
ABSTRACT
Colorectal cancer (CRC) is a leading cause of cancer-related deaths in the United States. We analyzed 26 MSI-High and 558 non-MSI-High CRC tumors. BRCA2 mutations were highly enriched (50%) in MSI-High CRC. Immunohistochemistry showed that BRCA2-mutated MSI-High CRC had high c-MET (64%) expression compared with BRCA-WT (17%). We hypothesized a mechanistic link between BRCA2-deficiency and c-MET overexpression and synergistic interaction between drugs that treat BRCA-deficient tumors (mitomycin C (MMC) or PARP inhibitors) and c-MET inhibitors (crizotinib). We tested CRC cell lines for sensitivity to MMC plus crizotinib or other drug combinations including PARP-inhibitors. Combined treatment of tumor cells with crizotinib and MMC led to increased apoptosis as compared with each drug alone. Additionally, combination treatment with increasing concentrations of both drugs demonstrated a synergistic anti-cancer effect (CI = 0.006–0.74). However, we found no evidence for c-MET upregulation upon effective BRCA2 knockdown in tumor cells −/+DNA damage. Although we found no mechanistic link between BRCA2 deficiency and c-MET overexpression, c-MET is frequently overexpressed in CRC and BRCA2 is mutated especially in MSI-H CRC. The combination of crizotinib with MMC appeared synergistic regardless of MSI or BRCA2 status. Using an in-vivo CRC xenograft model we found reduced tumor growth with combined crizotinib and MMC therapy (p = 0.0088). Our preclinical results support clinical testing of the combination of MMC and crizotinib in advanced CRC. Targeting cell survival mediated by c-MET in combination with targeting DNA repair may be a reasonable strategy for therapy development in CRC or other cancers.
Introduction
According to the American Cancer Society, colorectal cancer (CRC) is the third leading cause of cancer related-death in women in the United States and the second leading cause in men.Citation1 Microsatellite instability (MSI) accounts for approximately 15% of sporadic colorectal cancer cases and the majority of Lynch syndrome patients.Citation2 MSI has been correlated with deficiency in the mismatch repair (MMR) genes and therefore these tumors exhibit higher mutation rate than non-MSI tumors.Citation2,3 Only about 4% of advanced CRC tumors harbor mismatch repair defects and MSI. As a result of a better understanding of specific therapy targeted to molecular, genetic and biochemical changes, additional treatment options are becoming available for CRC patients with MSI, which exhibit a signature of high tumor mutation burden.Citation4
Mitomycin C (MMC) is an anti-cancer chemotherapeutic agent, which causes DNA damage by inducing double strand breaks (DSBs) through DNA cross-linking.Citation5 Tumors deficient in genes encoding proteins involved in DNA repair show hypersensitivity to mitomycin C.Citation6 BRCA2 is a crucial DNA repair gene involved in DNA homologous recombination.Citation7-9 Treatment of tumors with MMC induces DNA damage, which is normally repaired with the help of BRCA2. However, deficiency in BRCA2 results in the accumulation of damaged DNA, unable to be repaired and therefore apoptosis is induced within the dividing the tumor cells. Germ-line mutations in the BRCA genes are known to be associated with higher risk of breast and ovarian carcinomas.Citation10,11 There is also evidence for a correlation between mutations in these genes and susceptibility to other tumor types including CRC.Citation12 In addition, like other genes, the BRCA genes, especially BRCA2 that has numerous repetitive sequences, have higher mutation rates in MSI CRC.Citation13 Therefore, CRC patients that exhibit mutations in the BRCA genes might benefit from treatment with MMC.
Alternatively, PARP inhibitors have been shown to sensitize BRCA-deficient breast or ovarian tumor cells to killing by DNA damaging agents such as cisplatin.Citation14-16 Several PARP inhibitors have been approved by the FDA for cancer therapy including olaparib, rucaparib, and niraparib. As PARP inhibitors may have less toxicity versus MMC, we considered them as reasonable therapeutic agents for combination with other drugs for tumors with BRCA mutations or BRCA-deficiency.
Crizotinib is a small molecule inhibitor of several receptor tyrosine kinases (RTKs), targeting c-MET, anaplastic lymphoma kinase (ALK) and ROS1 receptors.Citation17 It was FDA-approved for the treatment of patients with ALK-positive or ROS1-positive locally advanced or metastatic non-small cell lung cancer (NSCLC) in 2013.Citation18 It is also under multiple clinical trials for advanced solid tumors and lymphoma. c-MET is a receptor tyrosine kinase activated by its ligand hepatocyte growth factor (HGF) to initiate RAS/mitogen activated protein kinase (MAPK), and phosphatidylinositol 3-kinase (PI3K)/AKT signaling as well as several other pathways.Citation19 c-MET plays a key role in biologic functions including cell proliferation, cell survival, cell motility, invasion/metastasis and angiogenesis.Citation20,21 It is frequently genetically altered or dysregulated in advanced cancers.Citation22 Overexpression of c-MET and its kinase activity contribute as a leading cause of tumor growth and metastasis in several types of cancers including CRC.Citation23 Consequently, c-MET is a potential candidate for anti-cancer targeted therapy and there is ongoing clinical development of anti-c-MET small molecule inhibitors.Citation24-26
In recent years, a growing number of small molecule inhibitors that specifically target RTKs have been developed.Citation27 These inhibitors are frequently combined with other drugs in multiple clinical trials and approved treatment options for advanced cancers.Citation28 Targeting multiple pathways may offer an optimal therapeutic approach and is becoming a new paradigm for anti-cancer treatment used in a variety of cancers including CRC. In the current study we first assessed the correlation between BRCA mutations and c-MET expression in CRC patients. We found that BRCA2-mutated MSI-H CRCs express higher levels of c-MET than BRCA2 WT CRCs. We tested the sensitivity of CRC cell lines to combination treatment with various drugs including MMC and crizotinib and provided the rationale for this combinatorial treatment of patients with advanced CRC.
Materials and methods
Cell culture and reagents
All cell lines were obtained from American Type Culture Collection (ATCC) and were authenticated. All cell lines were mycoplasma free and were regularly tested. HT-29, HCT-15 and HCT116 were maintained in McCoy's media, while SW480, SW620, DLD-1, RKO and Capan-1 were maintained in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin solution. Cells were cultured under a humidified atmosphere of 5% CO2 at 37c. Crizotinib was purchased from ApexBio (cat. no. A3020) and Medkoo (cat. no. 202222). MMC was purchased from Sigma (cat. no. M0440) and Medkoo (cat. no. 100630). Cisplatin was purchased from Sigma. Olaparib was purchased from Selleckchem (cat no. S1060).
Patient tissue samples and immunohistochemistry
CRC patient samples were identified and analyzed at Caris Life Sciences between 2009 and 2015. Samples were tested for MSI status and were divided into MSI (26) and non-MSI (558) groups. Samples were tested for BRCA mutations. IHC staining for a panel of cancer markers was then performed on the 26 samples that were identified as MSI-H. Correlation of expression of the markers and BRCA status was determined.
Immunoblotting
Cells were washed with PBS and lysed with RIPA buffer (Sigma) supplemented with protease inhibitor (Roche) and phosphatase inhibitor (Roche). The protein concentration of cell lysates was determined using BCA Protein Assay Kit (Thermo Fisher Scientific) and equal amounts of proteins were loaded onto NuPAGE 3–8% Tris-Acetate gel when BRCA2 was monitored or NuPAGE 4–12% Bis-Tris gel (Invitrogen). The separated proteins were transferred to PVDF membrane. After blocking with 5% bovine serum albumin in PBS, the membranes were incubated with primary antibody diluted 1:1000 in PBS/BSA overnight at 4°c. Membranes were then incubated with the appropriate secondary antibodies labeled with horseradish peroxidase followed by chemi-luminescence detection using ECL Reagents (Thermo Fisher Scientific). The primary antibodies used in this study were as follows: antibodies against BRCA2 (cat. no. ab90541), GHR (cat. no. ab134078), ALK (cat. no. ab140534) from abcam; c-MET (cat. no. 4560S), phospho-c-MET (cat. no. 3077S), PARP CLVD (cat. no. 9546), c-Kit (cat. no. 3074S), FGFR2 (cat. no. 11835S), PDGFR-β (cat. no. 3169S), γ-H2AX (cat. no. 2577S), IGF-1R (cat. no. 3027S), VEGFR (cat. no. 9698S) from Cell Signaling Technology. Secondary antibodies were acquired from Jackson Laboratories horseradish-peroxidase conjugated.
BRCA2 gene silencing
For transient knockdown of BRCA2 Cells were plated at 50% to 60% confluence in 6-well plates and incubated for 24 hours before transient transfection with siRNAs mixed with the Lipofectamine 2000 reagent (Invitrogen). The siRNA mix specific for BRCA2 was purchased from Santa Cruz, as well as the corresponding scrambled (control) siRNAs. BRCA2 KD was verified each time by western blot analysis of cell protein lysate using NuPAGE 3–8% Tris-Acetate gel (Invitrogen).
Cell viability assay
Briefly, cells (10,000) were seeded in a 96 well plate 24 hours before exposure to various concentrations of crizotinib, MMC or other drugs for 48 or 72 hours. Post-treatment luminescent-based cell viability was determined using the Cell-Titer Glo (CTG) assay according to the manufacturer's instructions (Promega). Percent of cell viability was determined by normalizing the luminescence signal to untreated control wells. All treatments were done in triplicates and reported as % Viability ± SEM. Dose response curves were generated to calculate the half-maximal growth inhibition concentration (GI50) in GraphPad Prism version 6.07. Combination indices were calculated using Compusyn software (ComboSyn, Inc.).
Xenografted mice
The protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of Fox Chase Cancer Center. Pathogens such as mouse hepatitis and c. Bovis were excluded. 6- to 7-week-old female athymic nu/nu mice were purchased from Taconic (Hudson, NY) and were acclimated for 1 week on arrival to the animal facility before in-vivo study was initiated. A total of 1 × 106 HT-29-Luc cells was suspended in 50 μl of PBS mixed with 50 μl of Matrigel (BD Biosciences) and subcutaneously injected into the rear flank of the mice. When tumor volume reached an average of 150 mm3, mice were randomly assigned to 4 treatment groups: control vehicle, crizotinib (10 mg/kg), MMC (2 mg/kg), or combination of crizotinib and MMC (each group containing 5 mice). Crizotinib was delivered in a solution of 10% DMSO, 20% Kolliphor® EL (Sigma cat. no. C5135) and 70% PBS daily by oral gavage. MMC was delivered in PBS containing less then 0.1% DMSO by intra-peritoneal injection on days 1,4,7,10 and 13. Vehicle consisted of 10% DMSO, 20% Kolliphor® EL (Sigma cat. no. C5135) and 70% PBS. The length (L) and width (W) of the tumor were measured twice a week using a digital caliper, and the volume of the tumor was calculated using the formula: 0.5* L*(W^2). Mice were also weighed twice a week to monitor signs of drug toxicity. For whole body bioluminescence imaging, mice were injected with 100 microliters of D-Luciferin (3.75 μg/μL) (Gold BioTechnology) and were anesthetized with isoflurane attached to a nose-cone mask within Xenogen IVIS 200 imaging system. On day 22, mice were killed and tumors were harvested, pictured and taken for IHC analysis.
Statistics
All statistical analyses were conducted using GraphPad Prism 6. Data are presented as means ± standard error of mean from at least 3 replicates. The Student's 2-tailed T test in GraphPad Prism 6 was used for pairwise analysis. Statistically significant changes are indicated in the figures with p-values.
Results
BRCA2 mutations correlate with high c-MET expression in MSI CRC
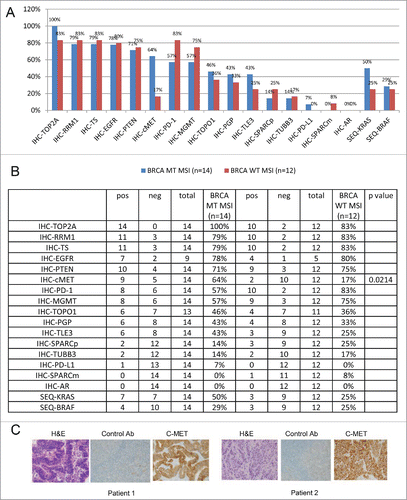
BRCA gene mutations have been previously noted in CRC.Citation29 We investigated a Caris Life Sciences cohort of 26 MSI and 558 non-MSI CRC samples for BRCA1/2 status.Citation30 MSI-H CRC had significantly higher mutations in BRCA1 (19% vs. 5% p = 0.02) and BRCA2 (50% vs. 14% p = 0.0001) as compared with non-MSI CRCs. MSI-H CRC samples were then divided to BRCA mutated and WT groups and the expression of a panel of cancer markers was measured by IHC. The comparison between BRCA mutant MSI-H to BRCA WT MSI-H samples revealed a correlation between mutated BRCA2 and high expression of the proto-oncogene c-MET (). The frequency of c-MET protein expression was significantly higher in BRCA2-mutant MSI-H than BRCA WT MSI-H CRC patient samples (). Other cancer markers such as TOP2A, EGFR and PTEN did not show a significant change in frequency between the 2 groups. IHC staining of c-MET in 2 patients with mutated BRCA2 is presented in to demonstrate the increased expression of c-MET. In addition, mutation analysis of KRAS and BRAF revealed significantly higher KRAS mutation rate in the BRCA mutated MSI-H in comparison to the WT MSI-H CRC patient samples while there was no significant difference in BRAF.
Figure 1. BRCA mutations in MSI-(H)CRCs correlate with high c-MET expression. (A) Immunohistochemistry analysis of a panel of cancer markers in BRCA mutated MSI-H (blue) compared with BRCA wild-type (WT) MSI-H (red) CRC samples graphed as biomarker frequency. The comparison revealed a correlation between mutated BRCA2 and high expression of c-MET. (B) Detailed analysis of the bar graphs presented in (A). (C) Representative IHC images of 2 cases of MSI-H BRCA mutant CRC with high c-MET expression.
CRC cell lines express BRCA2 when mutated in one allele and highly express RTKs including c-MET
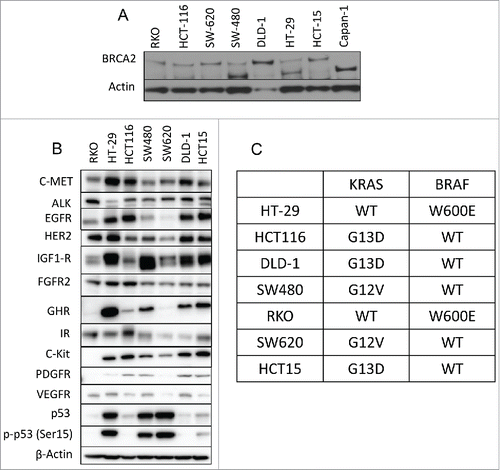
To further analyze the correlation between mutated BRCA2 and c-MET expression, we investigated a panel of 7 CRC cell lines. Supplementary table 1 describes the mutation status of the cell lines. As illustrated in all CRC cell lines including the BRCA2 mutated express BRCA2 in comparison to the control pancreatic cancer cell line Capan-1, that is BRCA2-deficient.Citation31 In addition, most cell lines highly express different receptor tyrosine kinases, among them c-MET is highly expressed in most CRC cell lines regardless of BRCA2 status (). All cell lines are mutated in either KRAS or BRAF genes. 5 out of the cell lines tested are KRAS mutated and 2 are BRAF mutated ().
Figure 2. Expression of BRCA2 and receptor tyrosine kinases in CRC cell lines. (A) Western blot analysis of BRCA2 protein expression levels in a panel of 7 CRC cell lines compared with the Capan1 pancreatic cell line that lacks BRCA2 expression. (B) Western blot analysis of RTKs protein expression levels in CRC cell lines. (C) KRAS and BRAF mutation status of the 7 cell lines used in this study.
BRCA2 knocked down did not correlate with higher expression of c-MET but sensitized CRC cell lines to MMC
We hypothesized there may be a functional link between BRCA2-defiency and c-MET overexpression, possibly mediated by NF-kB and that this may become apparent either in the absence or presence of DNA damage. To assess if the upregulation of c-MET is correlated with the lack of BRCA2 expression, we transiently knocked down (KD) BRCA2 using siRNA in HCT116 and SW480 cell lines. Subsequent western blot analysis of c-MET expression in BRCA2 KD cells showed no change in levels of c-MET protein (). In addition, HCT116 and SW480 CRC cell lines were treated +/− MMC (5 μg/ml) or cisplatin (5 μM) for 24h following BRCA2 siRNA-mediated KD.
Figure 3. BRCA2-knockdown showed no effect on c-MET expression but sensitized CRC cell lines to MMC. (A) Western blot analysis of HCT116 and SW480 CRC cell lines treated +/− MMC (5 μg/ml) or cisplatin (5 μM) for 24 hr following BRCA2 siRNA-mediated knockdown (KD). (B) MMC dose response curves of CRC cell lines measured by CTG cell viability assay. GI50 values (μM) were determined as 50% growth inhibition compared with untreated control.
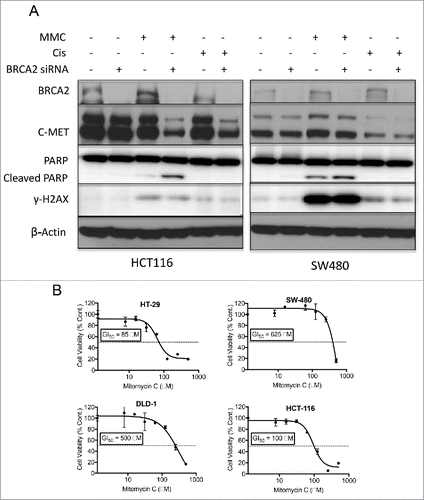
We expected that impaired DNA repair mediated by BRCA2 KD would sensitize CRC cell lines to DNA damaging agents. Activation of the DNA damage response was measured by monitoring the expression of γ-H2AXCitation32 using western blot analysis. Interestingly, MMC activated the DNA damage response more effectively than cisplatin. Upon BRCA2 siRNA-mediated knockdown, cells became more sensitive to MMC than cisplatin and underwent apoptosis as measured by cleaved-PARP. BRCA2 KD cells treated with MMC or cisplatin showed lower expression level of c-MET (). The sensitivity of CRC cell lines to MMC was further analyzed by the CTG cell-viability assay (, Supplementary Fig. 1). CRC cell lines were more sensitive to treatment with MMC as compared with the PARP inhibitor Olaparib. HT-29 (MSS, R273H mutant p53, WT KRAS, V600E mutant BRAF), HCT116 (WT p53, MSI-H), and RKO (MSI-H, WT p53, WT KRAS, V600E mutant BRAF) cell lines exhibited a higher sensitivity to MMC as compared with SW480 (MSS, mutant p53, mutant KRAS, WT BRAF) and DLD-1 (MSS, S246F mutant p53, G13D mutant KRAS, WT BRAF) cell lines. These results suggested MMC as a reasonable candidate to explore for combination therapy for CRC given its activity against CRC cell lines across different genetic subtypes (MSI-H vs MSS, mutant p53, mutant KRAS or BRAF).
Crizotinib inhibits the activation of c-MET in CRC cell line treated with HGF and shows therapeutic potential for CRC cell lines
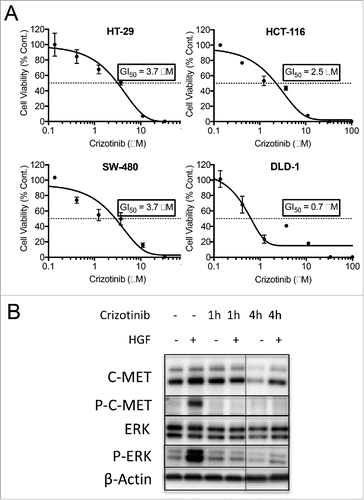
Although c-MET expression did not correlate with BRCA2 levels, we wanted to explore the therapeutic potential of crizotinib in CRC. Especially, since c-MET is highly expressed in CRC cell lines. As demonstrated in , CTG cell viability assay for all 4 CRC cell lines showed response to crizotinib treatment with GI50s being <4 μM. The inhibitory effect of crizotinib on c-MET activity was assessed by co-treatment of CRC cell lines with crizotinib and hepatocyte growth factor (HGF). HGF is the ligand for c-MET receptor, binding of which results in the receptor homo-dimerization, phosphorylation and consequent activation of c-MET receptor. Subsequently, signaling effectors are recruited to start a cascade of downstream signaling which involves the phosphorylation of downstream proteins including ERK.Citation33 HT-29 CRC cells were treated with or without HGF and crizotinib for 1 or 4 hours. Cells were then harvested, and lysates were analyzed for the phosphorylation of c-MET protein and the downstream molecule ERK (). Upon treatment with HGF the levels of phospho-c-MET and phospho-ERK were increased. Addition of crizotinib inhibited the phosphorylation of both proteins and therefore inhibited the activation of c-MET.
Figure 4. Crizotinib inhibits c-MET activation and shows in-vitro efficacy in CRC cell lines. (A) HT-29 CRC cells were treated with hepatocyte growth factor (HGF). Activation of c-MET was then monitored by western blot analysis of phospho-c-MET and phospho-ERK. Treatment of the cells with crizotinib inhibited the phosphorylation of c-MET and ERK. (B) Crizotinib dose response curves of CRC cell lines measured by the CTG cell viability assay. GI50 values (μM) were calculated as 50% growth inhibition compared with untreated control.
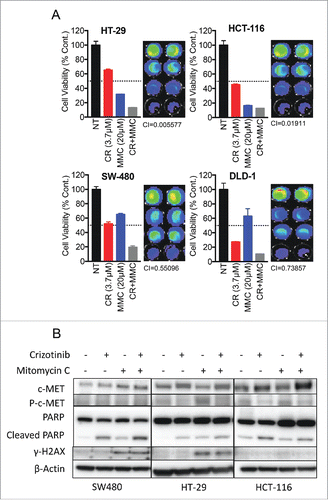
Crizotinib synergizes with MMC in combination treatments of CRC cell lines
Since CRC cell lines were sensitive to both MMC and crizotinib, we wanted to test the combination potential of the 2 drugs. We conducted CTG cell viability assay to first calculate the GI50 of each drug alone (, ). We then used various concentrations of each drug that were under the GI50 in combination and measured viability (). Our data indicate strong synergy between MMC and CR in all 4 CRC cell lines tested. Combination indices (CIs) of < 1 (indicating synergy) were observed at the MMC-crizotinib combination doses tested for each cell line. Furthermore, cells were treated with each drug alone or in combination, lysed and run for western blot analysis of apoptosis as measured by PARP cleavage (). This analysis revealed increased level of apoptosis in the combination treatment compared with each drug alone in SW480 and HT-29 cell lines. HCT-116 BRCA2 mutated cell line showed similar apoptosis when treated with MMC alone or in combination with crizotinib. Samples were also analyzed for the induction of DNA repair response as measured by upregulation of γ –H2AX. In this analysis, γ –H2AX levels were upregulated in all samples treated with MMC. We also tested the combination of the PARP inhibitor olaparib with crizotinib (Supplementary ). However, none of the combinations tested appeared superior to the MMC plus crizotinib combination in the cell lines tested. Thus, our combination therapy experiments showed a promising therapeutic potential for combining crizotinib and MMC for the treatment of CRC, independent of BRCA2 status.
Figure 5. In-vitro combination between crizotinib and MMC in CRC cell lines. (A) Combination of crizotinib and MMC was evaluated by the CTG cell viability assay. Combination index (CI) values were calculated using the program Compusyn. (B) Western blot analysis for apoptosis and DNA damage response of CRC cell lines treated with crizotinib, MMC or combination.
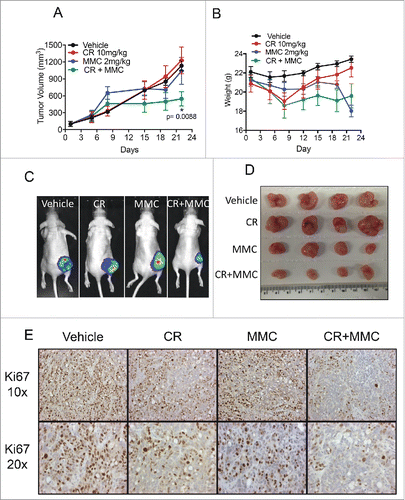
Crizotinib and MMC show synergistic potential in a CRC xenograft model
Combination experiments in CRC cell lines indicated that MMC in combination with crizotinib works independent of BRCA2 status, and that this combination has more efficacy against tumor cells than the PARP inhibitor plus crizotinib combination. We therefore decided to test the MMC plus crizotinib combination in-vivo using a luciferase expressing HT-29 xenograft model for CRC. HT-29 cells showed high sensitivity to both crizotinib and MMC (). In addition, in-vitro crizotinib and MMC combination treatment of HT-29 cells resulted in a synergistic effect and higher levels of apoptosis than either drug alone (). HT-29 is BRCA2 WT (Supplementary table 1) representing CRC regardless of BRCA2 status. Mice were treated with 10 mg/kg crizotinib by oral gavage daily or with 2 mg/kg MMC I.P. on days 1, 4, 7, 10 and 13 or treated with the combination of the 2 drugs. Biweekly measurements of tumor volume were taken with caliper and bio-luminescence imaging of tumors was recorded weekly (). demonstrates a significant reduction in tumor growth in the combination group as compared with the control group (p = 0.0088). Single agent MMC and crizotinib showed no statistically significant differences in the tumor growth compared with control vehicle group. Moreover, significantly lower tumor volumes were seen in the combination treatment group on the last day of the treatment by both bio-luminescence images as well as pictures of extracted tumors (, ). Ki67 immunohistochemical staining showed that combination of the 2 drugs reduced proliferation of HT-29 tumors more efficiently than each drug alone or control vehicle treatment (). The treated mice tolerated therapy as evidenced by lack of significant weight loss during the treatment period (). Our results provide a rationale for the clinical study of crizotinib and MMC combination as a novel therapy for patients with advanced CRC.
Figure 6. Crizotinib and MMC show synergistic efficacy in-vivo using the HT-29 CRC xenograft model. HT-29-luciferase xenografted tumors were established in nude mice. Mice were then treated with 10 mg/kg crizotinib oral gavage daily or with 2 mg/kg MMC I.P. on days 1, 4, 7, 10 and 13 or treated with the combination of the 2 drugs. (A) Relative tumor growth curve shows inhibition of tumor growth in the group of mice that received the drug combination in comparison to each drug alone or vehicle group (p = 0.0088). (B) Weight measurements of mice during the treatment period, show no significant weight loss in the treated groups compared with control vehicle group. (C) Representative bio-luminescence images of one mouse per group on the last day of treatment (day 26). (D) Photographs of extracted tumors on the last day of the experiment. (E) Ki67 immunohistochemistry staining of representative samples from each group.
Discussion
We found that targeting CRC with the combination of crizotnib and MMC has a promising potential for clinical evaluation based on our preclinical results including in-vivo experiments.
Mutations in BRCA1 or BRCA2 genes have been correlated with elevated risk of developing breast or ovarian cancers. However, these mutations have been also associated with increased risk of several other types of cancers including CRC. Preliminary genetic analysis has revealed significantly higher mutations in both BRCA1 and BRCA2 in MSI-H CRC samples as compared with non-MSI-H.Citation30 Consequently, the initial objective of our study was to evaluate the expression of a panel of genes that could be associated in MSI-H CRC clinical samples with or without BRCA mutations. The aim of this analysis was to reveal any correlation between BRCA mutations and expression of biologically-relevant biomarkers. Overexpression of a specific marker might suggest a therapeutic targeting strategy possibly in combination with targeting BRCA mutations. Our analysis suggested a correlation between BRCA mutations and c-MET overexpression (). High expression of c-MET was previously found in BRCA mutated DCIS as well as invasive BRCA-mutated breast cancer.Citation34 High c-MET expression or highly activated c-MET has also been reported in many types of tumors including CRC as c-MET is a proto-oncogene that is crucial for cancer progression.Citation35
To investigate any mechanistic or functional link between BRCA2 mutations and c-MET overexpression we used siRNA knockdown to effectively abrogate BRCA2. We hypothesized that BRCA2 deficiency would lead to increased double-strand DNA breaks in dividing cells or DNA damage exposed cells. Such damaged cells would be expected to trigger an NF-kB response and NF-kB is known to transcriptionally upregulate c-MET. However, to our surprise, c-MET expression levels were not affected by BRCA2-deficiency either in the absence or presence of DNA damage (), and we therefore disproved our hypothesis at least in the experimental model we used.
Although our study found no mechanistic or functional link between BRCA2 deficiency and c-MET overexpression, we nonetheless investigated the efficacy of targeting c-MET and BRCA mutations in CRC cell lines. We initially evaluated multiple single agents or combinations (Supp. Figures 1-2) including MMC, PARP inhibitor olaparib, chemotherapy agents used to treat CRC, and to target c-MET we used the small molecule inhibitor crizotinib. CRC cells lines were highly sensitive to crizotinib in-vitro (), however in-vivo treatment with crizotinib alone was not significantly efficacious (). Similarly, when cells were treated with single drug MMC in-vitro, some cell lines showed sensitivity () but treatment with MMC was not efficacious in our in-vivo xenograft model (). Ultimately, combination treatment with crizotinib and MMC reduced in-vitro cell viability (), induced apoptosis () and showed in-vivo efficacy using HT-29-Luc xenograft model (). The MMC-crizotinib combination was more efficacious against CRC cell lines than other combinations tested. HT-29 is not BRCA mutated but was highly sensitive to MMC and therefore was chosen for in-vivo study to include BRCA WT in our combination study.
Du et al.Citation36 recently showed that c-MET associates with and phosphorylate PARP to increase its enzymatic activity and reduced its binding to PARP inhibitor. Hence, combining c-MET and the PARP inhibitor olaparib, which is FDA approved for the treatment of BRCA mutated ovarian cancer,Citation37 showed efficacy in breast cancer and lung cancer in-vivo models. Since PARP and BRCA proteins are involved in different branches of DNA breaks repairCitation38 the reduced activity of PARP mediated by c-MET inhibitor may also explain the synergy we found with MMC. Additional follow up experiments to our findings may investigate whether reduced activity of PARP as mediated by c-MET inhibition might sensitize CRC cells to MMC. Zimmer and colleagues reported a cross talk between c-MET and the DNA damage response.Citation39-41 They showed that c-MET inhibition reduced the phosphorylation of RAD51 affecting the formation of the RAD51-BRCA2 complex thereby inhibiting double-strand break repair. They also showed that c-MET inhibition reduced survival when combined with ionizing radiation. These observations may also explain our findings showing that inhibition of c-MET sensitized cells to MMC.
In summary, the current study offers a promising combination approach for the treatment of advanced colorectal cancer based on preclinical results including an in-vivo xenograft model. Based on the results presented here we are pursuing a clinical protocol using combination treatment of advanced CRC patients using crizotinib and MMC. Targeting cell survival that may be mediated through c-MET in advanced or metastatic CRC in combination with targeting of DNA repair may be a reasonable strategy for therapy development in CRC or other cancers.
Conflict of interest
J.X. is an employee of Caris Life Sciences. The other authors did not declare any conflict of interest regarding this manuscript. Dr. El-Deiry serves as the Editor-in-Chief of Cancer Biology and Therapy. Manuscripts submitted for peer review by the Editor-in-Chief of Cancer Biology and Therapy are automatically redacted during peer review and not visible to the Editor-in-Chief for details of manuscript handling or decision processing. The Editor-in-Chief plays no role in decisions involving their own submitted manuscripts. For those manuscript submissions, the manuscripts are assigned to a senior editor who oversees the review process and decisions.
CBT1364323-s03.pptx
Download MS Power Point (1 MB)Acknowledgments
This work was presented in part at the 2015 annual ASCO and 2016 annual AACR meetings. W.S.E-D. is an American Cancer Society Research Professor.
References
- Howlader N, Noone AM, Krapcho M, Miller D, Bishop K, Kosary CL, et al. SEER Cancer Statistics Review, 1975-2014. National Cancer Institute.
- Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010;138(6):2073-2087 e3. doi:10.1053/j.gastro.2009.12.064. PMID:20420947
- Vilar E, Gruber SB. Microsatellite instability in colorectal cancer-the stable evidence. Nat Rev Clin Oncol. 2010;7(3):153-62. doi:10.1038/nrclinonc.2009.237. PMID:20142816
- Ciombor KK, Wu C, Goldberg RM. Recent Therapeutic Advances in the Treatment of colorectal cancer. Annual Rev Med. 2015;66(1):83-95. doi:10.1146/annurev-med-051513-102539
- Paz MM, Zhang X, Lu J, Holmgren A. A New Mechanism of Action for the Anticancer Drug Mitomycin C: mechanism-based inhibition of thioredoxin reductase. Chem Res Toxicol. 2012;25(7):1502-1511. doi:10.1021/tx3002065. PMID:22694104
- Wu K, Hinson SR, Ohashi A, Farrugia D, Wendt P, Tavtigian SV, Deffenbaugh A, Goldar D, Couch FJ. Functional evaluation and cancer risk assessment of BRCA2 Unclassified variants. Cancer Res. 2005;65(2):417-426. PMID:15695382
- Powell SN, Kachnic LA. Roles of BRCA1 and BRCA2 in homologous recombination, DNA replication fidelity and the cellular response to ionizing radiation. Oncogene. 2003;22(37):5784-5791. doi:10.1038/sj.onc.1206678. PMID:12947386
- Lee H. Cycling with BRCA2 from DNA repair to mitosis. Exp Cell Res. 2014;329(1):78-84. doi:10.1016/j.yexcr.2014.10.008. PMID:25447315
- Peng G, Lin SY. Exploiting the homologous recombination DNA repair network for targeted cancer therapy. World J Clin Oncol. 2011;2(2):73-9. doi:10.5306/wjco.v2.i2.73. PMID:21603316
- Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, Collins N, Gregory S, Gumbs C, Micklem G. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995;378(6559):789-792. doi:10.1038/378789a0
- Welcsh PL, King M-C. BRCA1 and BRCA2 and the genetics of breast and ovarian cancer. Hum Mol Genetics. 2001;10(7):705-713. doi:10.1093/hmg/10.7.705
- Sopik V, Phelan C, Cybulski C, Narod SA. BRCA1 and BRCA2 mutations and the risk for colorectal cancer. Clin Genetics. 2015;87(5):411-418. doi:10.1111/cge.12497
- Romanowicz-Makowska H, Smolarz B, Langner E, Kozlowska E, Kulig A, Dziki A. Analysis of microsatellite instability and BRCA1 mutations in patients from hereditary nonpolyposis colorectal cancer (HNPCC) family. Pol J Pathol. 2005;56(1):21-6. PMID:15921010
- Dziadkowiec KN, Gasiorowska E, Nowak-Markwitz E, Jankowska A. PARP inhibitors: review of mechanisms of action and BRCA1/2 mutation targeting. Prz Menopauzalny. 2016;15(4):215-219. PMID:28250726
- Lord CJ, Ashworth A. PARP inhibitors: synthetic lethality in the clinic. Science. 2017;355(6330):1152-1158. doi:10.1126/science.aam7344. PMID:28302823
- Seton-Rogers S. Targeted therapies: expanding the use of PARP inhibitors. Nat Rev Cancer. 2017;17(7):397
- Sahu A, Prabhash K, Noronha V, Joshi A, Desai S. Crizotinib: A comprehensive review. South Asian J Cancer. 2013;2(2):91-7. doi:10.4103/2278-330X.110506. PMID:24455567
- Qian H, Gao F, Wang H, Ma F. The efficacy and safety of crizotinib in the treatment of anaplastic lymphoma kinase-positive non-small cell lung cancer: a meta-analysis of clinical trials. BMC Cancer. 2014;14(1):683. doi:10.1186/1471-2407-14-683. PMID:25239305
- Organ SL, Tsao MS. An overview of the c-MET signaling pathway. Ther Adv Med Oncol. 2011;3(1 Suppl):S7-S19. doi:10.1177/1758834011422556. PMID:22128289
- Boccaccio C, Comoglio PM. Invasive growth: a MET-driven genetic programme for cancer and stem cells. Nat Rev Cancer. 2006;6(8):637-645. doi:10.1038/nrc1912. PMID:16862193
- Piguet AC, Medova M, Keogh A, Gluck AA, Aebersold DM, Dufour JF, Zimmer Y. Impact of MET targeting on tumor-associated angiogenesis and growth of MET mutations-driven models of liver cancer. Genes Cancer. 2015;6(7-8):317-27. PMID:26413215
- Sierra JR, Tsao MS. c-MET as a potential therapeutic target and biomarker in cancer. Ther Adv Med Oncol. 2011;3(1 Suppl):S21-35. doi:10.1177/1758834011422557. PMID:22128285
- Gayyed MF, Abd El-Maqsoud NMR, El-Heeny AAE-H, Mohammed MF. c-MET expression in colorectal adenomas and primary carcinomas with its corresponding metastases. J Gastrointest Oncol. 2015;6(6):618-27. PMID:26697193
- Mughal A, Aslam HM, Sheikh A, Khan AM, Saleem S. c-Met inhibitors. Infect Agent Cancer. 2013;8(1):13. doi:10.1186/1750-9378-8-13. PMID:23566349
- Sharma N, Adjei AA. In the clinic: ongoing clinical trials evaluating c-MET-inhibiting drugs. Ther Adv Med Oncol. 2011;3(1 Suppl):S37-50. doi:10.1177/1758834011423403. PMID:22128287
- Peruzzi B, Bottaro DP. Targeting the c-Met signaling pathway in cancer. Clin Cancer Res. 2006;12(12):3657-60. doi:10.1158/1078-0432.CCR-06-0818. PMID:16778093
- Hojjat-Farsangi M. Small-molecule inhibitors of the receptor tyrosine kinases: promising tools for targeted cancer therapies. Int J Mol Sci. 2014;15(8): 13768-801. doi:10.3390/ijms150813768. PMID:25110867
- Faivre S, Djelloul S, Raymond E. New paradigms in anticancer therapy: targeting multiple signaling pathways with kinase inhibitors. Seminars Oncol. 2006;33(4):407-420. doi:10.1053/j.seminoncol.2006.04.005.
- Phelan CM, Iqbal J, Lynch HT, Lubinski J, Gronwald J, Moller P, Ghadirian P, Foulkes WD, Armel S, Eisen A, et al. Incidence of colorectal cancer in BRCA1 and BRCA2 mutation carriers: results from a follow-up study. Br J Cancer. 2014;110(2):530-4. doi:10.1038/bjc.2013.741. PMID:24292448
- Deihimi S, Lev A, Slifker M, Shagisultanova E, Xu Q, Jung K, Vijayvergia N, Ross EA, Xiu J, Swensen J, et al. BRCA2, EGFR, and NTRK mutations in mismatch repair-deficient colorectal cancers with MSH2 or MLH1 mutations. Oncotarget. 2017;8(25):39945-62. PMID:28591715
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917-921. doi:10.1038/nature03445. PMID:15829967
- Scully R, Xie A. Double strand break repair functions of histone H2AX. Mutat Res. 2013;750(1-2):5-14. doi:10.1016/j.mrfmmm.2013.07.007. PMID:23916969
- Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. 2010;11(12):834-848. doi:10.1038/nrm3012. PMID:21102609
- Yang RL, Mick R, Lee K, Graves HL, Nathanson KL, Domchek SM, Kelz RR, Zhang PJ, Czerniecki BJ. DCIS in BRCA1 and BRCA2 mutation carriers: prevalence, phenotype, and expression of oncodrivers C-MET and HER3. J Translational Med. 2015;13:335. doi:10.1186/s12967-015-0698-3
- Zenali M, deKay J, Liu Z, Hamilton S, Zuo Z, Lu X, Bakkar R, Mills G, Broaddus R. Retrospective review of MET gene mutations. Oncoscience. 2015;2(5):533-41. doi:10.18632/oncoscience.161. PMID:26097886
- Du Y, Yamaguchi H, Wei Y, Hsu JL, Wang Hl, Hsu YH, Lin WC, Yu WH, Leonard PG, Lee GR, 4th, et al. Blocking c-Met-mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors. Nat Med. 2016;22(2):194-201. doi:10.1038/nm.4032. PMID:26779812
- Frampton JE. Crizotinib: a review of its use in the treatment of anaplastic lymphoma kinase-positive, advanced non-small cell lung cancer. Drugs. 2013;73(18):2031-51. doi:10.1007/s40265-013-0142-z. PMID:24288180
- Helleday T. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol. 2011;5(4):387-393. doi:10.1016/j.molonc.2011.07.001. PMID:21821475
- Medova M, Aebersold DM, Zimmer Y. MET inhibition in tumor cells by PHA665752 impairs homologous recombination repair of DNA double strand breaks. Int J Cancer. 2012;130(3):728-34. doi:10.1002/ijc.26058. PMID:21400509
- Medova M, Aebersold DM, Zimmer Y. The molecular crosstalk between the MET receptor tyrosine kinase and the DNA Damage Response-Biological and Clinical Aspects. Cancers (Basel). 2013;6(1):1-27. doi:10.3390/cancers6010001. PMID:24378750
- Medova M, Aebersold DM, Blank-Liss W, Streit B, Medo M, Aebi S, Zimmer Y. MET inhibition results in DNA breaks and synergistically sensitizes tumor cells to DNA-Damaging agents potentially by breaching a damage-induced checkpoint arrest. Genes Cancer. 2010;1(10):1053-62. doi:10.1177/1947601910388030. PMID:21779429