
ABSTRACT
The non-receptor cytoplasmic tyrosine kinase, Focal Adhesion Kinase (FAK) is known to play a key role in a variety of normal and cancer cellular functions such as survival, proliferation, migration and invasion. It is highly active and overexpressed in various cancers including Pancreatic Ductal Adenocarcinoma (PDAC) and Malignant Pleural Mesothelioma (MPM). Here, initially, we demonstrate that FAK is overexpressed in both PDAC and MPM cell lines. Then we analyze effects of two small molecule inhibitors PF-573228, and PF-431396, which are dual specificity inhibitors of FAK and proline rich tyrosine kinase 2 (PYK2), as well as VS-6063, another small molecule inhibitor that specifically inhibits FAK but not PYK2 for cell growth, motility and invasion of PDAC and MPM cell lines. Treatment with PF-573228, PF-431396 and VS-6063 cells resulted in a dose-dependent inhibition of growth and anchorage-independent colony formation in both cancer cell lines. Furthermore, these compounds suppressed the phosphorylation of FAK at its active site, Y397, and functionally induced significant apoptosis and cell cycle arrest in both cell lines. Using the ECIS (Electric cell-substrate impedance sensing) system, we found that treatment of both PF compounds suppressed adherence and migration of PDAC cells on fibronectin. Interestingly, 3D-tumor organoids derived from autochthonous KC (Kras;PdxCre) mice treated with PF-573228 revealed a significant decrease in tumor organoid size and increase in organoid cell death. Taken together, our results show that FAK is an important target for mesothelioma and pancreatic cancer therapy that merit further translational studies.
Introduction
Pancreatic Ductal Adenocarcinoma (PDAC) is the fourth leading cause of cancer related deathsCitation1 with excessive local invasion and systemic dissemination seen in 75% of patients.Citation2 Currently, there are no reliable screening tests and the treatment options are limited to surgery.Citation3 Despite the recent strides made in imaging and surgical techniques and other therapeutic approaches, the 5-year survival rate is a dismal 6–8%.Citation4,Citation5 The most likely reason underlying the poor prognosis of pancreatic cancer can be attributed to late diagnosis compounded by the highly aggressive nature. Treatments such as chemotherapy and radiation have no significant effect on survival and the patients invariably develop resistance. Redundant survival pathways in the tumor and stromal cells are likely to contribute to the observed drug resistance. In pancreatic cancer, tumor cell migration and invasion are seen in early stages; in addition, there is an intense desmoplastic fibrotic response, which is a hallmark of PDAC.Citation6 Diabetes, chronic inflammation (pancreatitis), the consumption of alcohol, and cigarette smoking are some of the associated risk factors for PDAC development. Indeed, smoking increases the incidence of PDAC by three- fold.Citation7 Furthermore, viral infections such as hepatitis B or C can also significantly enhance the incidence of PDAC.Citation8 Previously it has been shown that PDAC is a very complex genetic disease and could be due to successive accumulation of mutations in the KRAS, CDKN2A, TP53 and SMAD4 genes.Citation9 Among these, KRAS somatic mutations are observed in 90% of PDAC cases.Citation10
Malignant Pleural Mesothelioma (MPM) is mostly associated with asbestos exposure and the onset of MPM is linked to genetic predisposition, prior exposure to Simian Virus 40 (SV40) and radiotherapy. MPMs can be pleural (80%) or peritoneal (20%) in origin and very rarely, are localized to pericardium. The three main histological subtypes are epithelioid (60%), sarcomatoid (20%) and biphasic (20%). Frequently, tumors of mixed histology are also found. Due to the relatively long latency period (30-40 years), diagnosis of MPM is rather delayed thus contributing to the short median survival time of less than 12 months.Citation11 The recommended treatment is a combination of cisplatin and an anti-folate analog and the overall outcome remains poor. Due to the very low survival rates in both pancreatic and mesothelioma cancer patients, there is a pressing need for reliable prognostic markers and efficacious therapeutics. Toward this end, here, we have investigated intracellular focal adhesion kinase (FAK) as a potential therapeutic target for both PDAC and MPM.
FAK is a non-receptor tyrosine kinase localized to focal adhesions. It serves as a conduit to signals from extracellular matrix/integrin engagement. Several receptors including integrins, growth factor receptors, G protein coupled-receptors and cytokine receptors activate FAK, which then binds to and activates several downstream signaling molecules such as Src, p130 cas, Grb2, PI3K and paxillin. FAK plays a significant role in cell survival, proliferation, motility, migration and invasion.Citation12 Src-mediated phosphorylation of tyrosine-397 (Y397) in FAK results in its activation.Citation13,Citation14 FAK is essential for normal development and mice lacking FAK die in utero. FAK expression is relatively low in adult tissues; however, its activity is up regulated during wound healing and transformation process.Citation15-Citation17 FAK−/− embryonic fibroblasts revealed profound defects in migration, a harbinger of metastasis.Citation18 FAK is over-expressed in various tumor types including colon, breast, prostate, thyroid, neuroblastoma, ovarian, cervical, brain, head and neck, liver, esophageal, pancreatic and acute myeloid leukemia; high expression of FAK invariably bodes poor prognosis.Citation19,Citation20
In the present study, we demonstrate that FAK is overexpressed in both PDAC and MPM cell lines and describe the effects of the three inhibitors of FAK on tumor growth using in vitro and ex vivo models of MPM and PDAC. PF-573228 (Pfizer, New York City) is a highly specific, ATP competitor that binds with the kinase domain of FAK. Treatment with PF-573228 blocks FAK phosphorylation on Tyr397 as well as the phosphorylation of its downstream target, paxillin.Citation21 PF-431396 is an inhibitor of FAK and the proline-rich tyrosine kinase 2 (PYK2).Citation22 PYK2 is a cytoplasmic, non-receptor tyrosine kinase that was shown to be a negative regulator of osteogenesis and a viable drug target for developing osteoporosis therapies. Finally, the third small molecule inhibitor we used is Defactinib (VS-6063) which is a selective, orally active, competitive ATP inhibitor of FAK.Citation23
Materials and methods
Antibodies
Cleaved PARP (#5625), FAK (#130009), p-FAK (Y397) (#3283), and Cyclin D1 (#2922) were from Cell Signaling (Danvers, MA, USA). β-actin antibody (A2228) and fibronectin (AB1954) were from Sigma (St. Louis, MO, USA).
Cell lines
Mesothelioma cell lines (H2596, H513, H2461, H2052, H2452, H28, H2373) and one benign transformed mesothelial control cell line Met-5A and also Pancreatic cancer cell lines (PANC-1, COLO-357, CD18, AsPC-1, MiaPaca 2, and Capan 1) were obtained from American Type Culture Collection (ATCC) (Manassas, VA, USA). They were grown according to the recommended guidelines and were tested negative for mycoplasma contamination. While Met-5A cells were grown in M199 medium as per manufacturer's instructions, all other cells were cultured in RPMI 1640 medium (Gibco/BRL) supplemented with 10% (v/v) fetal bovine serum (FBS), L-glutamine and 1% penicillin-streptomycin at 37°C with 5% CO2.
Small molecule inhibitors and other reagents
Recombinant human HGF was purchased from R&D Systems (Minneapolis, MN, USA). PF-573228, PF-431396, and VS-6063 were purchased from Selleck Chemicals (Houston, TX, USA) and the stock solutions were prepared in DMSO and stored at -20°C.
Immunoblotting
Cells were treated with the indicated concentrations of inhibitors for the given time. Whole cell lysates were prepared using RIPA lysis buffer and proteins were detected by immunobloting as previously described.Citation24
Viability assays
Exponentially growing cells were plated in a 96 well flat bottom plates, left overnight and then treated with the inhibitors at indicated concentrations for 72 h. Cell viability was measured using Alamar Blue method as described previously.Citation24 Each experiment was repeated at least three times. IC50 values were generated for all the cell lines using GraphPad Prism software (GraphPad Software, Inc. La Jolla CA).
Cell Cycle analysis
The cells were treated with the inhibitors for 48 h at the indicated concentrations, and cell cycle analysis was done using propidium iodide as previously described.Citation24 Samples were analyzed by LSRII flow cytometer (BD Bioscience San Jose, CA, USA). The percentage of cells in different phases of the cell cycle was calculated using FlowJo 9.3.0 software (Tree Star Inc., Ashland, OR, USA).
Apoptosis assay
The cells were treated with the inhibitors at indicated concentrations for 48 h and apoptosis was evaluated using the Annexin V apoptosis kit from BD Biosciences as per the manufacture's protocol as previously described.Citation24
Anchorage-independent growth
The assay was carried out as previously described.Citation25 Briefly in a 24-well culture plate (Ibidi, Madison, Wisconsin, USA), a base layer of agar was applied followed by a top agar layer containing 2.5 × 103 cells per well. Cells were cultured for 4 to 5 weeks at 37°C in a humidified atmosphere containing 5% CO2. Viable colonies were photographed with a Zeiss Axiovert 200M with a Hammatsu Orca ER camera and counted using Image J software and custom written macros.
Electrical cell-based impedance sensing as a measure of cell adhesion
The resistance of cells in culture was measured using electrical cell-substratum impedance sensing (ECIS), as described previously.Citation26 Cells (7.5 × 104) were plated on the chambers of 96W1E+ well plate of a single-electrode ECIS arrays (Applied Biophysics, Troy, NY; www.biophysics.com) that were pre-coated with fibronectin. The data collection began half an hour after cell inoculation to the wells. Cell attachment, adhesion and proliferation were measured for 24–30 h. The cell resistance was measured at 40 kHz.
Organoid development from KrasG12D; Pdx-1 Cre- KC mouse tumor
We have recently developed tumor organoids from KrasG12D; Pdx-1 Cre- KC mouse tumor. In brief, tumor organoids were established after tumor resection, mechanical and enzymatic digestion of pancreatic tumor from 50th week of KC autochthonous mouse model with 0.012% (w/v) collagenase XI (Sigma, St Louis, MO) and 0.012% (w/v) dispase (GIBCO, Waltham, MA) in DMEM media containing 1% FBS (GIBCO) and were embedded in (GFR) Matrigel (BD Biosciences, San Jose, CA) and seeded in 48 well plate as described previously.Citation27 This plate was incubated at 37ᵒC for 15 minutes to allow the matrigel to set following which organoid media (described below) was added. Organoids we maintained in organoid media (AdDMEM/F12 medium supplemented with 1X HEPES [Invitrogen], 1X penicillin/streptomycin [Invitrogen], 1X Glutamax [Invitrogen], 1X B27 [Invitrogen], 1 mM N-acetyl-L-cysteine [Sigma], 1 μg/ml recombinant RSPO1[, 0.1μg/ml Noggin recombinant protein [Peprotech], 50 ng/ml epidermal growth factor [EGF, Peprotech], 10nM Gastrin [Sigma], 10 mM Nicotinamide [Sigma], and 0.5 μM A83-01 [Tocris]). Based on in vitro data we have selected 5 μM and 10 μM concentrations of PF-573228. Tumor organoids were treated once every 48 h, and images were taken at 20X on Day 0, 2 and 4. We have followed 15 tumoroids in each cohort and measured the size of each tumoroids using EVOS FL Auto Cell Imaging System and analyzed significant variation in the tumoroids area upon treatment.
Results
Expression of FAK at the protein and mRNA level in MPM and PDAC cell lines
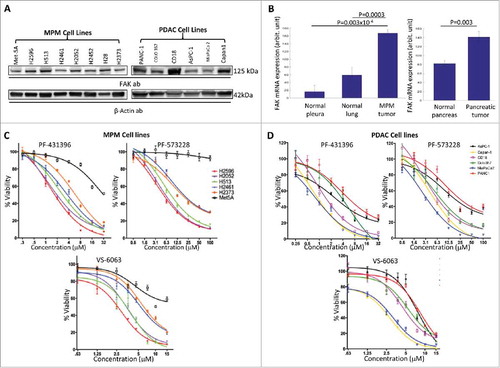
First, we assessed the relative expression of FAK protein in a panel of MPM cell lines listed in the Material & Methods as well as in the control cell line Met-5A, which are non-tumorigenic. In addition, we also determined FAK protein expression in a panel of PDAC cell lines also listed in the Materials & Methods. As shown in , FAK expression at protein level in both MPM and PDAC cell lines is relatively high compared with Met-5A control mesothelial cells. β-actin served as the loading control. To determine expression at the mRNA level, we mined gene expression microarray data from the National Center for Biotechnology Information Gene express Omnibus (GEO) (www.ncbi.nlm.nih.gov/geoprofiles) for FAK from mesothelioma (Reporter: GPL96, 207821_s_at (ID_REF), GDS1220, 5747 (Gene ID), NM_005607) and pancreatic cancer samples (Reporter: GPL570, 1559529_at (ID_REF), GDS4102, 5747 (Gene ID), BC043202) that were obtained in patients who underwent surgery and analyzed them. As shown in , consistent with the protein expression data, FAK mRNA was significantly upregulated in both MPM and PDAC compared to the respective adjacent normal tissue samples. The number of samples in each cohort is indicated in the figure legend.
Figure 1. Expression of FAK and effect of FAK inhibitors on the growth of MPM and PDAC cell lines. (A) Panel of MPM and PDAC cell lines analyzed for FAK using immunoblot analysis and β-Actin was used as loading control. (B) Gene expression microarray data from the National Center for Biotechnology Information Gene express Omnibus (GEO) (www.ncbi.nlm.nih.gov/geoprofiles) for FAK from mesothelioma (Reporter: GPL96, 207821_s_at (ID_REF), GDS1220, 5747 (Gene ID), NM_005607) and pancreatic cancer samples (Reporter: GPL570, 1559529_at (ID_REF), GDS4102, 5747 (Gene ID), BC043202). Number of samples for normal pleura, n = 5, normal lung, n = 4, and for malignant pleural mesothelioma, n = 42 (left). For pancreatic cancer, the number of samples for normal pancreas, n = 16, and for pancreatic cancer n = 36. (C) Viability curves of MPM cell lines and (D) PDAC cell lines after treatment with the FAK inhibitors PF-431396, PF-573228 and VS-6063 for 72 h and cell viability was then assessed using Almar blue assay. The results were normalized as a percentage of untreated controls and analyzed using graphpad Prism software (version 7.02) to calculate the EC50 value for each drug. All experiments were repeated at least twice and each concentration was assayed in triplicate for each experiment.
FAK inhibitors significantly suppress MPM and PDAC cell proliferation
Cell viability was determined in selected MPM cell lines and in the control Met-5A cells following treatment with increasing concentrations of PF-431396, PF-573228 and VS-6063 for 72 h. As shown in , cell viability was significantly decreased in all the MPM cell lines tested in response to all three inhibitors, but not in Met-5A. Of these, the H2596, H2052 and H513 cell lines were the most sensitive. In parallel, we also determined the effect of the inhibitors on PDAC cell lines and observed that most of the PDAC cell lines were also sensitive to the FAK inhibitors (). In particular, MiaPaca 2, Capan 1, CD18/HPAF and Colo357 were the most sensitive as compared to Panc-1 and AsPC-1 PDAC cell lines.
Effect of FAK inhibitors on apoptosis in MPM and PDAC cells
MPM and PDAC cells were treated with the three inhibitors separately for 48 h and the percentage of apoptotic cells was determined as described in the materials and methods. As shown in , at the lowest concentration of PF-431396 (1 μM) there was a two-fold increase in the number of apoptotic cells in H2596 cell line. However, both PF-573228 and VS-6063 at this concentration failed to demonstrate any discernable effect on apoptosis. The data clearly showed that, PF-431396 was more potent in inducing apoptosis (significant at all tested concentrations, P≤ 0.001) in MPM cells as compared to PF-573228 (significant at 5 and 10 μM) and VS-6063 (only at 5 and 7.5 μM concentrations).
Figure 2. FAK inhibitors, PF-431396, PF-573228 and VS-6063, induces apoptosis in both MPM and PDAC cells. (A, B) H2596 and (C, D) MiaPaca 2 cells were stained with Annexin V-FITC/PI after treatment with the inhibitors and were analyzed by flow cytometry. Results are expressed as the mean percentage of apoptotic cells ± SEM. (*p<0.05, **p<0.01, ***p<0.001).
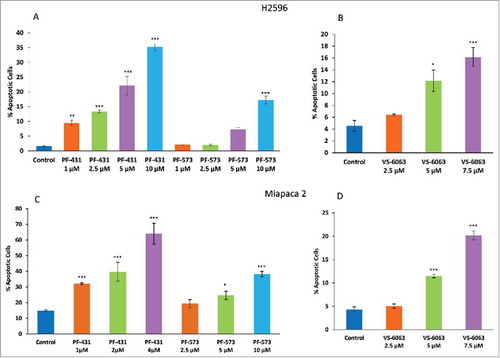
Similar results were observed with PDAC cells (Miapaca 2) () (For PF-431396 P<0.001 at all the concentrations, PF-573228 P<0.001 at 10μM and VS-6068 P<0.001 at 5 and 7.5 μM concentrations). However, in Capan 1 cells, we observed a significant increase in the number of apoptotic cells only at higher PF-431396 concentrations (2 and 4μM) (Supplementary Fig 1). Both Miapaca 2 and Capan 1 were more sensitive to PF-573228 compared to H2596 cells. As shown in , in the case of MiaPaca 2 cells, higher concentrations of VS-6063 (5.00 and 7.5 μM) induced a significant increase (three and four-fold P<0.001) in the number of apoptotic cells compared to untreated control cells. However, Capan 1 cells were highly susceptible even at the lowest concentration of VS-6063 tested. This clearly indicated that VS-6063 is much more effective in Capan 1 compared to MiaPaca 2 cells (Supplementary Fig 1).
Effect of FAK inhibitors on cell cycle arrest in MPM and PDAC cells
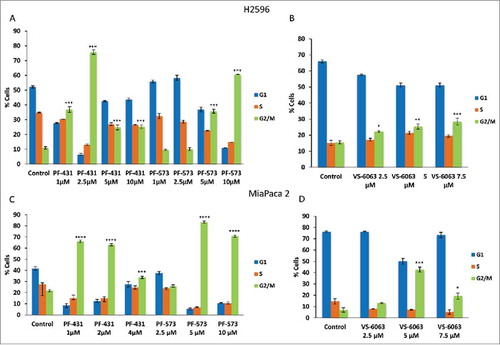
To investigate the underlying mechanism related to the loss of cell viability, we treated both MPM (H2596) and PDAC (MiaPaca 2 and Capan 1) cells with FAK inhibitors and determined cell cycle progression. Treatment of H2596 cells with PF-431396 significantly arrested the MPM cells in G2/M phase at all the concentrations tested (1 to 10 μM) but maximum arrest was seen at 2.5 μM concentration (P ≤ 0.001 for all concentrations). In comparison, PF-573228 arrested the MPM cells in G2/M phase only at higher concentrations (5 and 10 μM, P ≤ 0.001) (). PDAC cells revealed a similar trend (Miapaca 2 and Capan 1) (, and supplementary Fig 2A). Treatment with VS-6063 on the other hand, significantly arrested MPM cells in G2/M phase at all the concentrations tested (concentration-related P ≤ 0.05-0.001) (). The FAK inhibitors however, had no effect on G0/G1 phase in MPM cells (). MiaPaca 2 treated with VS-6063 succumbed to arrest at G2/M phase at higher concentrations (P ≤ 0.01 at 5 μM and P ≤ 0.001 at 7.5μM). However, in Capan 1 cells only the highest concentration was effective in bringing about G2/M arrest (P ≤ 0.001) (). In Miapaca2 and Capan 1 cell lines VS-6063 did not have a measurable effect on G0/G1 phase ( and Supplementary Fig 2A, B).
Figure 3. Treatment of MPM and PDAC cells with FAK inhibitors result in G2/M arrest. Summary of percentage of cells in each cell cycle phase after the treatment of both H2596 (A, B) and Miapaca 2 (C, D) cells with PF-431396, PF-573228, and VS-6063 for 48 h. Data is shown as the % of cells in G1, S, and G2-M phases ± SEM. (*p<0.05, **p<0.01, ***p<0.001).
Effect of FAK inhibitors on PARP cleavage and Cyclin D1 in MPM and PDAC cells
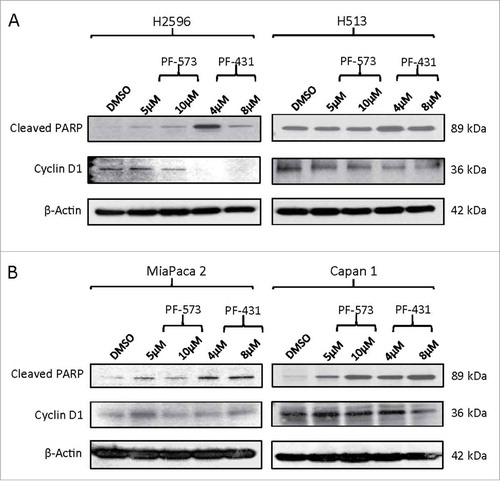
Since growth-arrest of cells, especially at G2/M phase can induce apoptosis, we investigated the effect of PF compounds on the levels of poly (ADP-ribose) polymerase (PARP) and its cleaved product, a marker of apoptosis in both MPM and PDAC cell lines. In addition, we also measured the levels of cyclin D1, a regulator of cell cycle and observed that they were decreased in MPM (H2596 and H513) cells treated with both the PF compounds. The effect of PF-431396 however was more intense than PF-573228 in both types of MPM cells tested. Treatment with PF-431396 resulted in increased levels of cleaved PARP in both MPM cells as compared to PF-573228. A similar trend was observed in PDAC (Miapaca 2 and Capan 1) cells (). This clearly suggested that PF-431396 is relatively more potent in triggering apoptosis and inhibiting cell cycle progression as compared to PF-573228.
Figure 4. FAK inhibition modulates PARP and Cyclin D1 proteins in MPM and PDAC cells. (A) MPM and (B) PDAC cells. Cells were treated with PF-573228 (5 and 10 µM) or PF-431396 (4 and 8 µM) FAK inhibitors for 48h, subjected to Western blot analysis, and analyzed for cleaved PARP and cyclin D1 protein levels.
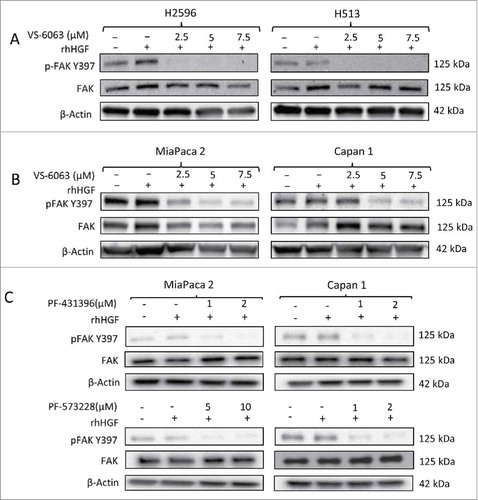
Effect of FAK inhibitors on HGF stimulated downstream signaling in MPM and PDAC cells
We next investigated the effect of PF-573228, PF-431396 and VS-6063 on FAK Y397 phosphorylation status, a reflection of its activity. VS-6063 significantly inhibited phosphorylation of FAK at Y397 in both the MPM cell lines H2596 and H513, in a dose-dependent manner (). Similar trends were also observed in both the PDAC cell lines MiaPaca 2 and Capan 1 following treatment with VS-6063 (). Next, we detected the effect of both PF-573228 and PF-431396 on the levels of p-FAK protein in Miapaca 2 and Capan 1 cells in a dose-dependent manner. PF-431396 decreased the levels of p-FAK in both Miapaca 2 and Capan 1 cells. However, PF-573228 had a dramatic suppressive effect only in Capan 1 cells, whereas in Miapaca 2 cells the same effect was observed only at a much higher concentration ().
Figure 5. Effect of FAK inhibitors on downstream signaling pathway in MPM and PDAC cells. (A) Immunoblots of MPM and (B) PDAC cells after treatment with indicated concentrations of VS-6063 for 24 h. (C) Immunoblots of PDAC cells treated with indicated concentrations of PF-431396 and PF-573228 for 24h. The cells were stimulated with human recombinant HGF (100 ng/ml) before preparing the cell lysates.
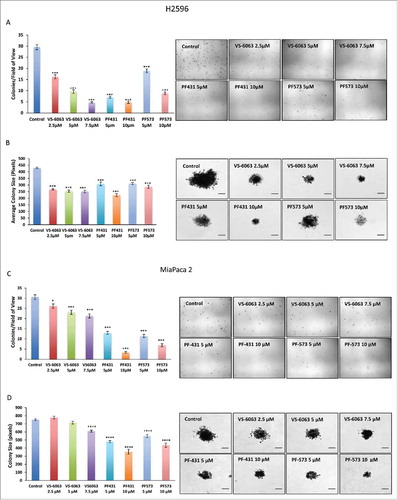
FAK inhibitors suppress anchorage-independent colony-forming ability of MPM and PDAC cells
The ability of tumor cells to form anchorage independent colonies in vitro is a reflection of their tumorigenic potential. We therefore determined the effect of all three FAK inhibitors on anchorage-independent colony formation in MPM and PDAC cells. H2596 cells were plated in soft agar and then treated with all three FAK inhibitors alone and allowed to form colonies over 4 to 5 weeks. Our results showed that treatment of H2596 cells with all the three FAK inhibitors resulted in a dose-dependent decrease in the number of colonies compared to control cells (P ≤ 0.001 for all the three FAK inhibitors). VS-6063 appeared to be the most effective at all the concentrations tested in inhibiting anchorage-independent growth of MPM cells followed by PF-431396. Compared to these, PF-573228 was least effective (). Also, we observed a significant decrease in the size of colonies formed by cancer cells in response to FAK inhibitors compared to untreated control. Once again, VS-6063 exhibited the maximum effect followed by PF-431396, and PF-573228 had the least effect (P ≤ 0.001 for all the three FAK inhibitors) (). Similarly, MiaPaca 2 cells when treated individually with the three FAK inhibitors resulted in significant decrease in the number of colonies in a dose-dependent manner as compared to untreated control (P ≤ 0.001 for PF compounds and VS-6063 except at lowest concentration). PF-431396 and PF-573228 inhibitors were more effective than VS-6063 (). Also, the size of the colonies was significantly reduced in the FAK inhibitor treated groups compared to the colonies in the control group (P ≤ 0.001 for PF compounds and highest concentration of VS-6063). () Similar results were also observed using Capan 1 cells (Supplementary Fig 3); however, Capan 1 cells were more sensitive than MiaPaca 2 cells. These results suggest that PF compound could be used to understand the FAK mediated mechanism(s) associated with proliferation and migration potential of MPM and PDAC cells. These in vitro studies appeared promising to initiate studies using a mice model or their derived tumoroids.
Figure 6. PF-431396, PF-573228, and VS-6063 inhibit the anchorage independent growth of MPM and PDAC cells in a soft agar assay. Colony formation assay showing cell growth inhibition of H2596 (A-D) and MiaPaca 2 (E-H) cells in response to treatment with FAK inhibitors. A, E show decrease in the number of colonies whereas C and G showed decrease in the size of colonies after the treatment of both the cell lines with the FAK inhibitors. Representative colony images are shown in B, D, F and H. Results are expressed as number of colonies per field of view ± SEM. (*p<0.05, **p<0.01, ***p<0.001). Scale bar = 20 μm.
FAK inhibitors suppress adherence and proliferation of PDAC cells on fibronectin
Electrical resistance measurement is a direct measure of cell spreading/adhesion, differentiation and maturation of cells into a confluent barrier.Citation28,Citation29 We investigated the effect of PF-573228 and PF-431396 on adhesion and proliferation of MiaPaca 2 and Capan 1 cells. Cell adhesion/spreading was measured using ECIS as described in the Materials & Methods. PDAC cells were treated with 1 μM of PF-431396 and 4 μM of PF-573228 for 24 h and inoculated to 96 well plate coated with fibronectin. Resistance measurements were recorded after 30 min of cell inoculation. MiaPaca 2 and Capan 1 cell lines was observed to slowly increase resistance over time (Supplementary Fig. 4A & C). During the first 10 h, cells treated with vehicle showed a robust increase in resistance and reached a maximum at 20 h for MiaPaca2 cells, and 30 h for Capan1 cells. Treatment of Miapaca 2 cells with FAK inhibitors dropped off the resistance over the 24 h and cells had poor initial attachment/spreading (Supplementary Fig. 4B). Similar results were observed in Capan 1 cells (Supplementary Fig. 4B & D). However, MiaPaca 2 and Capan 1 cells had different fibronectin adhesive characteristics. Capan 1 cells adhered more rapidly than MiaPaca 2 cells. Also, maximal normalized resistance for MiaPaca 2 cells was lower than that for Capan 1 cells. Bar graphs shown in Supplementary Fig. 4B & D, represent normalized resistance at indicated times for vehicle and PF-431396 and PF-573228 inhibitors for both the cell lines PF-573228 and PF-431396 significantly reduced maximum resistance of MiaPaca 2 (P ≤ 0.05) and Capan 1 (P ≤ 0.01) cells compared with vehicle (Supplementary Fig. 4).
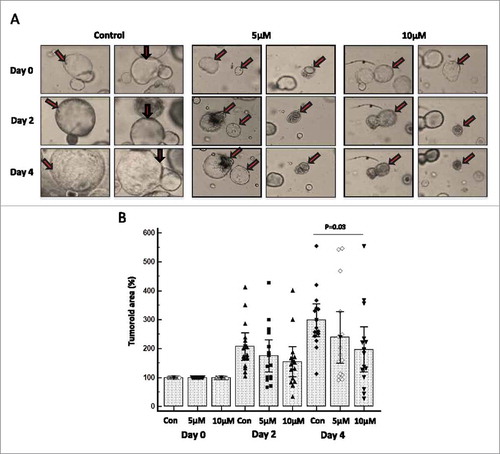
Effect of FAK specific inhibitor (PF-573228) on tumor organoids derived from KrasG12D; Pdx-1 Cre (KC) mouse model of pancreatic cancer
Tumor-organoids were derived from autochthonous KC mice model and subjected to FAK-specific inhibitor (PF-573228) treatment at 5 and 10 µM concentrations for four days, and then 15 organoids in each group were carefully followed for morphological or structural variations. FAK inhibitor treatment showed decreased tumor organoid size at both 5 and 10 µM treatments (). A statistically significant reduction in the growth of KC tumor organoids was observed at 10 µM concentration of PF-573228 (p = 0.03) as compared to untreated controls (). Furthermore, we also observed increased number of dead organoids when treated with FAK inhibitor compared to untreated organoids (Supplementary Fig. 4A, B, C). These ex-vivo experiments also support our contention that the FAK inhibitors deserve further translational studies to validate their therapeutic potential in treating MPM and PDAC.
Figure 7. FAK-specific inhibitor PF-573228 restricts the growth of murine pancreatic tumor organoids. We have generated and cultured tumor organoids derived from the well-established KrasG12D; Pdx-1 Cre (KC) mouse model of pancreatic cancer. Murine KC organoids grow with pancreatic ductal adenocarcinoma architectures when supplemented with specialized media. Tumoroids were treated with 5 and 10 μM concentrations of PF-573228 for 24 h and the effect of FAK inhibitor was followed for 0 to 4 days and images were taken using EVOS FL Auto Cell Imaging System once in every 48 h at 20X magnification. (A) Representative light microscopic images (Magnification at 20X) of KC organoids treated with no drug, 5 and 10 μM concentration of PF-573228 at days 0, 2 and 4. Arrows are pointing out the morphological resemblance of organoids at day 0, 2 and 4. (B) The effect of FAK inhibitor on tumoroids was determined by measuring the tumoroid area of each organoid in the micro square using the quantification software installed in the imaging system. Box and whisker plot shows the effect of FAK inhibitor PF-573228 at two different doses 5 and 10 μM on KC tumoroids. The growth of KC organoids was reduced at both lower (5 μM) and higher concentration (10 μM) on both days of 2 and 4. Statistical significance (P = 0.03) exists between control or no drug treatment and 10 μM concentration of PF-573228 treatment on KC organoids at day four (after drug exposure).
Discussion
MPM and PDAC are two of the most devastating diseases in dire need of highly effective therapies. Both cancers suffer from late diagnosis leaving limited therapeutic options. Understanding the role of key signaling molecules in the overall oncogenesis, growth, and metastasis, has propelled the discovery of novel cancer therapeutic targets. In this context, FAK is known to play a key role in a variety of cancers. It promotes both tumor growth and metastasis. Here, we have shown that FAK is overexpressed in a variety of PDAC and MPM cell lines and when treated with FAK inhibitors (PF-573228, PF-431396, and VS-6063) suppressed cell proliferation and anchorage independent colony formation in a dose-dependent manner. Further analysis revealed cell cycle arrest in G2/M phase and enhanced apoptosis. Using ECIS system-based analysis, we have demonstrated that treatment with the above FAK inhibitors revealed significant suppression of cell adherence and migration of select PDAC cells on fibronectin. Ex-vivo experiments using tumor-organoids derived from autochthonous KC mice treated with FAK-specific inhibitor PF-573228 resulted in a significant decrease in size and promoted tumor organoid death suggesting drug effectiveness in a relatively complex 3D environment.
Our results concur with several previous reports demonstrating FAK overexpression in a variety of cancers and a positive correlation between its overexpression and increased malignancy.Citation30 One of the underlying mechanisms could be amplification of the FAK gene or it's upregulation by tumor related transcription factors resulting in FAK overexpression and activation of FAK.Citation31,Citation32 In PDAC, a statistically significant correlation exists between FAK expression and tumor size and staging.Citation33,Citation34 Sawai H et al (2005) showed that, in PDAC, FAK is overexpressed with increased activity. Increased FAK activity could also be due to enhanced Y397 phosphorylation on FAKCitation35,Citation36 .We also have shown here using both MPM and PDAC cell lines, that the basal phosphorylated levels of FAK are much higher (). As shown by others, activation of FAK could, in turn, lead to activation of the downstream Ras/ERK signaling pathway, which is known to promote cell adhesion and invasion of PDAC cells.Citation35
In general, in case of MPM cell lines such as H2596 and H2052 that expressed relatively high levels of FAK responded with low IC50 values to all the three FAK inhibitors tested ( and Supplementary table 1), indicating that they were very sensitive. In contrast, H2373 and H2461 that expressed relatively low levels of FAK revealed much higher IC50 values to all the three FAK inhibitors suggesting that they were much less sensitive. High FAK expressers such as CD18 and CAPAN-1 were more sensitive to FAK inhibitors; however MiaPaCa2 that revealed much lower levels of FAK was the most sensitive. Although one can with some reservations say that higher FAK levels probably are predictive of higher sensitivity to FAK inhibitors, however there is significant variation between cell lines. One of the underlying reasons could be a significant variation in the expression of Merlin, a tumor suppressor. There appears to be a reciprocal relationship between the two.Citation37
Recently it was shown that hyperactivated FAK plays a key role in fibrosis and immunosuppression, which finally leads to progression of PDAC. FAK inhibition, on the other hand using VS-4718 significantly suppressed tumor progression.Citation38 In tumor cells, it has been reported that activated nuclear FAK enhances chemokine secretion and expansion of regulatory T cells, which in turn suppress cancer surveillance. In cancer cells treated with FAK inhibitor, this immune suppression is relieved thereby paving the way for PL1 based immunotherapy.Citation39
The FAK inhibitor, TAE-226, in a dose-dependent manner decreased FAK phosphorylation and cell viability. It is also known to inhibit the growth of breast cancer cells, their detachment and also promoted apoptosis.Citation40 In case of esophageal cancer cells, TAE-226 inhibited cell migration and proliferation. Interestingly, it induced apoptosis in vitro and inhibited tumor growth in vivo.Citation41 The results presented here using a different set of FAK specific inhibitors revealed similar effects on MPM and PDAC cell proliferation, migration and apoptosis. It is very likely that FAK inhibitors induced cell cycle arrest in G2/M phase () which is known to trigger enhanced apoptosis in MPM and PDAC cells (). Taken together, the above findings clearly indicate that FAK inhibitors can be potentially used for treating MPM and PDAC. An accurate and stringent in vitro assay for the detection of malignant transformation of cells is testing for anchorage-independent growth of cells on soft agar. Our results clearly demonstrate that treatment of MPM and PDAC cells with either of the FAK inhibitors significantly decreased the number and size of anchorage-independent colonies formed in a dose-dependent manner as compared to controls (). These findings are in agreement with those reported using PF-562271, a potent ATP competitive dual inhibitor of both FAK and Pyk2. Interestingly, unlike our observations made here, PF-562271 does not affect growth or apoptosis of several human cancer cell lines; however, it strongly inhibits anchorage-independent growth in soft agar and tumor-xenografts in mice.Citation42 GSK2256098, another FAK inhibitor that suppresses Y397 phosphorylation also decreased cell viability, anchorage-independent growth, AKT/ERK phosphorylation and motility in a dose-dependent manner and increased apoptosis in PDAC.Citation43 We are in the process of extending the studies to determine the effects of the three FAK inhibitors on in vivo MPM and PDAC mouse xenograft tumor growth and metastasis.
Several studies in a variety of cancers have shown that the levels of CD1 are elevated and contribute to the growth and development of various cancers. In a dividing cell, CD1 is synthesized in G0/G1 phase and the levels are maintained throughout the various phases of the cell cycle. Some of the chemotherapeutic agents are known to promote CD1 degradation.Citation44,Citation45 Our studies are in accordance; however low levels of CD1 should result in more of a G1/S arrest whereas our results show G2/M arrest. It is possible that the low levels of CD1 seen in this study are sufficient for the cell to cross G1/S checkpoint but not enough to surmount G2/M ( & ). It is also possible that apart from CD1, there may be other factors involved in the observed G2/M arrest.
The FAK inhibitors used here have been tested for their efficacy in cancer cell lines (in vitro) and mouse xenograft tumor models (in vivo) where a decrease in tumor growth and metastasis was observed.Citation20,Citation46 There are very few studies, which are focused on toxicity of these inhibitors in normal cells. In one such study, the effect of these inhibitors in neighboring endothelial cells residing in the tumor microenvironment was investigated using FAK inhibitors (PF-573228 and FAK Inhibitor 14). The endothelial cell viability, survival, migration and vessel formation were all adversely affected upon exposure to the above FAK inhibitors. Surprisingly endothelial cells appear to be more sensitive to FAK inhibition than tumor cells. Also PF-573228 additionally induces apoptosis of endothelial cells within 36 h of post-drug administration. It is therefore very likely that inhibition of FAK adversely affects tumor growth via suppression of angiogenesis.Citation47
Further validation of FAK inhibitors, either alone or in combination with first-line therapies such as platinum based compounds (MPM) or gemcitabine/albumin bound paclitaxel (Abraxane) (PDAC) that are currently under way in our laboratory should help translate these preclinical studies in the near future.
Author contributions
R.K. designed the study, performed the experiments, analyzed the data and wrote the manuscript. R.S. designed the study, analyzed the data and reviewed the manuscript. T.M. performed the experiments, analyzed the data and reviewed the manuscript. J.R. performed the experiments, analyzed the data. I.D. performed the experiments, analyzed the data. B.M. performed the experiments. J.W. performed the experiments. P.K. reviewed the manuscript. G.K. performed the experiments. P.S. performed the experiments. M.P. performed the experiments, analyzed the data and reviewed the manuscript. H.K. reviewed the manuscript. M.N. designed the study, analyzed the data and reviewed the manuscript. K.B. designed the study, analyzed the data and reviewed the manuscript.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
2017CBT10760R-file002.pdf
Download PDF (991.5 KB)Acknowledgments
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under award number P30CA033572. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Additional information
Funding
References
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA: A Cancer J Clin. 2015;65:5–29.
- Keleg S, Buchler P, Ludwig R, Buchler MW, Friess H. Invasion and metastasis in pancreatic cancer. Mol Cancer. 2003;2:14. doi:10.1186/1476-4598-2-14. PMID:12605717.
- Bilimoria KY, Bentrem DJ, Ko CY, Ritchey J, Stewart AK, Winchester DP, Talamonti MS. Validation of the 6th edition AJCC Pancreatic Cancer Staging System: report from the National Cancer Database. Cancer. 2007;110:738–44. doi:10.1002/cncr.22852. PMID:17580363.
- Jemal A, Center MM, Ward E. The convergence of lung cancer rates between blacks and whites under the age of 40, United States. Cancer Epidemiol Biomarkers Prev. 2009;18:3349–52. doi:10.1158/1055-9965.EPI-09-0740. PMID:19959681.
- Wong HH, Lemoine NR. Pancreatic cancer: molecular pathogenesis and new therapeutic targets. Nat Rev Gastroenterol Hepatol. 2009;6:412–22. doi:10.1038/nrgastro.2009.89. PMID:19506583.
- Mahadevan D, Von Hoff DD. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol Cancer Therapeutics. 2007;6:1186–97. doi:10.1158/1535-7163.MCT-06-0686..
- Herreros-Villanueva M, Hijona E, Banales JM, Cosme A, Bujanda L. Alcohol consumption on pancreatic diseases. World J Gastroenterol. 2013;19:638–47. doi:10.3748/wjg.v19.i5.638. PMID:23429423.
- Fiorino S, Cuppini A, Castellani G, Bacchi-Reggiani ML, Jovine E. HBV- and HCV-related infections and risk of pancreatic cancer. JOP. 2013;14:603–9. PMID:24216545.
- Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495–501. doi:10.1038/nature14169. PMID:25719666.
- Loosen SH, Neumann UP, Trautwein C, Roderburg C, Luedde T. Current and future biomarkers for pancreatic adenocarcinoma. Tumour Biol. 2017;39:1010428317692231. doi:10.1177/1010428317692231. PMID:28618958.
- Creaney J, Robinson BWS. Malignant Mesothelioma biomarkers: from discovery to use in clinical practice for diagnosis, monitoring, screening, and treatment. Chest. 2017;152:143–9. doi:10.1016/j.chest.2016.12.004. PMID:28007619.
- Peng X, Guan JL. Focal adhesion kinase: from in vitro studies to functional analyses in vivo. Curr Protein Pept Sci. 2011;12:52–67. doi:10.2174/138920311795659452. PMID:21190526.
- Calalb MB, Polte TR, Hanks SK. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol Cell Biol. 1995;15:954–63. doi:10.1128/MCB.15.2.954. PMID:7529876.
- Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003;116:1409–16. doi:10.1242/jcs.00373. PMID:12640026.
- Gates RE, King LE, Jr., Hanks SK, Nanney LB. Potential role for focal adhesion kinase in migrating and proliferating keratinocytes near epidermal wounds and in culture. Cell Growth Differ. 1994;5:891–9. PMID:7986754.
- Moissoglu K, Gelman IH. v-Src rescues actin-based cytoskeletal architecture and cell motility and induces enhanced anchorage independence during oncogenic transformation of focal adhesion kinase-null fibroblasts. J Biol Chem. 2003;278:47946–59. doi:10.1074/jbc.M302720200. PMID:14500722.
- Nagaharu K, Zhang X, Yoshida T, Katoh D, Hanamura N, Kozuka Y, Ogawa T, Shiraishi T, Imanaka-Yoshida K. Tenascin C induces epithelial-mesenchymal transition-like change accompanied by SRC activation and focal adhesion kinase phosphorylation in human breast cancer cells. Am J Pathol. 2011;178:754–63. doi:10.1016/j.ajpath.2010.10.015. PMID:21281808.
- Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995;377:539–44. doi:10.1038/377539a0. PMID:7566154.
- Golubovskaya VM, Zheng M, Zhang L, Li JL, Cance WG. The direct effect of focal adhesion kinase (FAK), dominant-negative FAK, FAK-CD and FAK siRNA on gene expression and human MCF-7 breast cancer cell tumorigenesis. BMC Cancer. 2009;9:280. doi:10.1186/1471-2407-9-280. PMID:19671193.
- Yoon H, Dehart JP, Murphy JM, Lim ST. Understanding the roles of FAK in cancer: inhibitors, genetic models, and new insights. J Histochem Cytochemistry. 2015;63:114–28. doi:10.1369/0022155414561498..
- Slack-Davis JK, Martin KH, Tilghman RW, Iwanicki M, Ung EJ, Autry C, Luzzio MJ, Cooper B, Kath JC, Roberts WG, et al. Cellular characterization of a novel focal adhesion kinase inhibitor. J Biol Chem. 2007;282:14845–52. doi:10.1074/jbc.M606695200. PMID:17395594.
- Mills RD, Mita M, Nakagawa J, Shoji M, Sutherland C, Walsh MP. A role for the tyrosine kinase Pyk2 in depolarization-induced contraction of vascular smooth muscle. J Biol Chem. 2015;290:8677–92. doi:10.1074/jbc.M114.633107. PMID:25713079.
- Jones SF, Siu LL, Bendell JC, Cleary JM, Razak AR, Infante JR, Pandya SS, Bedard PL, Pierce KJ, Houk B, et al. A phase I study of VS-6063, a second-generation focal adhesion kinase inhibitor, in patients with advanced solid tumors. Invest New Drugs. 2015;33:1100–7. doi:10.1007/s10637-015-0282-y. PMID:26334219.
- Kanteti R, Dhanasingh I, Kawada I, Lennon FE, Arif Q, Bueno R, Hasina R, Husain AN, Vigneswaran W, Seiwert T, et al. MET and PI3K/mTOR as a potential combinatorial therapeutic target in malignant pleural mesothelioma. PloS One. 2014;9:e105919. doi:10.1371/journal.pone.0105919. PMID:25221930.
- Kanteti R, Riehm JJ, Dhanasingh I, Lennon FE, Mirzapoiazova T, Mambetsariev B, Kindler HL, Salgia R. PI3 Kinase pathway and MET inhibition is efficacious in Malignant Pleural Mesothelioma. Sci Rep. 2016;6:32992. doi:10.1038/srep32992. PMID:27623107.
- Wegener J, Keese CR, Giaever I. Electric cell-substrate impedance sensing (ECIS) as a noninvasive means to monitor the kinetics of cell spreading to artificial surfaces. Exp Cell Res. 2000;259:158–66. doi:10.1006/excr.2000.4919. PMID:10942588.
- Boj SF, Hwang CI, Baker LA, Engle DD, Tuveson DA, Clevers H. Model organoids provide new research opportunities for ductal pancreatic cancer. Mol Cell Oncol. 2016;3:e1014757. doi:10.1080/23723556.2015.1014757. PMID:27308531.
- Szulcek R, Bogaard HJ, van Nieuw Amerongen GP. Electric cell-substrate impedance sensing for the quantification of endothelial proliferation, barrier function, and motility. J Vis Exp. 2014. doi:10.3791/51300. PMID:24747269.
- Wegener J, Janshoff A, Galla HJ. Cell adhesion monitoring using a quartz crystal microbalance: comparative analysis of different mammalian cell lines. Eur Biophys J. 1999;28:26–37. doi:10.1007/s002490050180. PMID:9933921.
- Kurenova E, Xu LH, Yang X, Baldwin AS, Jr., Craven RJ, Hanks SK, Liu ZG, Cance WG. Focal adhesion kinase suppresses apoptosis by binding to the death domain of receptor-interacting protein. Mol Cell Biol. 2004;24:4361–71. doi:10.1128/MCB.24.10.4361-4371.2004. PMID:15121855.
- Cance WG, Golubovskaya VM. Focal adhesion kinase versus p53: apoptosis or survival? Sci Signal. 2008;1:pe22. doi:10.1126/stke.120pe22. PMID:18493017.
- Corsi JM, Rouer E, Girault JA, Enslen H. Organization and post-transcriptional processing of focal adhesion kinase gene. BMC Genomics. 2006;7:198. doi:10.1186/1471-2164-7-198. PMID:16889663.
- Furuyama K, Doi R, Mori T, Toyoda E, Ito D, Kami K, Koizumi M, Kida A, Kawaguchi Y, Fujimoto K. Clinical significance of focal adhesion kinase in resectable pancreatic cancer. World J Surg. 2006;30:219–26. doi:10.1007/s00268-005-0165-z. PMID:16425085.
- Chatzizacharias NA, Giaginis C, Zizi-Serbetzoglou D, Kouraklis GP, Karatzas G, Theocharis SE. Evaluation of the clinical significance of focal adhesion kinase and SRC expression in human pancreatic ductal adenocarcinoma. Pancreas. 2010;39:930–6. doi:10.1097/MPA.0b013e3181d7abcc. PMID:20431421.
- Sawai H, Okada Y, Funahashi H, Matsuo Y, Takahashi H, Takeyama H, Manabe T. Activation of focal adhesion kinase enhances the adhesion and invasion of pancreatic cancer cells via extracellular signal-regulated kinase-1/2 signaling pathway activation. Mol Cancer. 2005;4:37. doi:10.1186/1476-4598-4-37. PMID:16209712.
- Li S, Hua ZC. FAK expression regulation and therapeutic potential. Adv Cancer Res. 2008;101:45–61. doi:10.1016/S0065-230X(08)00403-X. PMID:19055942.
- Shapiro IM, Kolev VN, Vidal CM, Kadariya Y, Ring JE, Wright Q, Weaver DT, Menges C, Padval M, McClatchey AI, et al. Merlin deficiency predicts FAK inhibitor sensitivity: a synthetic lethal relationship. Sci Transl Med. 2014;6:237ra68. doi:10.1126/scitranslmed.3008639. PMID:24848258.
- Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, Nywening TM, Hawkins WG, Shapiro IM, Weaver DT, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med. 2016;22:851–60. doi:10.1038/nm.4123. PMID:27376576.
- Serrels A, Lund T, Serrels B, Byron A, McPherson RC, von Kriegsheim A, Gómez-Cuadrado L, Canel M, Muir M, Ring JE, et al. Nuclear FAK controls chemokine transcription, Tregs, and evasion of anti-tumor immunity. Cell. 2015;163:160–73. doi:10.1016/j.cell.2015.09.001. PMID:26406376.
- Golubovskaya VM, Nyberg C, Zheng M, Kweh F, Magis A, Ostrov D, Cance WG. A small molecule inhibitor, 1,2,4,5-benzenetetraamine tetrahydrochloride, targeting the y397 site of focal adhesion kinase decreases tumor growth. J Med Chem. 2008;51:7405–16. doi:10.1021/jm800483v. PMID:18989950.
- Watanabe N, Takaoka M, Sakurama K, Tomono Y, Hatakeyama S, Ohmori O, Motoki T, Shirakawa Y, Yamatsuji T, Haisa M, et al. Dual tyrosine kinase inhibitor for focal adhesion kinase and insulin-like growth factor-I receptor exhibits anticancer effect in esophageal adenocarcinoma in vitro and in vivo. Clin Cancer Res. 2008;14:4631–9. doi:10.1158/1078-0432.CCR-07-4755. PMID:18628478.
- Zhao J, Guan JL. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009;28:35–49. doi:10.1007/s10555-008-9165-4. PMID:19169797.
- Zhang J, He DH, Zajac-Kaye M, Hochwald SN. A small molecule FAK kinase inhibitor, GSK2256098, inhibits growth and survival of pancreatic ductal adenocarcinoma cells. Cell Cycle. 2014;13:3143–9. doi:10.4161/15384101.2014.949550. PMID:25486573.
- Huang JW, Shiau CW, Yang J, Wang DS, Chiu HC, Chen CY, Chen CS. Development of small-molecule cyclin D1-ablative agents. J Med Chem. 2006;49:4684–9. doi:10.1021/jm060057h. PMID:16854074.
- Alao JP. The regulation of cyclin D1 degradation: roles in cancer development and the potential for therapeutic invention. Mol Cancer. 2007;6:24. doi:10.1186/1476-4598-6-24. PMID:17407548.
- Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer. 2014;14:598–610. doi:10.1038/nrc3792. PMID:25098269.
- Cabrita MA, Jones LM, Quizi JL, Sabourin LA, McKay BC, Addison CL. Focal adhesion kinase inhibitors are potent anti-angiogenic agents. Mol Oncol. 2011;5:517–26. doi:10.1016/j.molonc.2011.10.004. PMID:22075057.