
ABSTRACT
The FDA approved irreversible inhibitor of ERBB1/2/4, neratinib, was recently shown to rapidly down-regulate the expression of ERBB1/2/4 as well as the levels of c-MET and mutant K-RAS via autophagic degradation. In the present studies, in a dose-dependent fashion, neratinib reduced the expression levels of mutant K-RAS or of mutant N-RAS, which was augmented in an additive to greater than additive fashion by the HDAC inhibitors sodium valproate and AR42. Neratinib could reduce PDGFRα levels in GBM cells, that was enhanced by sodium valproate. Knock down of Beclin1 or of ATG5 prevented neratinib and neratinib combined with sodium valproate / AR42 from reducing the expression of mutant N-RAS in established PDX and fresh PDX models of ovarian cancer and melanoma, respectively. Neratinib and the drug combinations caused the co-localization of mutant RAS proteins and ERBB2 with Beclin1 and cathepsin B. The drug combination activated the AMP-dependent protein kinase that was causal in enhancing HMG Co A reductase phosphorylation. Collectively, our data reinforce the concept that the irreversible ERBB1/2/4 inhibitor neratinib has the potential for use in the treatment of tumors expressing mutant RAS proteins.
Abbreviations
| ca | = | constitutively active |
| CMV | = | empty vector plasmid or virus |
| dn | = | dominant negative |
| ER | = | endoplasmic reticulum |
| ERK | = | extracellular regulated kinase |
| HDAC | = | histone deacetylase |
| MAPK | = | mitogen activated protein kinase |
| mTOR | = | mammalian target of rapamycin |
| PI3K | = | phosphatidyl inositol 3 kinase |
| PTEN | = | phosphatase and tensin homologue on chromosome ten |
| ROS | = | reactive oxygen species |
| SCR | = | scrambled |
| si | = | small interfering |
| VEH | = | vehicle |
Introduction
Over-expression of the epidermal growth factor receptor (EGFR, HER1, ERBB1) is recognized as a biomarker for tumor cell growth, invasion and resistance to chemotherapy [1, and references therein]. Other members of this receptor family, ERBB2, ERBB3 and ERBB4, have also been linked to the oncogenic drug-resistant phenotype.Citation2–4 The recently FDA approved ERBB1/2/4 inhibitor neratinib was shown by our group to rapidly down-regulate the expression of ERBB1/2/4.Citation5 When we microscopically examined neratinib-treated cells we observed the appearance of large vesicles inside the cell, close to the plasma membrane. Others have made similar observations for neratinib and ERBB2.Citation6 The precise mechanisms by which neratinib causes receptor tyrosine kinase internalization and degradation are presently unknown.
As a negative control study, we also examined the impact of neratinib on receptor tyrosine kinases that are not a target for the drug, the HGF receptor c-MET as well as the kinase inactive ERBB family receptor, ERBB3. To our surprise, c-MET expression was reduced by neratinib. The expression of ERBB3 was also reduced. Whereas the down-regulation of ERBB1 required ubiquitination and autophagic degradation, the reduction in c-MET levels did not require ubiquitin. That an ERBB1/2/4 specific inhibitor reduced the expression of a receptor tyrosine kinase, c-MET, that is not catalytically inhibited by neratinib, and the kinase dead receptor ERBB3 that does not bind ATP, argued that the chemical modification of ERBB1/2/4 must trigger some sort of a seismic event in the plasma membrane where not only are ERBB family receptors internalized but “fellow-traveler” receptor tyrosine kinases in quaternary structures are also routed to lysosomal degradation.
We then reasoned, if neratinib could lower ERBB1/2/3/4 and c-MET expression, maybe the drug could also down-regulate other plasma membrane proteins, specifically the proto-oncogene RAS. There are three major subtypes of RAS that are known to be mutated / constitutively active; K- (Kirsten), H- (Harvey) and N- (Neuroblastoma), with the “K” and “N” types being the most common in human cancer (> 30%). Over 90% of pancreatic cancers express a mutant “K” isoform, and we demonstrated in pancreatic carcinoma cells that neratinib and to a greater extent, [neratinib + valproate], reduced mutant K-RAS levels within 6h by more than 50%. Mutant K-RAS co-localized with phosphorylated ATG13 in autophagosomes and with cathepsin B in autolysosomes. Neratinib also reduced N-RAS levels in a PDX ovarian cancer model.
The present studies were performed to determine whether concentrations of neratinib approximating to its safe plasma C max remained competent to down-regulate K-/N-RAS expression; whether other N-RAS transformed cells exhibited a similar biology to the previously tested mutant N-RAS ovarian cancer isolate, and whether N-RAS down-regulation required autophagy.
Results
Neratinib and the HDAC inhibitor sodium valproate interacted to kill PANC1 pancreatic carcinoma cells in a greater than additive fashion (A). Cell killing was blocked by knock down of Beclin1 or of ATG5. At 60X magnification, neratinib and significantly more so [neratinib + valproate] reduced the levels of K-RAS (B). Of note, vesicles staining for K-RAS could be observed in cells treated with neratinib (white arrows). The established safe C max of neratinib in human plasma is approximately 150 nM. Thus, we performed dose-response analyses with neratinib examining its effect on K-RAS expression. After a 6h incubation, at 100 nM and 200 nM, neratinib significantly reduced the expression of K-RAS, with at 10X magnification a trend for valproate enhancing this effect (C). Treatment of PANC1 cells with neratinib and to a greater extent [neratinib + valproate] reduced the expression of ERBB1 in a Beclin1-dependent manner (Figure S1). Treatment of cells with neratinib caused a disassociation of ERBB1 and K-RAS and in parallel an association of K-RAS with the lysosomal protease cathepsin B (D).
Figure 1. Neratinib and valproate combine to reduce the levels of K-RAS in pancreatic cancer cells. A. PANC1 cells were treated with vehicle control, neratinib (100 nM), sodium valproate (250 μM) or the drugs in combination for 24h. Cell viability was determined by trypan blue exclusion assay (n = 3 +/- SEM). B. PANC1 cells were treated with vehicle control, neratinib (100 nM), sodium valproate (250 μM) or the drugs in combination for 6h. The cells were fixed in place and immunostaining performed to determine the expression and localization of K-RAS at 60X magnification. Arrows indicate K-RAS in vesicular structures (data from 3 separate images +/- SEM) *p < 0.05 less than vehicle control; **p < 0.05 less than value in neratinib treated cells. C. PANC1 cells were treated with vehicle control, neratinib (100 nM – 500 nM), sodium valproate (250 μM) or the drugs in combination for 6h. The cells were fixed in place and immunostaining performed to determine the expression and localization of K-RAS at 60X magnification (data from multiple separate images & treatments +/- SEM) *p < 0.05 less than vehicle control; **p < 0.05 less than value in neratinib treated cells. D. PANC1 cells were treated with vehicle control, neratinib (100 nM), sodium valproate (250 μM) or the drugs in combination for 6h. The cells were fixed in place and immunostaining performed to determine the expression and localization of K-RAS at 60X magnification with either ERBB1 or with cathepsin B. E. PANC1 cells were treated with vehicle control or [neratinib (100 nM) and sodium valproate (250 μM)] in combination for 6h. The cells were fixed in place and immunostaining performed at 10X to determine the expression and localization of K-RAS. Arrows indicate K-RAS in vesicular structures.
![Figure 1. Neratinib and valproate combine to reduce the levels of K-RAS in pancreatic cancer cells. A. PANC1 cells were treated with vehicle control, neratinib (100 nM), sodium valproate (250 μM) or the drugs in combination for 24h. Cell viability was determined by trypan blue exclusion assay (n = 3 +/- SEM). B. PANC1 cells were treated with vehicle control, neratinib (100 nM), sodium valproate (250 μM) or the drugs in combination for 6h. The cells were fixed in place and immunostaining performed to determine the expression and localization of K-RAS at 60X magnification. Arrows indicate K-RAS in vesicular structures (data from 3 separate images +/- SEM) *p < 0.05 less than vehicle control; **p < 0.05 less than value in neratinib treated cells. C. PANC1 cells were treated with vehicle control, neratinib (100 nM – 500 nM), sodium valproate (250 μM) or the drugs in combination for 6h. The cells were fixed in place and immunostaining performed to determine the expression and localization of K-RAS at 60X magnification (data from multiple separate images & treatments +/- SEM) *p < 0.05 less than vehicle control; **p < 0.05 less than value in neratinib treated cells. D. PANC1 cells were treated with vehicle control, neratinib (100 nM), sodium valproate (250 μM) or the drugs in combination for 6h. The cells were fixed in place and immunostaining performed to determine the expression and localization of K-RAS at 60X magnification with either ERBB1 or with cathepsin B. E. PANC1 cells were treated with vehicle control or [neratinib (100 nM) and sodium valproate (250 μM)] in combination for 6h. The cells were fixed in place and immunostaining performed at 10X to determine the expression and localization of K-RAS. Arrows indicate K-RAS in vesicular structures.](/cms/asset/153bce66-c506-426c-a599-7d2e024b9281/kcbt_a_1394556_f0001_oc.gif)
Neratinib exposure down-regulated the expression of ERBB2 in Spiky ovarian and BT474 mammary carcinoma cells in a Beclin1 -dependent fashion (Figure S2, not shown). We have previously demonstrated that neratinib could rapidly reduce c-MET expression in NSCLC cells and in glioblastoma cells expressing PDGFRα, neratinib was also capable of reducing PDGFRα levels (Figure S3). Similar reduced expression findings were observed in GBM cells expressing a full-length mutated active ERBB1. Although exposure to 100 nM neratinib had only reduced K-RAS levels by ∼20% after 6h, the localization of K-RAS into intracellular vesicles was already apparent (E).
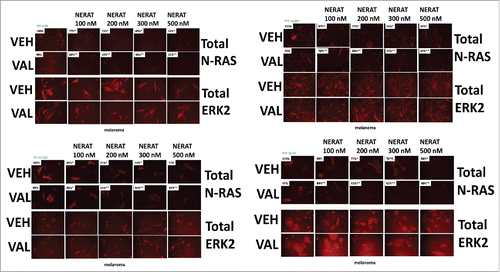
We next performed similar studies in ovarian cancer cells that express a mutant N-RAS; in their patient donor, these cells were completely chemotherapy resistant. Neratinib and the HDAC inhibitor sodium valproate interacted to kill ovarian carcinoma cells in a greater than additive fashion (A). Cell killing was blocked by knock down of Beclin1 or of ATG5. In a dose-dependent fashion neratinib and significantly more so [neratinib + valproate] reduced the levels of N-RAS (B). Similar data were obtained using the novel HDAC inhibitor AR42 (Figure S4). Vesicles staining for N-RAS could be observed in cells treated with neratinib (white arrows) (C). Treatment of cells with neratinib and a greater extent [neratinib + valproate] caused N-RAS expression to decline and for N-RAS to co-localize with the autophagosome protein Beclin1; N-RAS to co-localize with the autolysosome protein cathepsin B; and with increased association of N-RAS with LAMP2 (D). In BT474 mammary and in Spiky ovarian carcinoma cells both neratinib and the HDAC inhibitors sodium valproate and AR42 promoted the co-localization of ERBB2 with cathepsin B (Figures S5 and S6). HDAC inhibitors enhanced cathepsin B expression. Approximately 20% of malignant melanomas express a mutant N-RAS protein and using PDX isolates of melanomas expressing a mutant N-RAS we determined the impact of neratinib and [neratinib + valproate] on N-RAS expression. Within 6h, all melanoma isolates exhibited a dose-dependent reduction in N-RAS expression following neratinib or [neratinib + valproate] exposure (). It was apparent at between 100–200 nM neratinib that valproate sharply shifted the dose response curve of RAS down-regulation to the left.
Figure 2. Neratinib and valproate interact to reduce N-RAS levels in ovarian cancer cells and cause N-RAS to co-localize with autophagy regulatory proteins. A. Spiky ovarian cancer cells were treated with vehicle control, neratinib (100 nM), sodium valproate (250 μM) or the drugs in combination for 24h. Cell viability was determined by trypan blue exclusion assay (n = 3 +/- SEM). B. Spiky ovarian cancer cells were treated with vehicle control, neratinib (100 nM – 500 nM), sodium valproate (250 μM) or the drugs in combination for 6h. The cells were fixed in place and immunostaining performed to determine the expression and localization of N-RAS at 60X magnification (data from multiple separate images & treatments +/- SEM) *p < 0.05 less than vehicle control; **p < 0.05 less than value in neratinib treated cells. C. Spiky ovarian cancer cells were treated with vehicle control or [neratinib (100 nM) and sodium valproate (250 μM)] in combination for 6h. The cells were fixed in place and immunostaining performed at 60X to determine the expression and localization of N-RAS. Arrows indicate N-RAS in vesicular structures. D. Spiky ovarian cancer cells were treated with vehicle control, neratinib (100 nM), sodium valproate (250 μM) or the drugs in combination for 6h. The cells were fixed in place and immunostaining performed to determine the expression and localization of N-RAS at 60X magnification with Beclin1, LAMP2 or with cathepsin B.
![Figure 2. Neratinib and valproate interact to reduce N-RAS levels in ovarian cancer cells and cause N-RAS to co-localize with autophagy regulatory proteins. A. Spiky ovarian cancer cells were treated with vehicle control, neratinib (100 nM), sodium valproate (250 μM) or the drugs in combination for 24h. Cell viability was determined by trypan blue exclusion assay (n = 3 +/- SEM). B. Spiky ovarian cancer cells were treated with vehicle control, neratinib (100 nM – 500 nM), sodium valproate (250 μM) or the drugs in combination for 6h. The cells were fixed in place and immunostaining performed to determine the expression and localization of N-RAS at 60X magnification (data from multiple separate images & treatments +/- SEM) *p < 0.05 less than vehicle control; **p < 0.05 less than value in neratinib treated cells. C. Spiky ovarian cancer cells were treated with vehicle control or [neratinib (100 nM) and sodium valproate (250 μM)] in combination for 6h. The cells were fixed in place and immunostaining performed at 60X to determine the expression and localization of N-RAS. Arrows indicate N-RAS in vesicular structures. D. Spiky ovarian cancer cells were treated with vehicle control, neratinib (100 nM), sodium valproate (250 μM) or the drugs in combination for 6h. The cells were fixed in place and immunostaining performed to determine the expression and localization of N-RAS at 60X magnification with Beclin1, LAMP2 or with cathepsin B.](/cms/asset/9b474f2d-b6ca-4df4-a69f-f1686e77adc2/kcbt_a_1394556_f0002_oc.gif)
Figure 3. Neratinib and valproate down-regulate mutant N-RAS expression in PDX isolates of malignant melanoma. PDX isolates of N-RAS transformed melanoma cells were treated with vehicle control, neratinib (100 nM – 500 nM), sodium valproate (250 μM) or the drugs in combination for 6h. The cells were fixed in place and immunostaining performed to determine the expression and localization of N-RAS at 60X magnification (data from multiple separate images & treatments +/- SEM) *p < 0.05 less than vehicle control; **p < 0.05 less than value in neratinib treated cells.
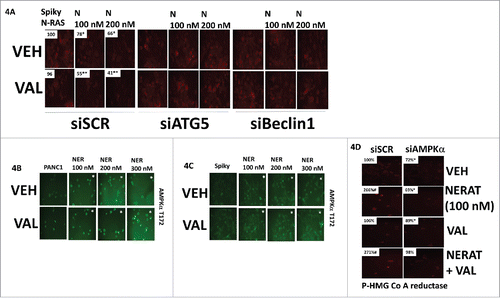
Prior studies had demonstrated that knock down of the autophagy regulatory proteins Beclin1 or ATG5 could prevent neratinib from reducing K-RAS expression. We also knew that neratinib activated the AMP-dependent protein kinase (AMPK).Citation5 Knock down of Beclin1 or ATG5 in ovarian cancer cells prevented the neratinib-induced reduction in N-RAS levels (A). Knock down of Beclin1 or ATG5 also prevented valproate from enhancing the reduction in mutant RAS expression. Neratinib activated the AMPK (B and C). Our prior studies had demonstrated that AMPK activation was essential for [neratinib + valproate] to stimulate toxic autophagosome formation.Citation5 In addition to autophagy, downstream of the AMPK are multiple enzymes whose functions play key roles in regulating cellular metabolism, e.g. HMG Co A reductase and mevalonate synthesis.Citation7 The AMPK phosphorylates and is known to inhibit HMG Co reductase activity; neratinib increased HMG Co A reductase phosphorylation, an effect that was blocked by knock down of AMPKα (D).Citation8
Figure 4. Neratinib and valproate reduce the expression of N-RAS via autophagic degradation. A. Spiky ovarian cancer cells were transfected with a scrambled siRNA control or with siRNA molecules to knock down the expression of Beclin1 or ATG5. Twenty-four h after transfection, cells were treated with vehicle control, neratinib (100 nM), sodium valproate (250 μM) or the drugs in combination for 6h. The cells were fixed in place and immunostaining performed to determine the expression and localization of N-RAS at 60X magnification. (data from multiple separate images & treatments +/- SEM) *p < 0.05 less than vehicle control; **p < 0.05 less than value in neratinib treated cells (for loading controls see Figure S6). B. and C. PANC1 and Spiky cells were treated with vehicle control, neratinib (100 nM), sodium valproate (250 μM) or the drugs in combination for 6h. The cells were fixed in place and immunostaining performed to determine the phosphorylation of AMPKα T172 at 60X magnification. (data from multiple separate images & treatments +/- SEM) *p < 0.05 less than vehicle control; **p < 0.05 less than value in neratinib treated cells (for loading controls see Figure S6). D. Spiky cells were transfected with a scrambled siRNA control or with an siRNA to knock down the expression of AMPKα. Twenty-four hours after transfection cells were treated with vehicle control, neratinib (100 nM), sodium valproate (250 μM) or the drugs in combination for 6h. The cells were fixed in place and immunostaining performed to determine the phosphorylation of HMG CoA reductase (data from multiple separate images & treatments +/- SEM) *p < 0.05 less than vehicle control; **p < 0.05 less than value in neratinib treated cells (for loading controls see Figure S6).
Discussion
Our group has shown that the levels of mutant K-RAS could be rapidly down-regulated by neratinib in pancreatic, colorectal and NSCLC cells.Citation5 The present studies were designed to further investigate the impact of the irreversible ERBB1/2/4 inhibitor neratinib on the expression levels of mutant RAS proteins in pancreatic, ovarian and melanoma cells. We determined that in all three tumor types neratinib reduced the expression of mutant K-/N-RAS proteins, an effect that was enhanced by the pan-HDAC inhibitors sodium valproate or AR42. Knock down of Beclin1 or of ATG5 prevented neratinib from reducing mutant N-RAS expression in ovarian cancer cells, and from killing the cells.
Neratinib was developed as an irreversible inhibitor of ERBB1/2/4, and was recently approved by the FDA for the treatment of breast cancer. Based on its development, the “on-target” biology of neratinib could simply be assumed to only target the ERBB1/2/4 growth factor receptors. However, we also observed that in addition to irreversibly inhibiting the catalytic site of the ERBB family receptors, the drug also caused the cell to rapidly degrade the receptors through a process requiring Beclin1 and ATG5.Citation5 This represents a novel unforeseen on-target biology for the drug. As negative controls, we also tested ERBB3, that lacks an active catalytic site, and c-MET, which is not part of the ERBB receptor family. To our surprise neratinib could reduce both ERBB3 and c-MET expression, and as noted in the present manuscript also the levels of PDGFRα. The logical progression from these discoveries was to examine the impact of neratinib on the levels of plasma membrane associated RAS proteins. Thus, our data argue that the degradation of other receptors and RAS proteins by neratinib is a secondary on-target effect of the drug and occurs because of the primary inhibition, internalization and degradation of ERBB family receptors.
Mutated RAS proteins have been considered for several decades to be one of the most important cancer-driving proteins to therapeutically target.Citation9 RAS proteins, to be active and plasma membrane localized, are prenylated, and this essential modification can be targeted using specific farnesyl-transferase inhibitors or inhibitors of HMG CoA reductase (Statins).Citation10 However, K-RAS and N-RAS proteins, in addition to farnesylation, can also be alternatively geranyl geranylated. As a result, farnesyl transferase inhibitors have largely been unsuccessful in the clinic at controlling RAS-dependent tumors. In our present studies, we demonstrated that neratinib and [neratinib + valproate] activate the AMPK which in turn phosphorylates and inactivates HMG Co A reductase. Reduced HMG Co A reductase activity will lower the levels of mevalonate that ultimately results in reduced levels of both the farnesyl and geranyl substrates. Thus, our use of [neratinib + valproate] attacks the activity of mutant RAS proteins by direct and indirect mechanisms. The drug combination directly reduces RAS protein levels through a process requiring autophagy and lysosomal degradation, and the combination indirectly reduces RAS function by impeding the ability of the cell to prenylate and facilitate plasma membrane localization of RAS proteins. Studies beyond the scope of the present manuscript will be required to determine whether the activity of [neratinib + valproate/AR42] can be further enhanced by FDA approved generic HMG Co A reductase inhibitors, e.g. lovastatin.
Materials and Methods
Materials. Sodium valproate was from Sigma (St. Louis, MO). Neratinib was supplied by Puma Biotechnology Inc. (Los Angeles, CA). AR42 was supplied by Selleckchem (Houston, TX). Trypsin-EDTA, DMEM, RPMI, penicillin-streptomycin were purchased from GIBCOBRL (GIBCOBRL Life Technologies, Grand Island, NY). PANC1 cells were purchased from the ATCC and were not further validated beyond that claimed by ATCC. Cells were re-purchased every ∼6 months. An established PDX model, Spiky ovarian cancer cells, were kindly provided by Dr. Karen Paz (Champions Oncology, NJ). Freshly established PDX melanoma isolates expressing a mutant N-RAS were provided by Dr. Kirkwood (University of Pittsburgh). Established PDX models of GBM were kindly provided by Dr. C.D. James (Northwestern University, Chicago, IL). Commercially available validated short hairpin RNA molecules to knock down RNA / protein levels were from Qiagen (Valencia, CA) (Figure S7). Reagents and performance of experimental procedures were described in refs: 1 and 5.
Methods
Culture and in vitro exposure of cells to drugs. All cell lines were cultured at 37 oC (5% (v/v CO2) in vitro using RPMI supplemented with dialyzed 5% (v/v) fetal calf serum and 10% (v/v) Non-essential amino acids. For short term cell killing assays, immune-staining studies, cells were plated at a density of 3 × 103 per cm2 and 24h after plating treated with various drugs, as indicated. In vitro drug treatments were generally from a 100 mM stock solution of each drug and the maximal concentration of Vehicle carrier (VEH; DMSO) in media was 0.02% (v/v). Cells were not cultured in reduced serum media during any study in this manuscript.
Transfection of cells with siRNA
Transfection for siRNA: Cells from a fresh culture growing in log phase as described above, and 24h after plating transfected. Prior to transfection, the medium was aspirated and serum-free medium was added to each plate. For transfection, 10 nM of the annealed siRNA, the positive sense control doubled stranded siRNA targeting GAPDH or the negative control (a “scrambled” sequence with no significant homology to any known gene sequences from mouse, rat or human cell lines) were used. Ten nM siRNA (scrambled or experimental) was diluted in serum-free media. Four μl Hiperfect (Qiagen) was added to this mixture and the solution was mixed by pipetting up and down several times. This solution was incubated at room temp for 10 min, then added drop-wise to each dish. The medium in each dish was swirled gently to mix, then incubated at 37 oC for 2h. Serum-containing medium was added to each plate, and cells were incubated at 37 oC for 24h before then treated with drugs (0-24h). Additional immuno-fluorescence / live-dead analyses were performed at the indicated time points.
Detection of cell viability, protein expression and protein phosphorylation by immuno-fluorescence using a Hermes WiScan machine. http://www.idea-bio.com/, Cells (4 × 103) are plated into each well of a 96 well plate, and cells permitted to attach and grow for the next 18h. Based on the experiment, after 18h, cells are then either genetically manipulated, or are treated with drugs. For genetic manipulation, cells are transfected with plasmids or siRNA molecules and incubated for an additional 24h. Cells are treated with vehicle control or with drugs at the indicated final concentrations, alone or in combination. Cells are then isolated for processing at various times following drug exposure. The 96 well plate is centrifuged / cyto-spun to associate dead cells (for live-dead assays) with the base of each well. For live dead assays, after centrifugation, the media is removed and cells treated with live-dead reagent (Thermo Fisher Scientific, Waltham MA) and after 10 min this is removed and the cells in each well are visualized in the Hermes instrument at 10X magnification. Green cells = viable; yellow/red cells = dying/dead. The numbers of viable and dead cells were counted manually from three images taken from each well combined with data from another two wells of separately treated cells (i.e. the data is the mean cell dead from 9 data points from three separate exposures). For immuno-fluorescence studies, after centrifugation, the media is removed and cells are fixed in place and permeabilized using ice cold PBS containing 0.4% paraformaldehyde and 0.5% Triton X-100. After 30 min the cells are washed three times with ice cold PBS and cells are pre-blocked with rat serum for 3h. Cells are then incubated with a primary antibody to detect the expression / phosphorylation of a protein (usually at 1:100 dilution from a commercial vendor) overnight at 37oC. Cells are washed three times with PBS followed by application of the secondary antibody containing an associated fluorescent red or green chemical tag. After 3h of incubation the antibody is removed and the cells washed again. The cells are visualized at either 10X or 60X in the Hermes machine for imaging assessments. All immunofluorescent images for each individual protein / phospho-protein are taken using the identical machine settings so that the levels of signal in each image can be directly compared to the level of signal in the cells treated with drugs. Similarly, for presentation, the enhancement of image brightness/contrast using PhotoShop CS6 is simultaneously performed for each individual set of protein/phospho-protein to permit direct comparison of the image intensity between treatments. All immunofluorescent images were initially visualized at 75 dpi using an Odyssey infrared imager (Li-Cor, Lincoln, NE), then processed at 9999 dpi using Adobe Photoshop CS6. For presentation, immunoblots were digitally assessed using the provided Odyssey imager software. Images have their color removed and labeled figures generated in Microsoft PowerPoint.
Data analysis. Comparison of the effects of various treatments (performed in triplicate three times) was using one-way analysis of variance and a two tailed Student's t-test. Statistical examination of in vivo animal survival data utilized both a two tailed Student's t-test and log rank statistical analyses between the different treatment groups. Differences with a p-value of < 0.05 were considered statistically significant. Experiments shown are the means of multiple individual points from multiple experiments (± SEM).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
supp_mat_1394556_KCBT.pdf
Download PDF (804.4 KB)Acknowledgments
Support for the present study was funded from philanthropic funding from Massey Cancer Center, the Universal Inc. Chair in Signal Transduction Research and PHS R01-CA192613. Thanks to Dr. H.F. Young and the Betts family fund for support in the purchase of the Hermes Wiscan instrument. The authors have no conflicts of interest to report.
References
- Booth L, Roberts JL, Tavallai M, Webb T, Leon D, Chen J, McGuire WP, Poklepovic A, Dent P. The afatinib resistance of in vivo generated H1975 lung cancer cell clones is mediated by SRC/ERBB3/c-KIT/c-MET compensatory survival signaling. Oncotarget. 2016;7:19620–30.
- Henson E, Chen Y, Gibson S. EGFR Family Members' Regulation of Autophagy Is at a Crossroads of Cell Survival and Death in Cancer. Cancers (Basel). 2017;9(4). pii: E27.
- Appert-Collin A, Hubert P, Crémel G, Bennasroune A. Role of ErbB Receptors in Cancer Cell Migration and Invasion. Front Pharmacol. 2015;6:283
- Arteaga CL, Engelman JA. ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell. 2014;25:282–303
- Booth L, Roberts JL, Poklepovic A, Avogadri-Connors F, Cutler RE, Lalani AS, Dent P. HDAC inhibitors enhance neratinib activity and when combined enhance the actions of an anti-PD-1 immunomodulatory antibody in vivo. Oncotarget. 2017; 8:90262-90277.
- Zhang Y, Zhang J, Liu C, Du S, Feng L, Luan X, Zhang Y, Shi Y, Wang T, Wu Y, et al. Neratinib induces ErbB2 ubiquitylation and endocytic degradation via HSP90 dissociation in breast cancer cells. Cancer Lett. 2016;382:176–85.
- Zhang X, Song Y, Feng M, Zhou X, Lu Y, Gao L, Yu C, Jiang X, Zhao J. Thyroid-stimulating hormone decreases HMG-CoA reductase phosphorylation via AMP-activatedprotein kinase in the liver. J Lipid Res. 2015;56:963–71.
- Soto-Acosta R, Bautista-Carbajal P, Cervantes-Salazar M, Angel-Ambrocio AH, Del Angel RM. DENV up-regulates the HMG-CoA reductase activity through the impairment of AMPK phosphorylation: A potential antiviral target. PLoS Pathog. 2017;13:e1006257. doi:10.1371/journal.ppat.1006257. eCollection 2017 Apr.
- McCormick, F. KRAS as a Therapeutic Target. Clin Cancer Res. 2015;21:1797–801.
- Hamed H, Mitchell C, Park MA, Hanna D, Martin AP, Harrison B, Hawkins W, Curiel DA, Fisher PB, Grant S, et al. Human chorionic gonadotropin (hCG) interacts with lovastatin and ionizing radiation to modulate prostate cancer cell viability in vivo. Cancer Biol Ther. 2008;7:587–93.