPC includes surgery, anti-cancer drugs and radiation. Among these, radiotherapy is regarded as the primary approach for NPC treatment.Citation10 Primary tumor control is important to reduce the later appearance of distant metastasis, however, side effects of radiotherapy cannot be ignored and it might trigger distant metastases and tumor progression. The previous study has shown that in patients with advanced carcinoma of cervix and bladder, failure of radiotherapy to control tumor is associated with an increasing subsequent incidence of metastasis.Citation11 In addition, radiotherapy may induces the distant metastases by modify the tumor microenvironment.Citation12,13 Moreover, some studies also demonstrated that accelerated development and proliferation of tumor cells may occur about 3 to 4 weeks after exposure to radition.Citation14 Thus, it is necessary to understand the molecular mechanisms of NPC radiotherapy treatment.Citation15-17 The induction of cell apoptosis and cell cycle arrest in cancer cells are the strategies of the radiotherapy.Citation18,19 Ataxia telangiectasia mutated (ATM) is a nuclear 350kDa protein kinase with a carboxylterminal phosphatidylinositol 3-kinase-like kinase domain.Citation20 The functions of ATM are multiple that it detects DNA breaks, arrests the cells at G1, S or G2/M phase temporarily, and activates DNA repair.Citation21 ATM is phosphorylated at specific sites and activated when DNA damage is sensed.Citation22 The activation of ATM subsequently phosphorylates the downstream proteins such as p53, checkpoint kinase 1 (CHK1) and CHK2. Among these proteins, the phosphorylated CHK1 and CHK2 are in response to replication stress or DNA damage.Citation23,24 The effects on the members of the cell division cycle 25 homolog (CDC25) phosphatase family (CDC25A, CDC25B and CDC25C) are understood functions of CHK1 and CHK2. The activation of the CHK1 and CHK2 through ATM pathway caused by DNA damage or replication stress, results in the inactivation of CDC25s and thereby inhibiting cyclin-dependent kinase (CDK)and inducing cell cycle arrest.Citation23 Apart from ATM pathway, Transforming growth factor (TGF)-β1 plays an important role in various biologic processes, such as cell proliferation, differentiation, and apoptosis. SMA- and MAD-related protein 3 (SMAD3) is the direct mediator of TGF-β signaling pathway. SMAD3 has been proven to mediate transcription of the CDK inhibitors p15 and p21. Therefore, it induces cell cycle arrest by regulating CDKs indirectly.Citation25-27
By considering the both ATM pathway and Smad pathway target on CDK family and lead to cell cycle arrest, we hypothesize that the 2 signaling pathways should be related and affected by the radiotherapy. In this study, we set up both in vitro assays using CNE-2 cell line and in vivo assays using nude mice to investigate the relationship between radiation therapy and the ATM and Smad signaling pathways.
Materials and methods
Ethical statement
This study was approved by the Ethical Committee of the Affiliated Cancer Hospital & Institute of Guangzhou Medical University.
Cell culture
Human poorly differentiated NPC cell line CNE-2 was purchased from the cell bank of Sun Yat-Sen University (Guangzhou, China). The cells were cultured at 37°C with 5% CO2 in RPMI-1640 medium (Life Technologies Inc., Gibco BRL, USA) supplemented with 10% fetal bovine serum (Life Technologies Inc., Gibco BRL, USA), 1% penicillin and streptomycin. The cells were incubated at humidified incubator, and the culture medium was changed routinely every 2 or 3 d. Cells were cultured, amplified and passaged. After 3 days, cells were digested and harvested. Cell morphology was viewed under a light microscope (Nikon, Japan), and suspended to concentration of 1 × 106/ml for further use.
Immunofluorescent staining for phosphorylated histone H2AX (γH2AX)
Cells were grown and treated in chamber slides. Cells were incubated with RPMI-1640 medium with or without TGF-β1 solution (10 ng/ml; Sigma-aldrich, USA). Cells were radiated using a Pantak (Solon, OH) X-ray source for 6 h at specific dose rate of 0, 2, 4, 6, 8 and 10 Gy/min. For 24 h incubation after radiation, medium was aspirated and cells were fixed in 4% paraformaldehyde for 10 min at room temperature. The paraformaldehyde was removed and the cells were treated with 0.2% NP40/PBS solution for 15min. Cells were washed by PBS twice, and then anti-γH2AX antibody (Cell Signaling Technology, USA) was added at dilution of 1:500 in 1% BSA and incubated overnight at 4°C. Cells were washed twice in PBS before incubating in the dark with an FITC-labeled secondary antibody at a dilution of 1:100 in 1% BSA for 1 h. The secondary antibody solution then was aspirated, and then cells were washed twice in PBS. Cells then were incubated in the dark with 4′, 6-diamidino-2-phenylindole (1 μg/ml) in PBS for 30 min and washed twice. The nuclear DNA was stained with 1 μM Hoechst. Slides were examined on a Leica DMRXA fluorescent microscope (Wetzlar, Germany).
Cell colony formation assay
Specific numbers of cells were first seeded into the wells of a 6-well tissue culture plate with RPMI-1640 medium, and radiation was delivered 6 h later. Plates were incubated for colony formation for 10–14 d. Colonies were stained with crystal violet, and the number of the surviving fractions was calculated.
Cell viability assay
Cells were seeded onto 96-well plates at a density of 1 × 104 per well. After overnight incubation, the culture medium was removed and cells were rinsed with PBS and treated with different dose rate of radiation. After 6 h treatment, MTT was added to each well and incubated for 4 h to allow mitochondrial dehydrogenase to convert MTT into insoluble formazan crystals. The medium was removed and formazan was solubilized by adding 150 μl of DMSO. The viability of the cells was measured at 490nm using an ELISA reader (BioTek, Winooski, VT, USA) according to the manufacturer's instructions. The cell viability was determined per 24 h.
Cell apoptosis assay
Cells were harvested, washed twice in ice-cold PBS, and re-suspended in buffer containing 7-AAD (7-Amino-actinomycin D) for 10 min, followed by the addition of Annexin V-PE. Cell apoptosis assay was performed using a flow cytometer (BD Biosciences, USA).
Cell cycle assay
Cells were harvested, washed with PBS twice and then fixed with 70% ethanol for 12 h. The fixed cells were spun down and re-suspended in PBS in 1 × 106/ml. After incubation with ribonuclease A at a final concentration of 3000 units/ml at 37°C for 30 min, cells were filtered through a nylon mesh (BD Bioscience, USA). The cells were stained with propidium iodide before analysis on a flow cytometer (BD Bioscience, USA).
Quantitative real-time PCR
Total RNA was prepared using TRIZOL (Invitrogen, Paisley, Scotland) following the manufacturer's protocol. Genomic DNA-free RNA was then converted into cDNA using the M-MLV Reverse Transcriptase (Promega, Melbourne, Australia) and PCR amplification was performed using GoTaq qPCR Master Mix (Promega, Melbourne, Australia) on CFX96 (Bio-Rad, CA, USA). Relative quantification of the mRNA level was calculated using the 2−△△Ct method. The primers used in this study was shown in .
Table 1. The primers used in this study.
Western blotting
Cell proteins were extracted and the concentration was determined using BCATM-Protein Assay Kit (Fulengen, Guangzhou, China). The protein samples were homogenized in the loading buffer and boiled for 5 min, then separated on 8% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Etten-leur, Netherlands). The membranes were blocked with 5% nonfat milk in phosphate-buffered saline containing 0.1% Tween 20 for 2 h, and then treated with primary antibodies in 1:200 duiltion for 12 h at 4°C. The membranes were washed with TBST followed by incubation with the secondary antibody for 45 min at room temperature. The complexes were detected using ECL (Forevergen, Guangzhou, China). The primary antibodies and the secondary antibodies were purchased from Cell Signaling Technology Company (Danvers, MA, USA).
Transfection
The Smad3 overexpression vector (LV-Smad3) and Smad7 suppression shRNA (LV-shSmad7) were designed and packaged in lenti-virus by Forevergen biotechnology company (Guangzhou, China). The CNE-2 cells (1 × 106/ml) were seeded in the 24-well plate overnight at 37°C. Lenti-virus was thawed at 37°C water bath and diluted in cell medium. The cell medium was removed and then incubated the cells with the lenti-virus solution. Polybrene was added and mixed gently several times. The cells were incubated overnight at 37°C. The culture medium was changed with the fresh one after incubation. The cells were cultured for 10–12 d. Fluorescence microscope was applied for observation and screening the cells with positive expression. Those cells were collected for the downstream applications.
Subcutaneous tumor formation of nude mouse
The 6–8 weeks old nude mice were purchased from Vitalriver, Beijing, China. The nude mice were assigned in different groups and injected with the cells and exposed to specific treatments: (1) blank CNE-2 cells (CNE-2); (2) CNE-2 cells with LV-Smad3 transfection (Smad3); (3) CNE-2 cells with LV-shSmad7 transfection (shSmad7); (4) CNE-2 cells with 6 Gy radiation (CNE-2+6 Gy); (5) CNE-2 cells with LV-Smad3 transfection and 6 Gy radiation (Smad3+6 Gy); (6) CNE-2 cells with LV-shSmad7 transfection and 6 Gy radiation (shSmad7+6 Gy). Each group was assigned with 5 animals. The cells in exponential phase were trypsinized and washed twice by PBS. The cells were re-suspended with PBS at 1 × 107/ml. The cell suspensions were injected into the axilla of the animals. The nude mice were then observed for survival and tumor formation. At 14 d after injection, the nude mice of group 4, 5 and 6 were radiated with 6 Gy radiation for 2 d. After 5 weeks, the mice were killed, and the cancerous tissues were removed by sterile surgery and were placed in RNase-free saline. The weight and size of the cancerous tissues were then examined. The collected cancerous tissues were fixated by formalin and embedded by paraffin.
Immunohistochemical Staining
The tumor tissues were deparaffinized by xylene and washed by PBS twice. The tissues were blocked with 3% H2O2 for 5–20 min and subsequently washed by PBS 3 times. The tissue was fixed by water incubation at 95°C and then washed by PBS. The tissues were then blocked with 5% BSA and incubated with primary antibody at 4°C overnight. The cells were washed by PBS 3 times, and incubated with secondary antibody for 1 h at room temperature. The cells were washed by PBS and were developed in freshly prepared diaminobenzidine solution (DAB) then counterstained with hematoxylin, dehydrated through graded ethanol, cleared with xylene, and coverslipped. The expression levels of the molecules were estimated by microscopy.
Statistical analysis
Comparisons were performed using independent t-tests. SPSS software was used for the statistical analysis. The significance was established as P < 0.05.
Results
Radiation inhibits the CNE-2 cell proliferation via ATM pathway and Smad3/7 cascades
To verify the efficacy of radiation as a treatment of nasopharyngeal carcinoma, CNE-2 cell line was applied and treated with different doses of radiation (0, 2, 4, 6, 8 and 10 Gy/min). The results suggested that radiation induced direct damage to the DNA of tumorous cells. The detected signal of γH2AX was presented in green and the optical density of γH2AX was increased positively related to the dose usage of radiation (). And the damage of the DNA thereby induced the decreased cell viability at 72 h after radiation (, * P < 0.05, ** P < 0.01). Compared with the cells without being irradiated, the cell viability was significantly decreased followed with the increased dose usage of radiation ( C, * P < 0.05, ** P < 0.01). According to the cell formation experiment, the decreased cell viability reduced the colony formation efficiencies of the cell line ( and ). The cells treated with radiation were significantly inhibited to form colonies and the efficiency of the colony formation was reduced when higher dose of radiation was used. Followed by the flow cytometry assays, the effect of the radiation to CNE-2 cells was verified. The analysis of the cell cycle obviously showed that after the cells received radiation treatment, the copies of cells were obviously maintained at G2/M phase (). However, the cells in G1 or S phase were decreased accordingly depending on the dose of radiation (). On the other hand, the cell apoptosis and necrosis percentage were dramatically increased after the treatment of radiation (, * P < 0.05, ** P < 0.01). It indicated that radiation prevented the cells from proliferation and also led to cell death via apoptosis and necrosis and therefore inhibited the oncogenesis.
Figure 1. Radiation induced DNA damage and inhibited cell proliferation in CNE-2 cells. (A) γH2AX signal presented the DNA damage was increased together with the increased doses of radiation. (B) the cell viability was significantly decreased followed with the increased dose usage of radiation. (C, D), the amounts of the cell colonies were significantly decreased followed with the increased doses of radiation. * P < 0.05 vs. 0 Gy; ** P < 0.01 vs. 0 Gy. Scale bar: 10 μm.
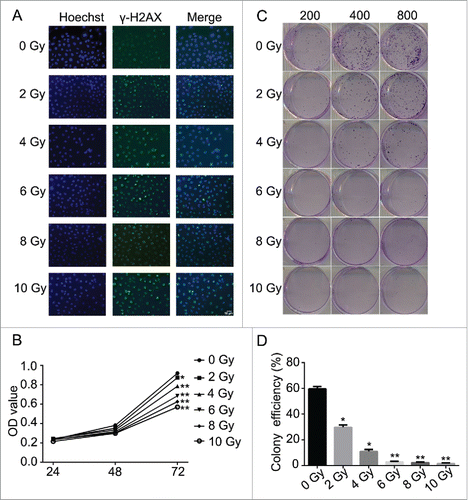
Figure 2. Radiation induced the cell cycle arrest and promoted cell apoptosis in CNE-2 cells. (A) radiation increased the percentage of the cells in G2/M phase. (B) the cell apoptosis and necrosis were enhanced after radiation. * P < 0.05 vs. 0 Gy; ** P < 0.01 vs. 0 Gy.
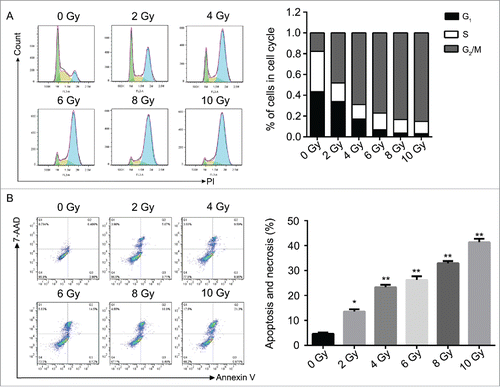
To further study the mechanism behind, we targeted on the cell proliferation related genes in ATM pathway and Smad3/7 cascade and evaluated the protein expression via western blot (). As shown in the results, the expression levels of the genes were changed accordingly depending of the dose of radiation. However, the expression levels of the genes were not shown further changes when the dose of radiation greater than 6 Gy. With the treatment of radiation, the expressions of p-ATM, p-CHK1, p-P53, P15, P21 and cleaved caspase 7 were upregulated, while the expression of CDC25A was downregulated. The expression of Smad3 was not changed. However, the expression level of p-Smad3 was detected significantly decreased while the expression level of Smad7 was increased significantly (). It indicated that radiation activated the ATM pathway and also the Smad3/7 cascade that should be related to the decreased amounts of CNE-2 cells.
Figure 3. Western blotting analysis. Radiation activated the ATM pathway, increased the expression of Smad7 and inhibited the phosphorylation of Smad3.
TGF-β1 activates the ATM pathway and induces CNE-2 cell cycle arrest and cell apoptosis
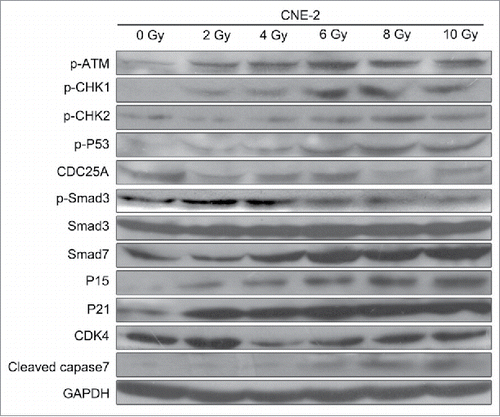
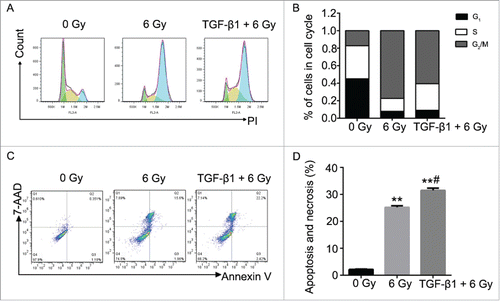
According to the previous study, TGF-β1 is an activator of the ATM pathway in regulating the cell apoptosis manner.Citation28 Therefore, we performed the pretreatment to CNE-2 cells using TGF-β1 (10 ng/ml in medium containing 1% FBS) to investigate the relationship between ATM pathway and radiation. As shown in the results, with the supplement of TGF-β1, it was detected the highest signal of γH2AX compared with the 0 Gy group and 6 Gy group (). In this case, the 6 Gy radiation with TGF-β1 treatment presented significant decrease of the cell viability compared with the 0 Gy group (, ** P < 0.01 vs. 0 Gy). Cell colony formation assay was performed to verify the direct effect of radiation and TGF-β1. The amount of the cell colonies was obviously decreased and hardly observed in the 6 Gy and TGF-β1+6 Gy groups (). The colony formation efficiency of the TGF-β1+6 Gy group was dramatically decreased compared with the 0 Gy group (, ** P < 0.01 vs. 0 Gy). At the same moment, flow cytometry was applied to determine the cell cycle and cell apoptosis. The results of the cell cycle assay suggested that both of the 6 Gy and TGF-β1+6 Gy groups were presented fewer cells in G1 phase but more cells in S and G2/M phase compared with the 0 Gy group ( and ). On the other hand, it proved the highest cell apoptosis and necrosis percentage in TGF-β1+6 Gy group in the cell apoptosis assays ( and ). Although the 6 Gy treatment induced significant increase of the apoptosis and necrosis percentage compared with the 0 Gy group (** P < 0.01), the TGF-β1+6 Gy group presented significant higher percentage no matter compared with 0 Gy group or 6 Gy group (** P < 0.01 vs. 0 Gy, #P < 0.05 vs. 6 Gy).
Figure 4. Application of TGF-β1 increased the sensitivity of CNE-2 cells to radiation. (A) the DNA damage was enhanced by TGF-β1, compared with 0 Gy and 6 Gy group. (B) TGF-β1 treatment significantly decreased of the cell viability of CNE-2 cells compared with the 0 Gy and 6 Gy radiation groups. (C, D) the amounts of the cell colonies were significantly decreased after the TGF-β1 treatment.** P < 0.01 vs. 0 Gy.
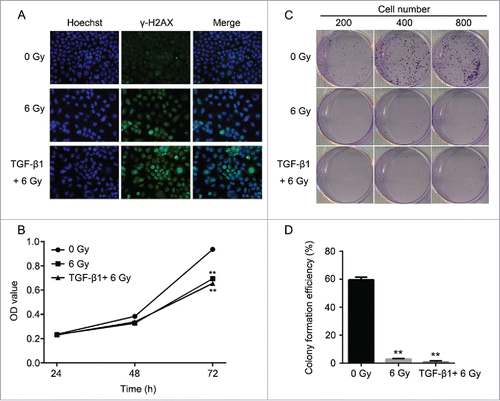
Figure 5. The effect to the cell cycle and cell apoptosis of CNE-2 cells after TGF-β1 treatment. (A) the percentage of the cells was increased in G2/M phase. (B) the cell apoptosis and necrosis were significantly enhanced. ** P < 0.01 vs. 0 Gy; # P < 0.05 vs. 6 Gy.
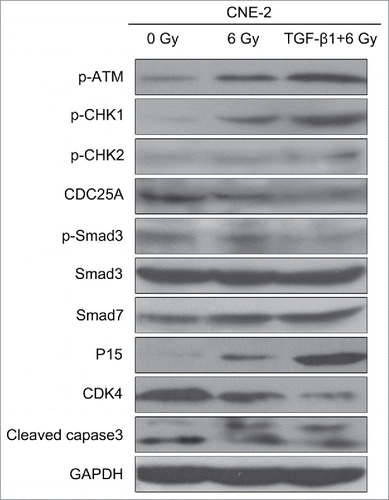
To further investigate the mechanism behind, western blot was performed to determine the expression levels of the genes in the ATM pathway and Smad cascade (). The results of 0 Gy and 6 Gy treatment were similar as the previous radiation dose assays. Comparing the TGF-β1+6 Gy group with 6 Gy group, the expression levels of p-ATM, p-CHK1, p-CHK2, P15, and cleaved caspase3 were obvious higher in the TGF-β1+6 Gy group. It indicated that the ATM pathway was activated by the application of TGF-β1. At the same time, the expression of Smad3 presented nearly no difference among all groups. However, the expression level of p-smad3 was decreased and smad7 was increased further in the TGF-β1+6 Gy group. These results above proved that the ATM pathway should be related to the Smad3/7 cascade. TGF-β1 as an agonist activated the ATM pathway and regulated the balance of p-Smad3 and Smad7. As a result, the CNE-2 cells became vulnerable to radiation and present higher apoptosis.
Figure 6. TGF-β1 activated the ATM pathway, increased the expression of Smad7 and inhibited the phosphorylation of Smad3.
Smad3/7 cascade is the regulator of the cell sensitivity to radiation
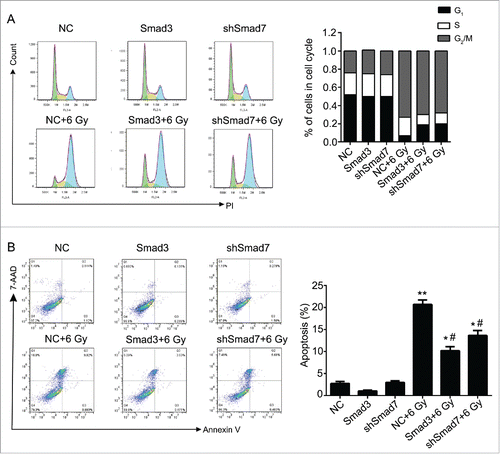
According to the results above, the Smad3/7 cascade indicated its potential role in regulating the cell cycle and apoptosis when CNE-2 cells were exposed to radiation. We hypothesized that Smad3/7 cascade should have relationship with the cell sensitivity to radiation. Therefore, we performed transfection of Smad3 overexpression vector (CNE-2 Smad3 group) and Smad7 suppression vector (CNE-2 shSmad7 group) to the CNE-2 cells. The transfection efficiency was shown in . It can be obviously observed that green fluorescence was detected when the vectors were transfected into the CNE-2 cells. Compared with negative control (NC) group, the Smad3 expression was significantly increased in the Smad3 group but decreased in the shSmad7 group (, ** P < 0.01 vs. NC), indicating that the vectors have been transfected into the cells successfully. Under the condition of radiation, it obviously showed that the transfection of each vector induced significant γH2AX signal under the treatment of radiation, while it was almost not different from the NC group when the cells were not treated with radiation (). However, when compared with the group of cells treated 6 Gy radiation only, the γH2AX signal was slightly weaker in both transfection treatment groups. Therefore, neither the overexpression of Smad3 nor suppression of Smad7 induced the damage to the cells. The transfection treatments attenuated the sensitivity of the cells to radiation and thereby the cells became less vulnerable when exposed to radiation. As expected, the damage to the cells came decreased cell viability () and weaker capacity of cell colony formation (). But the radiation treated cells presented weaker viability and slightly fewer cell colony amounts when compared with the Smad3/7 transfection and radiation treatment groups. However, the difference was not significant. By examining the cell cycle, the 6 Gy radiation induced more cells maintained in S and G2/M phase. However, the transfection of the Smad3/7 related vector induced a bit more cells in G1 phase compared with the NC+6 Gy group (). Meanwhile, the cell apoptosis in the NC+6 Gy group presented the highest percentage among the groups (). The cells transfected with Smad3/7 related vector presented resistance to the radiation and thereby the apoptosis percentage in both vector transfection plus radiation groups was significantly lower than the NC+6 Gy group.
Figure 7. Overexpression of Smad3 or suppression of Smad7 decreased the damage to CNE-2 cells after radiation. (A) the Smad3 overexpression vector and Smad7 suppression shRNA was transfected to CNE-2 cells successfully. (B) Smad3 mRNA expression was significantly increased in the Smad3 group but significantly decreased in the shSmad7 group. ** P < 0.01 vs. NC group. (C) compared with the NC+6 Gy group, the DNA damage was decreased in Smad3+6 Gy group and shSmad7+6 Gy group.
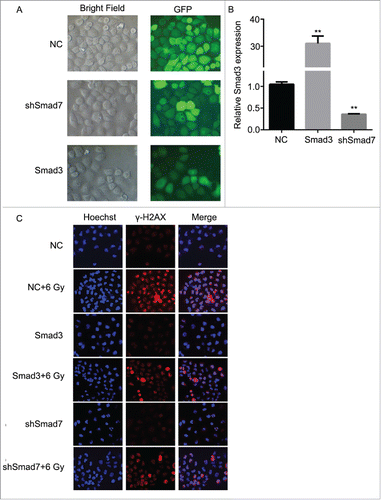
Figure 8. Overexpression of Smad3 or suppression of Smad7 affected the cell proliferation and colony formation. (A) the cell viability was slightly increased in Smad3+6 Gy group and shSmad7+6 Gy group. * P < 0.05 vs. NC group; #P < 0.05 vs.Smad3 group; & P < 0.05 vs. shSmad7 group. (B) overexpression of Smad3 or suppression of Smad7 increased the cell colony formation; ** P < 0.01 vs. NC group; # P < 0.05 vs. NC+6 Gy group.
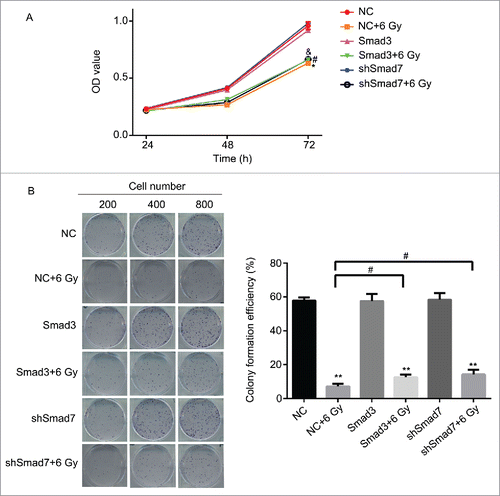
Figure 9. Overexpression of Smad3 or suppression of Smad7 affected the cell cycle and cell apoptosis. (A) the cells in G2/M phase were decreased, while the cells in G1 and S phase were increased. (B) Cell apoptosis and necrosis were significantly decreased. Compared Smad3 group, the cell apoptosis was increased in Smad3+6 Gy group; Compared with shSmad7 group, the cell apoptosis was increased in shSmad7+6 Gy group. * P < 0.05, ** P < 0.01 vs. NC group; #P < 0.05 vs. NC+6 Gy group.
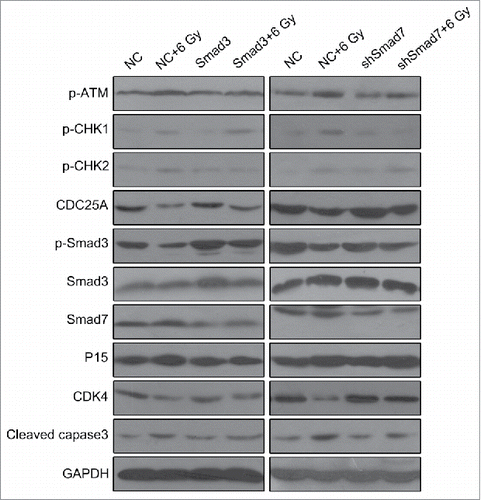
For understanding the mechanism behind, we determined the expression ATM pathway and Smad3/7 cascade related genes via western blotting (). By comparing with the 6 Gy radiation group, the genes including p-ATM and cleaved caspase3 in Smad3, Smad3+6 Gy, shSmad7 and shSmad7+6 Gy groups were downregulated expressed, while CDK4 and CDC25A were upregulated expressed. In the Smad3/7 cascade, the expression of the p-Smad3 was increased and expression of Smad7 was decreased. By concluding the results above, the levels of the Smad3 and Smad7 were maintained in a balance, and the changes of the expression level affect the robustness of the ATM pathway. As a result, the cell sensitivity to radiation could be enhanced or suppressed that affect the cell apoptosis.
Figure 10. Overexpression of Smad3 or suppression of Smad7 attenuated the activation of ATM pathway.
In vivo assays indicated the ATM pathway and Smad3/7 cascade affects the cell sensitivity to radiation
Nude mice were applied to verify the exact functions of the ATM pathway and Smad3/7 cascade in vivo. We conducted the subcutaneous tumor formation assay for the purpose. The results clearly proved that the radiation did effect to inhibit the cells proliferation that the volume and weight of the tumor were smaller compared with the animals without radiation treatment ( and ). However, for those mice injected with the CNE-2 cells with Smad3/7 vector transfection, the tumor grew faster than those animals injected with normal CNE-2 cells when exposed to 6 Gy radiation. By extracting the total RNA from the tumorous tissues and conducted with RT-PCR, the mRNA level of both Smad3 and Smad7 was determined (). The results were in correspondence with the in vitro assays. The 6 Gy radiation induced increased level of the Smad7 but did not induce any change to Smad3. However, when the CNE-2 cells transfected with the Smad3 overexpression vector or Smad7 suppression vector, the expression of Smad3 was increased while the expression of Smad7 was decreased. Immunohistochemical staining was performed to determine the robustness of the ATM pathway and Smad3/7 cascade in the tumorous tissues. The expression of the genes in the ATM pathway including the p-ATM, p-P53, cleaved caspase3 was increased after treated with radiation indicating the ATM pathway was activated (, and ). In this case, the level of the p-Smad3 was decreased and Smad7 was increased accordingly (, ). However, compared with the tumorous tissue that accepted 6 Gy radiation treatment only, in the Smad3/7 vector transfected CNE-2 cells formed tumorous tissues, the changes were slightly attenuated. In another word, the expression of p-ATM, p-P53, caspase3 was slightly lower. Meanwhile, the p-Smad3 presented higher expression but Smad7 was in slightly lower expression level. By detecting the level of cleaved caspase3, western blot was performed (). The results presented that, when the CNE-2 formed tumor exposed to the 6 Gy radiation, the cleaved caspase3 was increased in expression and thereby induced the cell apoptosis. However, the expression of cleaved caspase3 was inhibited when Smad3 was overexpressed or Smad7 was suppressed in expression as it was observed in the image, although the animals were treated with 6 Gy radiation. By summarizing the results, the radiation induced cell apoptosis was regulated by the ATM pathway and Smad3/7 cascade. The mechanism is valid in the in vivo conditions. When the expression of Smad7 is raised and p-Smad3 is decreased, the cell will become more sensitive to radiation and consequently induces cell apoptosis.
Figure 11. Overexpression of Smad3 or suppression of Smad7 suppressed the activation of ATM pathway, and decreased the sensitivity of tumor to radiation in nude mice. (A, B), compared with CNE-2+6 Gy group, the volume and the weight of the tumor was increased obviously in Smad3+6 Gy group and shSmad7+6 Gy group. (C) the mRNA level of Smad3 and Smad7 was detected by RT-PCR. * P < 0.05,** P < 0.01.
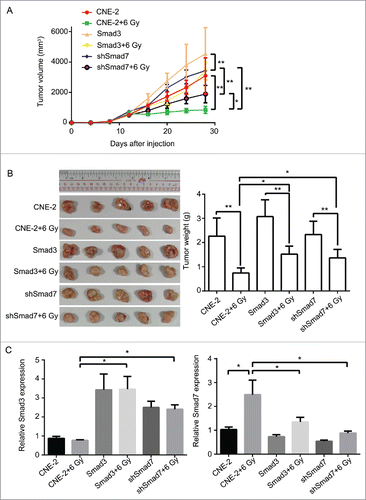
Figure 12. Immunohistochemical staining in evaluating the robustness of ATM pathway and Smad3/7 cascade before and after treated with radiation.
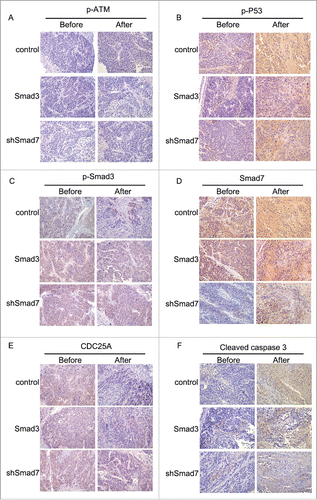
Figure 13. Western blotting to determine the expression level of cleaved caspase3 in formed tumors.

Discussion
Radiotherapy as one of the effective therapy to treat NPC, it causes the cell cycle arrest and cell apoptosis and thereby slow down the metastasis progress. In our study, radiation induced DNA damage and activated the ATM pathway and subsequently altered the expression of downstream proteins including the p15, p21, CHK1 and CDC25A. The Smad3/7 cascade was also activated in the CNE-2 cells after radiation. It indicated that the ATM and Smad pathway contribute to the cell cycle arrest and cell apoptosis. The percentage of apoptotic cells was gradually increased in a dose-dependent manner after radiation. This is consistent with other published data.Citation29,30 ATM is the crucial factor in the ATM pathway, and also the mediator for double-strand breaks responses. It is activated by auto-phosphorylation upon double-strand breaks induction and critical for phosphorylating a couple of proteins involved in DNA repair and damage signaling pathways.Citation31 The double-strand breaks sensing and processing proteins induced by radiation can be observed by the immunofluorescence and are referred to the special foci.Citation32 γH2AX was used in this study to detect the double-strand breaks. Many studies have revealed monitoring γH2AX as a fairly accurate means to estimate the formation and loss of double-strand breaks formation at different times.Citation33-35 Redon et al reported that detection of γH2AX could be used to be a biomarker of DNA damage induce by ionizing radiation in human blood and skin tests.Citation36 The signal of the γH2AX presented the immediate damage to the DNA after the radiation,Citation37 which may lead to better understanding of mutagenic pathways in cancer cells and provide a useful biomarker for monitoring the effectiveness of DNA damaging anticancer agents or therapy.Citation38 Accompanied with the DNA damage, ATM pathway was activated and the expression of the p-ATM was increased accordingly. For the regular process, the activation of ATM resulted in the subsequent activation of the downstream proteins such as p53, CHK1 and CHK2 but degradation of CDC25A that leads to the cell cycle arrest. For this process, it is corresponded to our results. Normally, during this process, Smad3 will be activated and leads to an increase of CDK inhibitors p15 and p21 along with a decrease CDC25A and leads to the cell cycle arrest.Citation25-27,39 However, the results in our study presented a bit different. The expression of Smad3 did not present any change and the expression of p-Smad3 was also declined after the treatment of radiation. As observed, the expression of Smad7 was obviously increased when the cells were exposed to radiation. According to studies in previous, Smad7 is an inhibitory Smad that interferes with Smads signaling by preventing the phosphorylation of Smads2/3, which normally occurs following binding of TGF-β/activin to their cell surface receptors.Citation40-43 Therefore, Smad7 inhibited the phosphorylation of Smad3 and inhibited the downstream genes activation. In another word, it can predict that the radiation performed its function to induce the cell cycle arrest and cell apoptosis via the activation of ATM pathway and inhibition of the Smad3/7 cascade. On the other hand, the 2 pathways might interact each other during radiation treatment. According to the Smad3 overexpression and Smad7 suppression assays, we can also observe that the expression of p-ATM, p-CHK1, p-CHK2, CDK4 and CDC25A were almost no difference although it is expression changes of Smad3 and Smad7. However, when the cell received radiation treatment, the expression change of the Smad3/7 induced the expression decline of p-ATM and other ATM pathway related genes. The role of the Smad3/7 in TGF-β signaling pathway should be complicated.
Actually, the functions of the TGF-β signaling in radiation is still a matter of debate, with some studies showing that it acts as a radiosensitizer Citation44 while others report it functioning as a radioprotector.Citation45 It was also not previously clear whether TGF-β signaling regulates the DNA damage response network. We originally performed assay using TGF-β1 solution to activate the Smad3/7 cascade in the study. Eventually, we noticed that the TGF-β signaling pathway was inhibited by the increased expression of Smad7 that was out of our expectation. However, the expression of the ATM pathway related genes were increased suggesting that the ATM pathway was activated. On the other hand, if we purposely raise level of Smad3 so that the Smad3 related pathway could be activated, both processes of cell cycle arrest and cell apoptosis were attenuated. Under the conditions, the ATM pathway related genes were decreased in expression. Therefore, the mechanism of the CNE-2 cells in radiation should be complicated. We hypothesized that the radiation induced cell cycle arrest and cell apoptosis was relied on the ATM pathway but inactivation of TGF-β and Smad3/7 signaling pathway might perform functions to stimulate the anti-tumor functions of the activated ATM pathway. Actually, the previous studies also suggested that ATM might be the first main target of TGF-β signaling in response to radiation. The researchers proposed that extracellular TGF-β1 could be a mediator of intracellular responses to DNA damage through modifying ATM cascade that is important in regulating cellular responses to radiation.Citation46-48 For NPC, the activation of TGF-β Smad signaling might act as an inhibitor to the ATM pathway activation. However, the ATM pathway was activated mainly depending on the DNA damage. As a result, when the cells were without any radiation treatment, the Smad3/7 did not bring any interference to the ATM pathway related genes. Thus, it should be a recent unknown system to regulate and balance these 2 pathways.
In conclusion, we herein revealed that the radiotherapy in treating NPC mainly by activating the ATM pathway and inhibiting the Smad3 activation.
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
This work was supported by Funding of Science and Technology Planning Project of Guangdong Province, China (2014A020212345); Funding of Medical Scientific Research Foundation of Guangdong Province, China (C2015038); Guangzhou Key Medical Discipline Construction Project.
References
- Janvilisri T Omics-based identification of biomarkers for nasopharyngeal carcinoma. Disease markers. 2015;2015:762128. doi:10.1155/2015/762128. PMID:25999660
- Wu Z, Weng D, Li G. Quantitative proteome analysis of overexpressed Cripto-1 tumor cell reveals 14-3-3 gamma as a novel biomarker in nasopharyngeal carcinoma. Journal of proteomics. 2013;83:26-36. doi:10.1016/j.jprot.2013.03.001. PMID:23500129
- Arnold M, Wildeman MA, Visser O, Karim-Kos HE, Middeldorp JM, Fles R, Bing Tan I, Coebergh JW. Lower mortality from nasopharyngeal cancer in The Netherlands since 1970 with differential incidence trends in histopathology. Oral oncology. 2013;49:237-43.
- Wei KR, Zheng RS, Zhang SW, Liang ZH, Ou ZX, Chen WQ. Nasopharyngeal carcinoma incidence and mortality in China in 2010. Chinese journal of cancer. 2014;33:381-7. PMID:25096544
- Bray F, Ferlay J, Laversanne M, Brewster DH, Gombe Mbalawa C, Kohler B, Piñeros M, Steliarova-Foucher E, Swaminathan R, Antoni S, et al. Cancer Incidence in Five Continents: Inclusion criteria, highlights from Volume X and the global status of cancer registration. International journal of cancer. 2015;137:2060-71. doi:10.1002/ijc.29670. PMID:26135522
- Razak AR, Siu LL, Liu FF, Ito E, O'Sullivan B, Chan K. Nasopharyngeal carcinoma: the next challenges. European journal of cancer. 2010;46:1967-78. doi:10.1016/j.ejca.2010.04.004. PMID:20451372
- Ben Chaaben A, Busson M, Douik H, Boukouaci W, Mamoghli T, Chaouch L, Harzallah L, Dorra S, Fortier C, Ghanem A, et al. Association of IL-12p40 +1188 A/C polymorphism with nasopharyngeal cancer risk and tumor extension. Tissue antigens. 2011;78:148-51. doi:10.1111/j.1399-0039.2011.01702.x. PMID:21623733
- Li M, Dai W, Zhou H. Cyclin D1 G870A polymorphism and risk of nasopharyngeal carcinoma: a meta-analysis. TheScientificWorldJournal. 2013;2013:689048. doi:10.1155/2013/689048. PMID:24222746
- Hildesheim A, Wang CP. Genetic predisposition factors and nasopharyngeal carcinoma risk: a review of epidemiological association studies, 2000–2011: Rosetta Stone for NPC: genetics, viral infection, and other environmental factors. Seminars in cancer biology. 2012;22:107-16. doi:10.1016/j.semcancer.2012.01.007. PMID:22300735
- Lee AW, Lin JC, Ng WT. Current management of nasopharyngeal cancer. Seminars in radiation oncology. 2012;22:233-44. doi:10.1016/j.semradonc.2012.03.008. PMID:22687948
- Anderson P, Dische S. Local tumor control and the subsequent incidence of distant metastatic disease. International journal of radiation oncology, biology, physics 1981;7:1645-8. doi:10.1016/0360-3016(81)90186-3. PMID:7037703
- Mlecnik B, Bindea G, Kirilovsky A, Angell HK, Obenauf AC, Tosolini M, Church SE, Maby P, Vasaturo A, Angelova M, et al. The tumor microenvironment and Immunoscore are critical determinants of dissemination to distant metastasis. Sci Transl Med. 2016;8:327ra26. doi:10.1126/scitranslmed.aad6352. PMID:26912905
- Lunt SJ, Chaudary N, Hill RP. The tumor microenvironment and metastatic disease. Clinical & Experimental Metastasis. 2009;26:19-34. doi:10.1007/s10585-008-9182-2
- Kim JJ, Tannock IF. Repopulation of cancer cells during therapy: an important cause of treatment failure. Nature Reviews Cancer. 2005;5:516. doi:10.1038/nrc1650. PMID:15965493
- Alajez NM, Shi W, Hui AB, Bruce J, Lenarduzzi M, Ito E, Yue S, O'Sullivan B, Liu FF. Enhancer of Zeste homolog 2 (EZH2) is overexpressed in recurrent nasopharyngeal carcinoma and is regulated by miR-26a, miR-101, and miR-98. Cell death & disease. 2010;1:e85. doi:10.1038/cddis.2010.64
- Lin YH, Chang KP, Lin YS, Chang TS. Evaluation of effect of body mass index and weight loss on survival of patients with nasopharyngeal carcinoma treated with intensity-modulated radiation therapy. Radiation oncology. 2015;10:136. doi:10.1186/s13014-015-0443-3. PMID:26122711
- Lee SW, Chen TJ, Lin LC, Li CF, Chen LT, Hsing CH, Hsu HP, Tsai CJ, Huang HY, Shiue YL. Overexpression of thymidylate synthetase confers an independent prognostic indicator in nasopharyngeal carcinoma. Experimental and molecular pathology. 2013;95:83-90. doi:10.1016/j.yexmp.2013.05.006. PMID:23726796
- Abdulkarim B, Deutsch E. Endothelial-cell apoptosis and tumour response to radiotherapy. Lancet Oncology. 2004;5:9. doi:10.1016/S1470-2045(03)01317-2. PMID:14700602
- Roa W, Zhang X, Guo L, Shaw A, Hu X, Xiong Y, Gulavita S, Patel S, Sun X, Chen J, et al. Gold nanoparticle sensitize radiotherapy of prostate cancer cells by regulation of the cell cycle. Nanotechnology. 2009;20:375101. doi:10.1088/0957-4484/20/37/375101. PMID:19706948
- Shiloh Y. ATM (ataxia telangiectasia mutated): expanding roles in the DNA damage response and cellular homeostasis. Biochemical Society transactions. 2001;29:661-6. doi:10.1042/bst0290661. PMID:11709050
- Sommer SS, Jiang Z, Feng J, Buzin CH, Zheng J, Longmate J, Jung M, Moulds J, Dritschilo A. ATM missense mutations are frequent in patients with breast cancer. Cancer genetics and cytogenetics. 2003;145:115-20. doi:10.1016/S0165-4608(03)00119-5. PMID:12935922
- Abraham RT Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes & development. 2001;15:2177-96. doi:10.1101/gad.914401
- Chen Y, Poon RY. The multiple checkpoint functions of CHK1 and CHK2 in maintenance of genome stability. Frontiers in bioscience: a journal and virtual library. 2008;13:5016-29. PMID:18508566
- Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes & development. 2000;14:1448-59.
- Feng XH, Liang YY, Liang M, Zhai W, Lin X. Direct Interaction of c-Myc with Smad2 and Smad3 to Inhibit TGF-beta-Mediated Induction of the CDK Inhibitor p15(Ink4B). Molecular cell. 2016;63:1089. doi:10.1016/j.molcel.2016.08.027. PMID:27618489
- Pardali K, Kurisaki A, Moren A, ten Dijke P, Kardassis D, Moustakas A. Role of Smad proteins and transcription factor Sp1 in p21(Waf1/Cip1) regulation by transforming growth factor-beta. The Journal of biological chemistry. 2000;275:29244-56. doi:10.1074/jbc.M909467199. PMID:10878024
- Millet C, Zhang YE. Roles of Smad3 in TGF-beta signaling during carcinogenesis. Critical reviews in eukaryotic gene expression. 2007;17:281-93. doi:10.1615/CritRevEukarGeneExpr.v17.i4.30. PMID:17725494
- Zhang S, Ekman M, Thakur N, Bu S, Davoodpour P, Grimsby S, Tagami S, Heldin CH, Landström M. TGFβ1-Induced Activation of ATM and p53 Mediates Apoptosis in a Smad7-Dependent Manner. Cell Cycle. 2006;5:2787-95. doi:10.4161/cc.5.23.3523. PMID:17172861
- Dunne AL, Price ME, Mothersill C, Mckeown SR, Robson T, Hirst DG. Relationship between clonogenic radiosensitivity, radiation-induced apoptosis and DNA damage/repair in human colon cancer cells. British Journal of Cancer. 2003;89:2277. doi:10.1038/sj.bjc.6601427. PMID:14676806
- Xu L, Yang D, Wang S, Tang W, Liu M, Davis M, Chen J, Rae JM, Lawrence T, Lippman ME. (−)-Gossypol enhances response to radiation therapy and results in tumor regression of human prostate cancer. Mol Cancer Ther. 2005;4:197-205. PMID:15713891
- Riballo E, Kuhne M, Rief N, Doherty A, Smith GC, Recio MJ, Reis C, Dahm K, Fricke A, Krempler A, et al. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Molecular cell. 2004;16:715-24. doi:10.1016/j.molcel.2004.10.029. PMID:15574327
- Fernandez-Capetillo O, Celeste A, Nussenzweig A. Focusing on foci: H2AX and the recruitment of DNA-damage response factors. Cell cycle. 2003;2:426-7. doi:10.4161/cc.2.5.509. PMID:12963833
- Anderson JA, Harper JV, Cucinotta FA, O'Neill P. Participation of DNA-PKcs in DSB repair after exposure to high- and low-LET radiation. Radiation research. 2010;174:195-205. doi:10.1667/RR2071.1. PMID:20681786
- Cucinotta FA, Pluth JM, Anderson JA, Harper JV, O'Neill P. Biochemical kinetics model of DSB repair and induction of gamma-H2AX foci by non-homologous end joining. Radiation research. 2008;169:214-22. doi:10.1667/RR1035.1. PMID:18220463
- Kinner A, Wu W, Staudt C, Iliakis G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic acids research. 2008;36:5678-94. doi:10.1093/nar/gkn550. PMID:18772227
- Redon CE, Dickey JS, Bonner WM, Sedelnikova OA. γ-H2AX as a biomarker of DNA damage induced by ionizing radiation in human peripheral blood lymphocytes and artificial skin. Advances in Space Research the Official Journal of the Committee on Space Research. 2009;43:1171-8. doi:10.1016/j.asr.2008.10.011
- Sato K, Nishikino M, Okano Y, Ohshima S, Hasegawa N, Ishino M, et al. gamma-H2AX and phosphorylated ATM focus formation in cancer cells after laser plasma X irradiation. Radiation research. 2010;174:436-45. doi:10.1667/RR2178.1. PMID:20718602
- Ji J, Zhang Y, Redon CE, Reinhold WC, Chen AP, Fogli LK, Holbeck SL, Parchment RE, Hollingshead M, Tomaszewski JE, et al. Phosphorylated fraction of H2AX as a measurement for DNA damage in cancer cells and potential applications of a novel assay. PLoS One. 2017;12:e0171582. doi:10.1371/journal.pone.0171582. PMID:28158293
- Ray D, Terao Y, Nimbalkar D, Chu LH, Donzelli M, Tsutsui T, Zou X, Ghosh AK, Varga J, Draetta GF, et al. Transforming growth factor beta facilitates beta-TrCP-mediated degradation of Cdc25A in a Smad3-dependent manner. Molecular and cellular biology. 2005;25:3338-47. doi:10.1128/MCB.25.8.3338-3347.2005. PMID:15798217
- Moustakas A, Pardali K, Gaal A, Heldin CH. Mechanisms of TGF-beta signaling in regulation of cell growth and differentiation. Immunology letters. 2002;82:85-91. doi:10.1016/S0165-2478(02)00023-8. PMID:12008039
- Ten Dijke P, Goumans MJ, Itoh F, Itoh S. Regulation of cell proliferation by Smad proteins. Journal of cellular physiology. 2002;191:1-16. doi:10.1002/jcp.10066. PMID:11920677
- Massague J, Wotton D. Transcriptional control by the TGF-beta/Smad signaling system. The EMBO journal. 2000;19:1745-54. doi:10.1093/emboj/19.8.1745. PMID:10775259
- Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577-84. doi:10.1038/nature02006. PMID:14534577
- Wiegman EM, Blaese MA, Loeffler H, Coppes RP, Rodemann HP. TGFbeta-1 dependent fast stimulation of ATM and p53 phosphorylation following exposure to ionizing radiation does not involve TGFbeta-receptor I signalling. Radiotherapy and oncology: journal of the European Society for Therapeutic Radiology and Oncology. 2007;83:289-95. doi:10.1016/j.radonc.2007.05.013. PMID:17560675
- Lee J, Kim MR, Kim HJ, An YS, Yi JY. TGF-beta1 accelerates the DNA damage response in epithelial cells via Smad signaling. Biochemical and biophysical research communications. 2016;476:420-5. doi:10.1016/j.bbrc.2016.05.136. PMID:27237972
- Kirshner J, Jobling MF, Pajares MJ, Ravani SA, Glick AB, Lavin MJ, Koslov S, Shiloh Y, Barcellos-Hoff MH. Inhibition of transforming growth factor-beta1 signaling attenuates ataxia telangiectasia mutated activity in response to genotoxic stress. Cancer research. 2006;66:10861-9. doi:10.1158/0008-5472.CAN-06-2565. PMID:17090522
- Lobrich M, Jeggo PA. The two edges of the ATM sword: co-operation between repair and checkpoint functions. Radiotherapy and oncology: journal of the European Society for Therapeutic Radiology and Oncology. 2005;76:112-8. doi:10.1016/j.radonc.2005.06.027. PMID:16026874
- Zgheib O, Huyen Y, DiTullio RA, Jr., Snyder A, Venere M, Stavridi ES, et al. ATM signaling and 53BP1. Radiotherapy and oncology: journal of the European Society for Therapeutic Radiology and Oncology. 2005;76:119-22. doi:10.1016/j.radonc.2005.06.026. PMID:16024119