
ABSTRACTS
This research aimed to explore effects of SIX1 and DACH1 on hepatocellular carcinoma (HCC) cell proliferation, apoptosis and cell cycle. Fifty paired hepatocellular carcinoma tissues were screened for differentially expressed genes. SIX1 and DACH1 expressions were subjected to qRT-PCR and western blot in tumor tissues and cells. The knockdown efficiency of siRNAs and transfection efficiency of cDNAs and siRNAs were validated by qRT-PCR and western blot as well. Then colony formation assay and flow cytometry were applied to observe cell proliferation, cell apoptosis and cell cycle changes. Immunofluorescence co-localization and immunoprecipitation were used to analyze the interaction between proteins which was quantified using western blot. Effects of SIX1 and DACH1 on tumor growth and their expressions in tumors were confirmed in vitro in nude mice model. Results of these experiments showed that SIX1 was overexpressed while DACH1 was suppressed in HCC tissues and cells. The suppression of SIX1 and overexpression of DACH1 not only inhibited cell proliferation, but also induced cell apoptosis and arrested cell cycle in G2/M phase compared with control group. Results of immunofluorescence co-localization suggested that SIX1, p53 and DACH1 were significantly overlapped. Immunoprecipitation showed that DACH1 (marked with Flag tag) could pull down p53 and SIX1, but SIX1 (marked with His tag) could only pull down DACH1, which indicated that an indirect regulation between SIX1 and p53. Validated with western blot afterwards, DACH1 overexpression suppressed tumorigenesis in vivo by up-regulating p53 expression while SIX1 overexpression accelerated tumor growth by down-regulating p53 expression. Therefore, the decrease of SIX1 facilitated the expression of DACH1, thus activated the expression of p53 and suppressed the progression of HCC both in vitro and in vivo.
Introduction
Hepatocellular carcinoma (HCC) is the fourth most commonly diagnosed cancer and the second primary cause of cancer-related death.Citation1 Numerous advances have been achieved in the understanding of the molecular basis of HCC. However, the molecular mechanisms that define the relationships between early environmental cues and disease phenotypes are poorly understood. Main reasons are that these interactions are complex, difficult to quantify accurately, and often occur over long periods of time.Citation2
SIX1, a member of the SIX families of homeodomain transcription factors, is essential for the development of numerous organs.Citation3 In fact, it is considered as an oncofetal protein because dysregulation and inappropriate re-expression of SIX1 can result in genomic instability, malignant transformation, and metastasis in animal models and humans.Citation2 Overexpression of SIX1 has been found in various human cancers and is associated with increased tumor progression, metastasis, and decreased survival.Citation3 SIX1 can promote colorectal cancer growth and metastasis through increasing features of cancer stem cells, and stimulate angiogenesis by up-regulating vascular endothelial growth factor (VEGF).Citation3 Additionally, increased SIX1 level is associated with poor survival outcome of osteosarcoma patients.Citation4 However, the biological function of SIX1 in HCC is rarely investigated.
Dachshund homolog 1 (DACH1), a fundamental component of the Retinal Determination Gene Network, is frequently expressed in epithelial cells. DACH1 abundance is decreased in a variety of malignancies, involving organs including breast, prostate, liver, lung, and brain. Massive evidence suggested that DACH1 might function as a new type of tumor suppressor.Citation5 Ke Chen et.al identified DACH1 as a novel p53 binding partner that participated in p53-mediated induction of p21 and cell cycle arrest.Citation6 The knockdown of DACH1 arrested the cell cycle progression in myeloid progenitor cells,Citation7 which also suggested its regulation on cell cycle changes in HCC.
Studies have found that DACH1 was correlated with the expression of SIX1.Citation8,Citation9 For instance, Miller et al. identified the protein-protein interaction of SIX and DACH in malignant peripheral nerve sheath tumors.Citation10 P53 is another important molecule in tumor apoptosis and can bind to DACH1 which therefore blocks the propagation in lung adenocarcinoma cells.Citation11,Citation6 In spite of current researches, discussions of the relationship between SIX1 and p53 remain in shortage.
MDM2 interacts with p53 is common in HCC. MDM2 binds p53 at its transactivation domain and blocks p53-mediated transcriptional regulation, and p53 regulates MDM2 transcription through p53-specific response elements in the promoter region of MDM2, thus forming an auto-regulatory feedback loop.Citation12,Citation13 Therefore, MDM2 expression together with p53 expression was evaluated in this study.
Here we evaluated SIX1 and DACH1 expressions in HCC and their functional mechanism on tumor growth. The possible mechanism behind the regulation was explored by monitoring p53 protein expressions. These findings may provide new references for the study on the molecular mechanism of hepatocellular carcinoma, contributing to the therapeutic strategy and the reduction of morality rates of HCC in the future.
Results
High expression of SIX1 and low expression of DACH1 in HCC were detected
To screen out aberrantly expressed genes, we examined 50 cases of hepatocellular carcinoma tissue samples and 50 adjacent tissue samples. Among all differentially expressed genes, five high expressed genes and five low expressed genes respectively were selected to draw the heat map. SIX1 was high expressed in hepatocellular carcinoma tissues and DACH1 was low expressed in hepatocellular carcinoma tissues (). Both genes showed significant statistical significance (P < 0.05) (). Log fold-change value and P value of SIX1 and DACH1 expressions were shown in . A positive log fold-change value of SIX1 indicated a higher expression in tumor tissues compared with adjacent ones. A negative log fold-change value of DACH1 indicated a lower expression in tumor tissues compared with adjacent ones. In order to further validate the results of microarray analysis, we carried out RT-qPCR on 50 pairs of samples. RT-qPCR results also showed that SIX1 was high expressed and DACH1 was low expressed in hepatocellular carcinoma tissues (both P < 0.001, ). Five pairs of patient samples were randomly selected, and protein expression differences between SIX1 and DACH1 was detected by Western Blot, confirming the high expression of SIX1 protein and the low expression of DACH1 protein (). These results suggested that SIX1 and DACH1 may be potential predictors of HCC. In order to study the effects of SIX1 and DACH1 on the function of HCC cells, we examined the expression of SIX1 and DACH1 in three HCC cell lines (SK-HEP-1, Huh-7 and HepG2) and normal cell line HL-7702[L-02] (). Compared with normal HCC cell line HL-7702 [L-02], SIX1 was high expressed in HCC while DACH1 was low expressed in HCC. HepG2 cells displayed generally the most significant changes and therefore were selected for further functional experiment study.
Figure 1. SIX1 was high expressed and DACH1 was low expressed in HCC tissues and cells. (A) Heat map showed that SIX1 was high expressed and DACH1 was low expressed in HCC tissues compared with adjacent tissues. (B) Volcano plot of SIX1 overexpression and DACH1 suppression showed statistical significance (P < 0.05). (C-D) RT-qPCR analysis showed high expression of SIX1 mRNA and low expression of DACH1 mRNA in 50 samples (P < 0.001). (E) WB results showed the high expression of SIX1 and the low expression of DACH1 in HCC tissues of five patients. (F) SIX1 was high expressed and DACH1 was low expressed in three kinds of HCC cell lines (SK-HEP-1, Huh-7 and HepG2) compared with normal human hepatoma cell line (HL-7702 [L-02]).
![Figure 1. SIX1 was high expressed and DACH1 was low expressed in HCC tissues and cells. (A) Heat map showed that SIX1 was high expressed and DACH1 was low expressed in HCC tissues compared with adjacent tissues. (B) Volcano plot of SIX1 overexpression and DACH1 suppression showed statistical significance (P < 0.05). (C-D) RT-qPCR analysis showed high expression of SIX1 mRNA and low expression of DACH1 mRNA in 50 samples (P < 0.001). (E) WB results showed the high expression of SIX1 and the low expression of DACH1 in HCC tissues of five patients. (F) SIX1 was high expressed and DACH1 was low expressed in three kinds of HCC cell lines (SK-HEP-1, Huh-7 and HepG2) compared with normal human hepatoma cell line (HL-7702 [L-02]).](/cms/asset/f6a8fb95-828c-428a-8779-0acaaf38f71e/kcbt_a_1423920_f0001_oc.gif)
SIX1 promoted but DACH1 inhibited HCC progression
Three siRNAs for SIX1 and DACH1 respectively were pre-tested for the choice of most suitable siRNA to knock down the expression of SIX1 and DACH1. According to protein expressions shown in , si-SIX1−2, si-SIX1−3 and si-DACH1−1, si-DACH1−3 presented better knockdown efficiency and therefore were chosen for following experiments. Overexpression (cDNA) and downregulation (siRNAs) of SIX1 and DACH1 () were confirmed by qPCR and western blot. cDNA-SIX1 greatly up-regulated SIX1 expression and si-SIX1 markedly down-regulated SIX1 mRNA and protein expressions. Similarly, cDNA-DACH1 significantly enhanced DACH1 expression while si-DACH1 drastically suppressed DACH1 mRNA expression and protein expression (). Following the validation of transfection efficiency, colony formation assay further tested influences of SIX1 and DACH1 on the cell proliferation ability. Results indicated that SIX1 overexpression increased colony numbers and SIX1 knockdown decreased colony numbers. On the contrary, high expression of DACH1 inhibited colony formation and low expression of DACH1 promoted colony formation () (P < 0.05). These results suggested that SIX1 could promote whereas DACH1 could inhibit HCC proliferation. Besides, si-SIX1−2 and si-SIX1−3 could efficiently suppress the proliferation and si-DACH1−1 and si-DACH1−3 could reversely improve the proliferation (). Flow cytometry results further evaluated influences of SIX1 and DACH1 on cell apoptosis as well as on cell cycle. Cell apoptosis assay showed that overexpression of SIX1 reduced cell apoptosis rate but overexpression of DACH1 increased cell apoptosis rate (P < 0.05), this suggested that SIX1 acted as an inhibitor while DACH1 played a role of promoter in cell apoptosis. The inhibition of SIX1 and DACH1 validated the above results. The knockdown of SIX1 could promote cell apoptosis rate while the inhibition of DACH1 expression efficiently restrained cell apoptosis progress (). Cell cycle assay showed that cells were retarded at G2 / M phase when SIX1 was inhibited and when DACH1 was overexpressed. Besides, cells arrested in G2/M phase were reduced in cDNA-SIX1 group and si-DACH1−1 and si-DACH1−3 group (, P < 0.05). Cell cycle related proteins were tested as well to further confirm cell cycle changes. P21 expression was increased and CDK1 expression was decreased when SIX1 was inhibited in si-SIX1−2 and si-SIX1−3 group. However, p21 expression was inhibited and CDK1 expression was elevated in cDNA-SIX1 group. Similarly, p21 expression was suppressed and CDK1 expression was elevated in si-DACH1−1 group and si-DACH1−3 group while p21 was increased and CDK1 expression was decreased in cDNA-DACH1 group ().
Figure 2. SiRNAs could significantly inhibit SIX1 and DACH1 expressions. (A-B) WB results showed that si-SIX1-2 had the best inhibition efficiency of SIX1 expression and si-DACH1-1 had the best inhibition efficiency. (C&E) QPCR and WB verified high expression of SIX1 in cDNA-SIX1 group and low expression of SIX1 in si-SIX1−2 group and si-SIX1-3 group. (D&F) QPCR and WB verified high expression of DACH1 in cDNA-DACH1 group and low expression of DACH1 in si-DACH1−1 and si-DACH1-3 group.
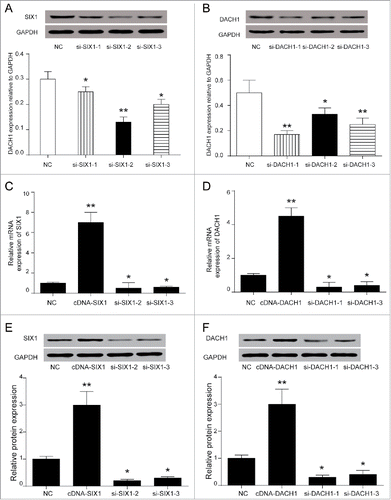
Figure 3. SIX1 promoted proliferation and inhibited apoptosis while DACH1 had an opposite effect. (A-B) SIX1 promoted proliferation while DACH1 inhibited proliferation. The overexpression of SIX1 and suppression of DACH1 promoted cell proliferation by colony formation assay. On the contrary, the suppression of SIX1 and overexpression of DACH1 inhibited cell proliferation. *P < 0.05 indicated significant difference compared with NC group. (C-D) SIX1 inhibited cell apoptosis and DACH1 promoted cell apoptosis. The overexpression of SIX1 and the inhibition of DACH1 suppressed cell apoptosis. Oppositely, the inhibition of SIX1 and overexpression of DACH1 promoted cell apoptosis.
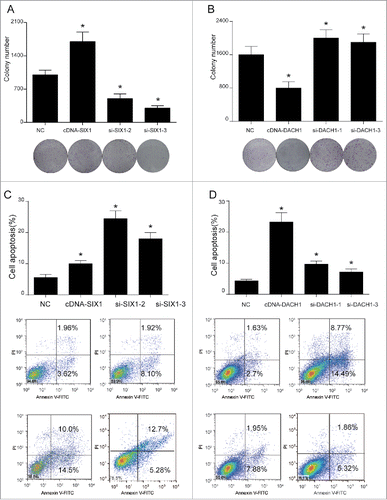
Figure 4. SIX1 promoted cell cycle while DACH1 retarded cell cycle. (A-B) SIX1 suppression and DACH1 overexpression retarded cell cycle at G2/M phase. However, SIX1 overexpression and DACH1 suppression greatly reduced cells that retarded at G2/M phase. (C-D) p21 expression was elevated and CDK1 expression was inhibited when cell cycle was retarded. p21 expression was suppressed and CDK1 expression was improved when SIX1 was overexpressed and DACH1 was suppressed. Reversely, p21 expression was elevated and CDK1 expression was suppressed when SIX1 was inhibited and DACH1 was overexpressed. *P < 0.05 indicated significant difference compared with NC group.
SIX1/DACH1 regulated p53 expression
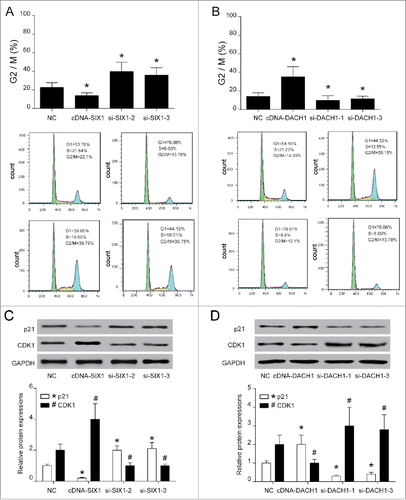
Immunofluorescence co-localization experiments ensured that antibodies bound to the correct target proteins. For instance, perfect co-localization was observed for proteins DACH1 and p53 whereas co-localization of SIX1 revealed that SIX1 could bind to DACH1 as well (). Co-immunoprecipitation was used to analyze protein-protein interactions. The results showed that DACH1 labeled by Flag tag co-precipitated p53 and SIX1, while SIX1 marked by HIS tag co-precipitated DACH1 protein. These results indicated that DACH1 interacted with SIX1 and p53 while SIX1 could only interact with DACH1 (). Interactions among DACH1, SIX1, p53 and MDM2 were further validated with western blot. DACH1 and p53 protein expressions were inversely correlated with the expression of SIX1 (P < 0.05). SIX1 overexpression suppressed expressions of DACH1 and p53. MDM2, which attributed to p53 stability, was elevated when p53 expression was suppressed. However, overexpression or silence of DACH1 had no significant effect on SIX1 protein expression. DACH1 overexpression elevated expressions of p53, inhibited expressions of MDM2 and had no marked influence of SIX1 expression (, P < 0.05). The combination of overexpressed SIX1 and decreased expression of DACH1 further decreased p53 expression and induced hepatocellular carcinoma.
Figure 5. SIX1/DACH1 regulated p53 expression. (A-B) Immunofluorescence co-localization observed that the relative position of p53 protein and SIX1 protein overlap significantly, indicating possible combination. (C) SIX1 co-precipitated with DACH1 and DACH1 co-precipitated with SIX1 and p53. In overexpression group, DACH1, p53 and SIX1 were detected. In Flag IP group, DACH1 directly interacted with SIX1 and p53. In His IP group, SIX1 directly interacted with p53 only. (D-E) SIX1 suppressed DACH1 and p53 expression but DACH1 could only influence p53 expression. SIX1 overexpression inhibited DACH1 and p53 protein expression and promoted MDM2 expression. SIX1 suppression promoted DACH1 and p53 protein expression and inhibited MDM2 expression. DACH1 overexpression promoted p53 expression and inhibited MDM2 expression. DACH1 suppression inhibited p53 expression and promoted MDM2 expression. *P < 0.05 indicated significant difference compared with NC group. *, #, &, $ P < 0.05 indicated statistical significance compared with NC group.
Effects of SIX1/DACH1 on HCC cell growth in vivo
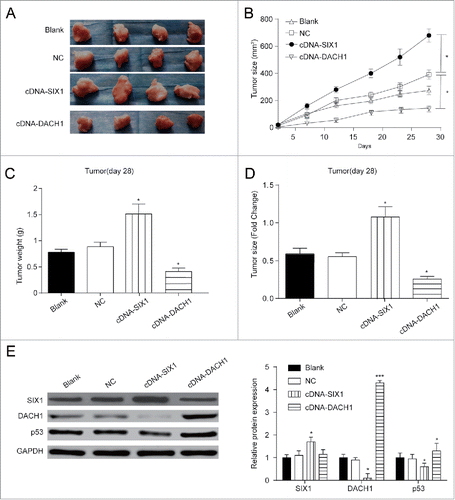
In order to further observe the effect of SIX1 and DACH1 overexpression on tumor growth in vivo, stably infected cells were injected into nude mice. In blank group, HepG2 cells without any treatment were injected into mice; in negative control group, HepG2 cells infected with pc3.1 empty vector were injected into mice; in cDNA-SIX1 group and in cDNA-DACH1 group respectively, HepG2 cells infected with cDNA-SIX1 vector or cDNA-DACH1 were injected into mice. showed tumor growth in different groups after resection. Changes in tumor size () and tumor weight () validated that SIX1 overexpression promoted tumor growth and DACH1 overexpression alleviate tumor growth. In cDNA-SIX1 group, tumors had the faster growth rate and larger tumor size as compared with NC group. In cDNA-DACH1 group, tumors displayed the lower growth rate and smaller tumor size as compared with NC group (, P < 0.05). Tumor weight showed significant increase at day 28 in cDNA-SIX1 group and great decrease in cDNA-DACH1 group (, P < 0.05). Protein expressions were tested in resected tumors to validate that tumor growth changes were caused by SIX1/DACH1/p53 expression fluctuations. DACH1 and p53 were negatively correlated with SIX1; DACH1 exerted no effect on the expression of SIX1 but positively regulated the expression of p53 (, P < 0.05). Therefore, SIX1 could inhibit the expression of DACH1 which could positively regulated p53 protein and inhibit tumor growth primarily.
Figure 6. Effects of SIX1/DACH1 on tumor growth in vivo. (A&B&D) SIX1 promoted tumor growth and DACH1 inhibited tumor growth. Tumor size in cDNA-SIX1 group was the largest, and in cDNA-DACH1 group was the smallest. (C) Tumor in cDNA-SIX1 group was the heaviest, and in cDNA-DACH1 group was the lightest on day 28. (E) SIX1 was highly expressed, DACH1 was lowly expressed and p53 was lowly expressed in large tumors. On the contrary, SIX1 was lowly expressed, DACH1 was highly expressed and p53 was highly expressed in small tumors. *P < 0.05 indicated significant difference compared with Blank and NC group.
Discussion
In the present study, we screened out SIX1, highly expressed in HCC tissues and cells, and DACH1 with low expression in HCC tissues and cells. SIX1 promoted cell proliferation and inhibited cell apoptosis while DACH1 impeded cell proliferation, promoted cell apoptosis and retarded cell cycle. Additionally, we found that SIX1 overexpression could inhibit p53 by suppressing DACH1, which also promoted tumor growth. Therefore SIX1/DACH1/p53 axis may be the underlying modulation mechanism of HCC progression.
As for SIX1, it is a member of SIX families (SIX1-6) of homeobox genes and is an important regulator in cancer. Studies have found that SIX1 stimulated tumor progression. For instance, Zeng et al. found that the overexpression of SIX1 positively correlated with the growth of prostate cancer.Citation14 Lerbs demonstrated the inhibition of SIX1 suppressed pancreatic cancer.Citation15 What's more, SIX1 contributes to the progression of glioblastoma cell and tumor growth.Citation16 In this study, SIX1 overexpression could promote cell proliferation and inhibit cell apoptosis, which is consistent with the findings of previous studies.
DACH1 belongs to Retinal Determination Gene Network (RDGN), which mainly includes Dach, Eya and Six family members. Studies have shown that DACH1 actively participated in tumor inhibition. For example, Chu et al. demonstrated that DACH1 inhibited renal cancer cell propagation and tumor growth.Citation17 Han et al. reported that DACH1 could also restrain lung adenocarcinoma aggressiveness by blocking CXCL5 signaling.Citation18 Wu et al. found that the silencing of DACH1 was also vital for esophageal cancer growth.Citation19 Here, same as previous discoveries, we found the tumor inhibition role of DACH1 in HCC.
It is known that p53 is highly connected with the pathway of apoptosis and contributes to anticancer progresses.Citation11 CD147 promoted cell proliferation in HCC cells by inhibiting the p53-dependent signaling pathway.Citation20 MiR-221 sustained cell-cycle progression and apoptotic response to doxorubicin in hepatocellular carcinoma–derived cell lines by modulating p53/mdm2 feedback loop.Citation21 Therefore, p53 acts as an important modulator in tumor progression, which was found to be regulated by SIX1/DACH1in this study.
However, the limitation of this study should be taken into consideration. For example, the cell lines chosen for colony-formation assays are not so representative; although we have examined the correlation between SIX1, DACH1 and p53 and their effects on HCC, the specific interaction mechanism, especially the correlation between SIX1 and p53 has not been elucidated and needs further study.
In conclusion, we have found that SIX1 was high expressed and DACH1 was low expressed in hepatocellular carcinoma tissues and cells; SIX1 overexpression and DACH1 suppression accelerated HCC cells progression. Moreover, we have excavated the mechanism of SIX1, DACH1 and p53 regulation: SIX1 could bind to DACH1, which further decreased p53 expression in hepatocellular carcinoma, and induced tumor progression. Our findings provided a new sight for mechanisms of SIX1/DACH1/p53 regulation and may have potential clinical significance in hepatocellular carcinoma.
Materials and methods
Patient samples
Fifty hepatocellular carcinoma tissue and para-cancerous tissue specimens were collected at Affiliated Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology. Patient tissue samples were surgically resected without radiotherapy and chemotherapy. The tissues were immediately frozen in liquid nitrogen and stored at −80°C until total RNAs or proteins extraction. The study was approved by the Ethics Committees at Affiliated Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, and informed consent on revised version of Declaration of Helsinki was obtained from each patient.
Cell culture and transfection
Non-metastatic human HCC cell lines HepG2, Huh-7, SK-HEP-1 and human normal hepatocytes HL-7702 [L-02], HEK293T cells were purchased from BNCC (Beijing, China). HL-7702, HepG2 and Huh-7 cells were cultured in 90% FBS and 10% DMSO. SK-HEP-1 Cells were cultured in 50% RPMI-1640, 40% FBS and 10% DMSO. All reagents were purchased from BeNa Culture Collection (Beijing, China). For a transient transfection, siRNAs were designed and commercially obtained from to target SIX1 and DACH1 as shown in . The pcDNA3.1 plasmid was double-digested by HpaI and XhoI enzymes followed by inserting human SIX1 or DACH1 cDNA into nuclear acids of the pc3.1 vector to develop the cDNA-SIX1 vector and cDNA-DACH1 vector. Cells were seeded in six-well plates at 2× 104 cells/well and cultured to 80% confluence. Transfection was performed by using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instruction.
Table 1. siRNA design.
Microarray analysis
RNA for gene expression analysis was obtained from fifty hepatocellular carcinoma tissue and para-cancerous tissue specimens. GeneChip Human Genome U133 Plus 2.0 Array (Affymetrix) were used for these studies and detailed information was indicated in . Assays of triplicate samples were performed at the core facility at the Affiliated Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology.
Table 2. SIX1 and DACH1 expressions in hepatocarcinoma.
RT-qPCR
Total RNA was isolated using TRIzol™ Plus RNA Purification Kit (Invitrogen, USA). First-strand cDNA for real-time quantitative PCR analysis was synthesized from five micrograms of total RNA using SuperScriptTM III Reverse Transcriptase kit (Invitrogen). Reverse transcription of mRNA was carried out using TaqMan high-capacity cDNA kit (Thermo Fisher Scientific, USA) with GAPDH as internal control. Applied Biosystems StepOne real-time PCR was used for testing. Cycling parameters were as follows: initial denaturation for 3 min at 95°C, followed by 45 cycles of 5 s at 95°C and 30 s at 60°C. Calculations of relative gene expression in treatment samples versus controls were performed using the 2−ΔΔCt method. Primers are listed in .
Table 3. Oligonucleotide primer sequences.
Western blot
Frozen tissue or HCC cells were lysed using RIPA buffer (Cell Signaling, USA, #9806). The collected protein lysates were quantitatively prepared to a consistent concentration using a BCA kit (Beyotime Biotechnology, Shanghai, China, Lot # P0012S). Protein extracts were separated by 12% SDS–PAGE and transferred to PVDF membranes (Millipore, Billerica, MA, USA). After being blocked with 5% nonfat milk at room temperature for 1 h, properly diluted primary antibodies (Abcam, USA, #ab211359, #ab176718, #ab9485) were hybridised with the membranes at 4°C overnight. The membrane was washed for three times with TBST for 10 min and incubated with secondary antibodies (Abcam, #ab97095) at room temperature for over 1 h. Band signals were determined using ECL Plus system (GE Healthcare UK Ltd., Buckinghamshire, UK).
Colony-formation assays
Transfected cells were seeded onto a 6-well plate and incubated in normal condition. After 2 weeks of cultivation, cells were fixed by ice-cold methanol for 30 min and stained by 0.04% crystal violet for 10 min in methanol for 30 min. Colonies (more than 50 cells) were counted directly on the plate. A light microscope was used to observe the number of cell colonies and statistical significance was calculated from three independent experiments.
Cell apoptosis analysis
Quantification of apoptotic cells was performed according to the Annexin-V-fluorescein isothiocyanate (FITC) manufacturer's instructions (KeyGen Biotech, Nanjing, China). Cells were harvested and fixed overnight with 70% ethanol at 4°C, followed by resuspension in 500 μL of PBS. Then 2 μL Annexin-V-FITC and 5μL of PI were added. Analyses were performed by a flow cytometer (BD FACScan) with Ex = 488 nm, Em = 530 nm. FITC-positive and PI-negative cells were regarded as apoptotic cells. The sample was incubated for 5 minutes in the dark before analysis by a flow cytometer (BD Biosciences, San Jose, CA).
Immunofluorescent localization
HepG2 cells counting 3 × 104 were plated onto slides for 48 h growth in advance of the experiment. Cells were then washed with phosphate buffered saline (PBS) prior to fixation for 15 min with 4% ice-cold paraformaldehyde (PFA). Following the fixation, cells were permeated using 0.1% (w/v) Triton X-100 (Sigma-Aldrich) in PBS for 3× 5 min and blocked by 5% BSA at 37°C for 30 min, incubated with primary antibodies anti-DACH1 (Abcam, #ab176718), anti-SIX1 (Abcam, #ab211359) or anti-p53 (Santa Cruz Biotechnology, USA, #SC-126) at 37°C for 2 h, and stained with Alexa Fluor 488-conjugated or 568-conjugated IgG at 37°C for 1 h. Meanwhile, the nuclei were stained with DAPI, and the images were collected using fluorescence microscopy.
Immunoprecipitation
Amplified DACH1 cDNA was inserted into pCMV-flag plasmids (Riobio, Guangzhou, China) and SIX1 cDNA was subcloned into pCMV-his plasmid (Riobio). HEK 293T cells were transfected with pFLAG-DACH1, pCMV-his-SIX1 and pCMV-p53 (Riobio) one by one to overexpress flag-DACH1, his-SIX1 and p53. Selection with G418 was performed after each transfection. Transfected 293T cells were harvested in Tris-NaCl-EDTA (TNE) buffer (10 mM Tris–HCl, pH 7.8, 0.15 M NaCl, 1 mM ethylenediaminetetraacetic acid and 1% Nonidet P-40) supplemented with a protease inhibitor cocktail (Roche Diagnostics). Cell lysates were pre-absorbed with mouse IgG-agarose and subsequently incubated with an anti-His antibody (Invitrogen) for 2 h at 4°C and then for 1 h with anti-Flag affinity gel (Sigma-Aldrich). The beads were then added and the incubation was performed at 4°C for 3 hours. Afterwards, each tube was centrifuged at 5,000 rpm for 5 minutes at 4°C. The supernatant was obtained and stored in microcentrifuge tubes at −80°C. The beads were then washed five times with TNE buffer, and the immunoprecipitated proteins were examined by western blot analysis. All experiments were performed at least twice.
Nude mice study
Animal handling and experimental procedures were approved by the Affiliated Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology. We inoculated transfected cells subcutaneously into nude mice aging 4–6 weeks purchased from the National Cancer Institute, NIH. The tumor growth was measured every 7 days for 4 times by using a digital caliper since day 7 and graphs were drawn. Tumor weight was measured when mice were sacrificed on day 28 after cell implantation. We measured tumor diameters every other day, and calculated tumor volume (mm3) as follows: volume = (shortest diameter)Citation2 × (longest diameter) × 0.5.
Statistics analysis
All statistical analysis was performed using Graphpad statistical software, and the data was expressed as mean standard deviation (mean±SD). The Student's t-test and one-way ANOVA were applied to evaluate the differences in groups as appropriate and the significance level was set at 0.05.
Ethical approval
The study was approved by the Ethics Committees at Affiliated Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, and informed consent on revised version of Declaration of Helsinki was obtained from each patient.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contribution
Research conception and design: Deng Ning
Data analysis and interpretation: Jin Chen and Xue Li
Statistical analysis: Xue Li and Qi Cheng
Drafting of the manuscript: Qi Cheng
Critical revision of the manuscript: Xiaoping Chen and Li Jiang
Receiving grant: Li Jiang
Approval of final manuscript: all authors.
Acknowledgments
None.
Additional information
Funding
References
- Zhu H, Wu K, Yan W, Hu L, Yuan J, Dong Y, Li Y, Jing K, Yang Y, Guo M. Epigenetic silencing of DACH1 induces loss of transforming growth factor-beta1 antiproliferative response in human hepatocellular carcinoma. Hepatology. 2013;58(6):2012–22. doi.org/10.1002/hep.26587 PMID:23787902.
- Suen AA, Jefferson WN, Wood CE, Padilla-Banks E, Bae-Jump VL, Williams CJ. SIX1 oncoprotein as a biomarker in a model of hormonal carcinogenesis and in human endometrial cancer. Mol Cancer Res. 2016;14(9):849–58. doi.org/10.1158/1541-7786.MCR-16-0084 PMID:27259717.
- Xu H, Zhang Y, Pena MM, Pirisi L, Creek KE. Six1 promotes colorectal cancer growth and metastasis by stimulating angiogenesis and recruiting tumor-associated macrophages. Carcinogenesis. 2017;38(3):281–92. doi.org/10.1093/carcin/bgw121 PMID:28199476.
- Chao L, Liu J, Zhao D. Increased Six1 expression is associated with poor prognosis in patients with osteosarcoma. Oncol Lett. 2017;13(5):2891–96. doi.org/10.3892/ol.2017.5803 PMID:28521394.
- Wu K, Chen K, Wang C, Jiao X, Wang L, Zhou J, Wang J, Li Z, Addya S, Sorensen PH, et al. Cell fate factor DACH1 represses YB-1-mediated oncogenic transcription and translation. Cancer Res. 2014;74(3):829–39. doi.org/10.1158/0008-5472.CAN-13-2466 PMID:24335958.
- Chen K, Wu K, Cai S, Zhang W, Zhou J, Wang J, Ertel A, Li Z, Rui H, Quong A, et al. Dachshund binds p53 to block the growth of lung adenocarcinoma cells. Cancer Res. 2013;73(11):3262–74. doi.org/10.1158/0008-5472.CAN-12-3191 PMID:23492369.
- Lee JW, Kim HS, Kim S, Hwang J, Kim YH, Lim GY, Sohn WJ, Yoon SR, Kim JY, Park TS, et al. DACH1 regulates cell cycle progression of myeloid cells through the control of cyclin D, Cdk 4/6 and p21Cip1. Biochem Biophys Res Commun. 2012;420(1):91–5. doi.org/10.1016/j.bbrc.2012.02.120 PMID:22405764.
- Martik ML, McClay DR. Deployment of a retinal determination gene network drives directed cell migration in the sea urchin embryo. Elife. 2015;4:e08827. doi.org/10.7554/eLife.08827 PMID:26402456.
- Liu Y, Han N, Zhou S, Zhou R, Yuan X, Xu H, Zhang C, Yin T, Wu K. The DACH/EYA/SIX gene network and its role in tumor initiation and progression. Int J Cancer. 2016;138(5):1067–75. doi.org/10.1002/ijc.29560 PMID:26096807.
- Miller SJ, Lan ZD, Hardiman A, Wu J, Kordich JJ, Patmore DM, Hegde RS, Cripe TP, Cancelas JA, Collins MH, et al. Inhibition of eyes absent Homolog 4 expression induces malignant peripheral nerve sheath tumor necrosis. Oncogene. 2010;29(3):368–79. doi.org/10.1038/onc.2009.360 PMID:19901965.
- Kuo PL, Lin TC, Lin CC. The antiproliferative activity of aloe-emodin is through p53-dependent and p21-dependent apoptotic pathway in human hepatoma cell lines. Life Sci. 2002;71(16):1879–92. PMID:12175703
- Meng X, Franklin DA, Dong J, Zhang Y. MDM2-p53 pathway in hepatocellular carcinoma. Cancer Res. 2014;74(24):7161–7. doi.org/10.1158/0008-5472.CAN-14-1446 PMID:25477334.
- Lahav G, Rosenfeld N, Sigal A, Geva-Zatorsky N, Levine AJ, Elowitz MB, Alon U. Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat Genet. 2004;36(2):147–50. doi.org/10.1038/ng1293 PMID:14730303.
- Zeng J, Shi R, Cai CX, Liu XR, Song YB, Wei M, Ma WL. Increased expression of Six1 correlates with progression and prognosis of prostate cancer. Cancer Cell Int. 2015;15:63. doi.org/10.1186/s12935-015-0215-z PMID:26161040.
- Lerbs T, Bisht S, Scholch S, Pecqueux M, Kristiansen G, Schneider M, Hofmann BT, Welsch T, Reissfelder C, Rahbari NN, et al. Inhibition of Six1 affects tumour invasion and the expression of cancer stem cell markers in pancreatic cancer. BMC Cancer. 2017;17(1):249. doi.org/10.1186/s12885-017-3225-5 PMID:28388884.
- Tian T, Li A, Lu H, Luo R, Zhang M, Li Z. Six1 promotes glioblastoma cell proliferation and invasion by upregulation of connective tissue growth factor. Am J Cancer Res. 2015;5(5):1823–30. PMID:26175950
- Chu Q, Han N, Yuan X, Nie X, Wu H, Chen Y, Guo M, Yu S, Wu K. DACH1 inhibits cyclin D1 expression, cellular proliferation and tumor growth of renal cancer cells. J Hematol Oncol. 2014;7:73. doi.org/10.1186/s13045-014-0073-5 PMID:25322986.
- Han N, Yuan X, Wu H, Xu H, Chu Q, Guo M, Yu S, Chen Y, Wu K. DACH1 inhibits lung adenocarcinoma invasion and tumor growth by repressing CXCL5 signaling. Oncotarget. 2015;6(8):5877–88. doi.org/10.18632/oncotarget.3463 PMID:25788272.
- Wu L, Herman JG, Brock MV, Wu K, Mao G, Yan W, Nie Y, Liang H, Zhan Q, Li W, et al. Silencing DACH1 promotes esophageal cancer growth by inhibiting TGF-beta signaling. PLoS One. 2014;9(4):e95509. doi.org/10.1371/journal.pone.0095509 PMID:24743895.
- Huang Q, Li J, Xing J, Li W, Li H, Ke X, Zhang J, Ren T, Shang Y, Yang H, et al. CD147 promotes reprogramming of glucose metabolism and cell proliferation in HCC cells by inhibiting the p53-dependent signaling pathway. J Hepatol. 2014;61(4):859–66. doi.org/10.1016/j.jhep.2014.04.035 PMID:24801417.
- Ren ZJ, Nong XY, Lv YR, Sun HH, An PP, Wang F, Li X, Liu M, Tang H. Mir-509-5p joins the Mdm2/p53 feedback loop and regulates cancer cell growth. Cell Death Dis. 2014;5:e1387. doi.org/10.1038/cddis.2014.327 PMID:25144722.