
ABSTRACT
Lung cancer is the leading cause of cancer-related death worldwide. Bromodomain and extraterminal domain (BET) proteins act as epigenome readers for gene transcriptional regulation. Among BET family members, BRD4 was well studied, but for its mechanism in non-small cell lung carcinoma has not been elucidated. eIF4E regulates gene translation and has been proved to play an important role in the progression of lung cancer. In this study, we first confirmed that BET inhibitors JQ1 and I-BET151 suppressed the growth of NSCLCs, in parallel with downregulated eIF4E expression. Then we found that knockdown of BRD4 expression using siRNAs inhibited the growth of NSCLCs as well as decreased eIF4E protein levels. Moreover, overexpression of eIF4E partially abrogated the growth inhibitory effect of JQ1, while knockdown of eIF4E enhanced the inhibitory effect of JQ1. Furthermore, JQ1 treatment or knockdown of BRD4 expression decreased eIF4E mRNA levels and inhibited its promoter activity by luciferase reporter assay. JQ1 treatment significantly decreased the binding of eIF4E promoter with BRD4. Finally, JQ1 inhibited the growth of H460 tumors in parallel with downregulated eIF4E mRNA and protein levels in a xenograft mouse model. These findings suggest that inhibition of BET by JQ1, I-BET151, or BRD4 silencing suppresses the growth of non-small cell lung carcinoma through decreasing eIF4E transcription and subsequent mRNA and protein expression. Considering that BET regulates gene transcription epigenetically, our findings not only reveal a new mechanism of BET-regulated eIF4E in lung cancer, but also indicate a novel strategy by co-targeting eIF4E for enhancing BET-targeted cancer therapy.
Introduction
Lung cancer is the leading cause of cancer death among men and the second leading cause of cancer death among women, even though it is not the most prevalent malignancy worldwide.Citation1 Lung cancer includes non-small cell lung carcinoma (NSCLC) and small cell lung carcinoma (SCLC). NSCLC accounts for 83% of lung cancers, and it includes adenocarcinoma, squamous cell carcinoma, and large cell carcinoma.Citation2 Due to decreased smoking and advance in cancer diagnosis and treatment, the incidence rate and the motility of lung cancer decreased in United States in recent years.Citation3 However, the 5-year survival rate was only about 21%.Citation2 For the reason that most of the patients were diagnosed at middle or late stage with local invasion or metastasis, a combined treatment including chemotherapy and molecular-targeted therapy are commonly used, except to surgery and radiotherapy, to benefit the patients.Citation2 Therefore, identifying the molecules that play an important role in cancer progression and clarifying their mechanism will provide novel therapeutic targets or rationales for lung cancer treatment.
Bromodomain and extraterminal domain (BET) proteins are a highly conserved class of proteins that act as important epigenome readers for gene transcriptional regulation.Citation4 The family members contain BRD2, BRD3, BRD4, and BRDT, among which BRD4 has been well-studied. It recognizes acetylated histone or other proteins in the chromatin, and recruits transcriptional complex, containing p-TEFb, RNA polymerase Ⅱ, and other mediators to promote transcription elongation.Citation5,Citation6 BRD4 has been considered to generally regulate gene's expression; however, accumulating studies suggest that it selectively activates a set of gene transcription in different diseases, especially in cancer.Citation7 Some oncogenes, such as c-Myc, IL-7R, FOSL1, and E2F have been proved to be its downstream targets.Citation4,Citation8,Citation9 Targeting BET has shown therapeutic effects for hematological malignancies, inclusing acute myeloid leukemia, multiple myoloma, and lymphoma, as well as solid tumors such as prostate cancer, ovarian cancer, and lung cancer.Citation7 Numerous BET inhibitors has been developed to target BET proteins for cancer therapy, and some of them such as JQ1, I-BET151, and I-BET762 showed potency in pre-clinical studies and clinical trials.Citation10,Citation11 To date, the effect and mechanism of BET inhibition haven't been clarified in lung cancer. Therefore, identifying the downstream signals that mediated its effects in different context and elucidating the mechanism are urgently needed.
The eukaryotic translation initiation factor 4E (eIF4E) is a component of translational initiation complex eIF4F.Citation12 Previous studies proved that eIF4E specifically regulates the translation of some cancer-related mRNAs with a long 5’ cap structure, such as c-Myc, cyclin D1, Bcl-2, Mcl-1, snail, MMP-9, VEGF to promote cell growth, survival, epithelial-mesenchymal transition (EMT), and angiogenesis.Citation13,Citation14 It promotes tumorigenesis, metastasis, recurrence, and drug resistance in numerous cell lines and animal models.Citation15 However, for the upstream regulators that promote eIF4E expression are quite limited. c-Myc was reported to upregulate eIF4E transcription by binding to the E-box of its promoter.Citation16 To date, epigenetic regulation of specific genes transcription attracts more and more interests. Zhang et al. reported that PRMT5 (Protein arginine methyltransferase 5) knockdown decrease the methylation levels of H4R3me2s (symmetric dimethylation of histone 4) and H3R8me2s on the promoter of eIF4E, and decreased eIF4E expression in colorectal cancer.Citation17 This study suggests that eIF4E expression could be regulated epigenetically. Thus, we suspect that BRD4 may regulate eIF4E transcription.
In this study, we first evaluated the growth inhibitory effects and the regulation of eIF4E by targeting BET using two BET inhibitors, JQ1 and I-BET151, and the BRD4 knockdown. Then we examined the inhibitory rate changes of JQ1 after overexpression or knockdown of eIF4E expression to further clarify the role of eIF4E in JQ1-induced growth inhibition. Moreover, we explore the mechanism of eIF4E downregulation by JQ1. Finally, we accessed the effect of JQ1 on tumor growth and the expression eIF4E in a xenograft mouse model. These findings will further clarify the mechanism of BRD4 in lung cancer, and provide new therapeutic strategy for BRD4-targeted cancer therapy.
Results
JQ1 inhibited the growth of NSCLCs in parallel with downregulation of eIF4E expression
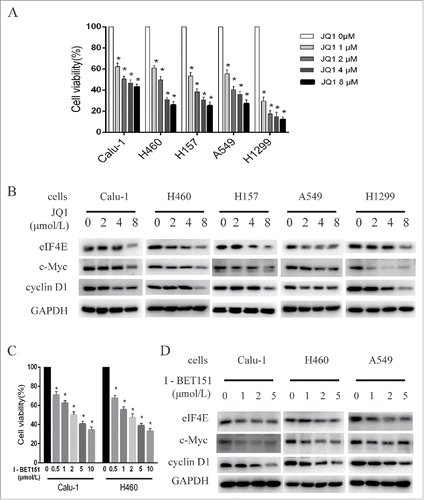
JQ1 has been reported to inhibit the growth of lung cancer cells, we first detected the effect of JQ1 on several NSCLC cells in our system. SRB assay showed that JQ1 inhibited the growth of Calu-1, H460, H157, A549, and H1299 cells in a dose-dependent manner, with an IC50 of 1.8, 2, 1.43, 1.79, and 0.56 μmol/L, respectively (). To explore the downstream signaling pathways that mediated JQ1's anticancer effect, we detected the changes of some key molecules that determine cell growth after JQ1 treatment for 24h by Western blot assay. To induce significant changes of signals in short-time treatment, concentrations equal to or higher than IC50 in our 3-day SRB assay were used. c-Myc is a well-known downstream target of JQ1. Western blot assay showed that JQ1 decreased c-Myc expression in a dose-dependent manner (). We also found that eIF4E and its downstream target cyclin D1 were decreased by JQ1 treatment dose-dependently, suggesting that eIF4E may be a downstream target of JQ1 in lung cancer (). Then we evaluated the effect of another BRD4 inhibitor I-BET151 in Calu-1 and H460 cells. As shown in , I-BET151 inhibited the growth of NSCLCs dose dependently. As well, I-BET151 decreased protein levels of c-Myc, eIF4E, and cyclin D1 dose dependently, just as JQ1 did (). These results demonstrate that eIF4E was downregulated by BRD4 inhibitor JQ1 and I-BET151.
Figure 1. BET inhibitors suppressed the growth of the NSCLCs as well as decreased eIF4E expression. A, NSCLC cells, including Calu-1, H460, H157, A549, and H1299, were treated with different concentrations of JQ1 for 3 days and subjected to SRB assay. B, the NSCLC cells were treated with 0–8 μmol/L JQ1 as indicated for 24h, then the whole-cell lysates were prepared and subjected to western blot assay. C, Calu-1 and H460 cells were treated with I-BET 151 for 3 days and subjected to SRB assay. D, Calu-1 and H460 cells were treated with 0–5μmol/L I-BET 151 as indicated for 24h, then the whole-cell lysates were prepared and subjected to western blot assay. Columns, means of four replicate determinations; bars, SD. *, P < 0.05 vs control. The data are representatives of three independent experiments.
Knockdown of BRD4 expression inhibited cell growth as well as downregulated eIF4E expression in NSCLCs
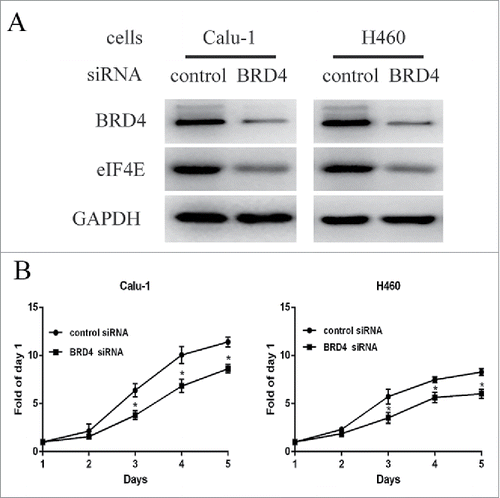
JQ1 and I-BET151 are BET inhibitors that mainly block BRD4, but also block other BET family members, such as BRD2, BRD3, and BRDT.Citation11,Citation18 To further clarify the mechanism of JQ1, we assessed the regulation of eIF4E by interfering BRD4 expression. Calu-1 and H460 cells were transiently transfected with a pool of 3 siRNA sequences that targeting BRD4 or control siRNAs. Western blot assay showed that BRD4 protein levels decreased significantly, suggesting a successful silencing (). We also found that eIF4E protein expression levels greatly decreased by BRD4 knockdown (). Moreover, SRB assay showed that knockdown of BRD4 expression inhibited the growth of Calu-1 and H460 cells, suggesting that targeting BRD4 mimics the effect of JQ1 and I-BET151 (). These findings indicate that downregulation of eIF4E expression maybe a mechanism of targeting BRD4 by JQ1 and I-BET151.
Figure 2. Knockdown BRD4 expression inhibited the growth of NSCLCs in parallel with downregulated eIF4E expression. A, Calu-1 and H460 cells were transiently transfected with a pool of 3 different sequences of BRD4 siRNAs or the control siRNAs for 48h using lipofectamine 2000. The whole-cell lysates were prepared and subjected to western blot assay. B, the two cell lines were seeded to 6-well plates and transiently transfected with the pool of 3 BRD4 siRNAs and the control siRNAs for 24h. Then the cells were re-seeded to 96-well plates for another 5 days and subjected to SRB assay. Points, means of four replicate determinations; bars, SD. *, P<0.05. The data are representatives of three independent experiments.
Manipulating eIF4E expression eliminated the effect of JQ1
Then, we evaluated the role of eIF4E in the growth inhibitory effect of JQ1 in NSCLCs. Calu-1, H460, and A549 cells were transfected with constructs encoding full length eIF4E and the control vector. Western blot assay showed that eIF4E protein levels increased, suggesting the successful overexpression (). Then we found that overexpression of eIF4E partially abrogated the growth inhibitory effect of JQ1 in these cell lines by SRB assay (). The IC50 increased from 1.7, 1.9, and 1.5 μmol/L to 3.4, 7.0, and 3.6 μmol/L in Calu-1, H460, and A549, respectively.
Figure 3. Overexpression or knockdown of eIF4E expression partially abrogated or enhanced the growth inhibitory effect of JQ1 in NSCLCs, respectively. A, Calu-1, H460, and A549 cells were transiently transfected with eIF4E plasmid and the control vector for 48h using lipofectamine 2000, then subjected to western blot assay. B, cells transfected with plasmids as aforementioned for 24 h were re-seeded to 96-well plates, treated with different concentrations of JQ1 as indicated for another 3 days, and subjected to SRB assay. C, Calu-1 and H460 cells were transfected with 2 sequences of eIF4E siRNAs or the control siRNAs for 48h, then subjected to western blot assay. D, cells transfected with the pool of eIF4E siRNAs or the control siRNAs for 24h were re-seeded to 96-well plates and treated with JQ1 as indicated, and then subjected to SRB assay. Points, means of four replicate determinations; bars, SD. *, P < 0.05. The data are representatives of three independent experiments.
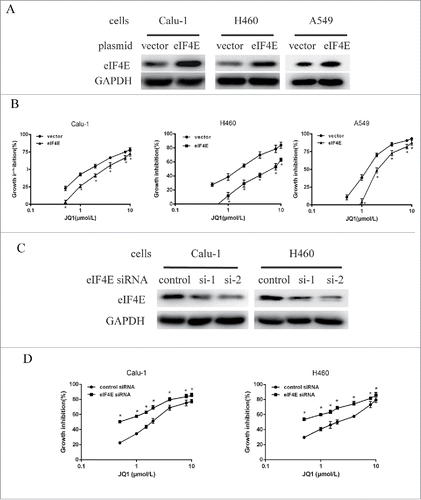
Moreover, we performed an opposite experiment, which evaluated the growth inhibitory effects of JQ1 after knockdown of eIF4E expression. Calu-1 and H460 cells were transiently transfected with 2 different sequences of siRNAs that targeting eIF4E, or the control siRNAs. Western blot assay showed that eIF4E protein levels decreased more than 70% compared to the control, suggesting a successful silencing (). The SRB assay showed that the inhibition of JQ1 on these two cell lines increased significantly in eIF4E knockdown group compared with that in control group (). These results suggest that JQ1 inhibited the growth of NSCLCs through downregulation of eIF4E expression.
JQ1 directly downregulated transcriptional expression of eIF4E
Since downregulation of eIF4E expression played an important role in mediated growth inhibitory effect of JQ1, we further evaluated whether eIF4E was a direct downstream target of BRD4 in NSCLCs. We first detected the mRNA levels of eIF4E regulated by JQ1. We found that JQ1 treatment decreased eIF4E mRNA levels at 6h in H460, A549, and Calu-1 cells, indicating a rapid and direct regulation of eIF4E transcription (). Moreover, eIF4E mRNA levels decreased significantly after 24h JQ1 treatment in these cells (). As well, qRT-PCR assay showed that knockdown of BRD4 expression using siRNA decreased eIF4E mRNA levels significantly (). Then, we performed promoter activity assay of eIF4E by dual-luciferase reporter assay. The pGL3-eIF4E promoter plasmid and the control vector pGL3 were transfected to Calu-1 and H460 cells for 24h, and then treated with JQ1 for another 24h. The renilla plasmid was co-transfected to normalize the transfection efficiency. The ratio of firefly luciferase vs. renilla luciferase indicated the eIF4E promoter activity. We found that eIF4E promoter activity increased significantly when cells were transfected with pGL3-eIF4E promoter plasmid. Moreover, JQ1 treatment decreased eIF4E promoter activity in both cell lines, suggesting that JQ1 inhibited the transcription of eIF4E (). These results indicate that JQ1 downregulated the transcription of eIF4E through inhibition of BRD4.
Figure 4. JQ1 decreased eIF4E mRNA expression, the promoter activity, and the binding of eIF4E promoter with BRD4. A and B, cell lines as indicated were treated with 8 μmol/L JQ1 for 6h (A) or 24 h (B), then subjected to qRT-PCR assay. C, Calu-1 and H460 were transiently transfected with the pool of 3 BRD4 siRNAs and the control siRNAs for 24h. Then total RNAs were purified and subjected to qRT-PCR assay. D, Calu-1 and H460 cells were transiently transfected with eIF4E promoter plasmid (pGL3-eIF4E) or the vector (pGL3-basic) using lipofectamine 2000, and treated with 8 μmol/L JQ1 for 24h. Renilla plasmids were co-transfected as loading control. Then the cell lysates were prepared and subjected to luciferase reporter assay. Relative expression of BRD4 and eIF4E mRNA were calculated using the 2−ΔΔCt method. E, H460 cells were treated with 8 μmol/L JQ1 for 24h, then subjected to ChIP assay using BRD4 antibody. The eIF4E promoter was detected using primers designed from -648 to -489bp upstream of the start site. The PCR products of eIF4E promoter was subjected to quantitative PCR (left), and agarose gel (right). Columns, means of three replicate determinations; bars, SD. *, P < 0.05. The data are representatives of three independent experiments.
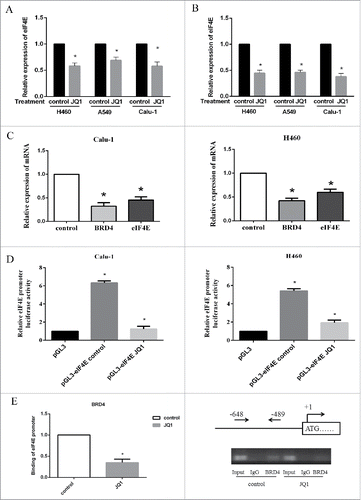
To further identify the mechanism of how JQ1 inhibited eIF4E promoter activity, we performed a chromatin immunoprecipitation (CHIP) assay using BRD4 antibody. H460 cells were treated with 8 μmol/L JQ1 for 24h, and then subjected for CHIP assay. As shown in , the quantitative PCR products of eIF4E promoter was significantly decreased in BRD4 antibody immunoprecipitation group than that in control group, suggesting that JQ1 decreased the binding of eIF4E promoter with BRD4 (, left). The primer design and the products of eIF4E promoter PCR after BRD4 immunoprecipitation were also shown (, right). These results suggest that JQ1 inhibited the binding of BRD4 to eIF4E promoter, and suppressed subsequent eIF4E transcription expression.
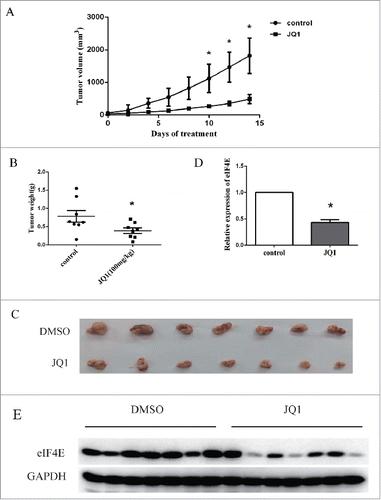
JQ1 inhibited the xenograft growth of NSCLCs in a nude mouse model in parapllel with downregulation of eIF4E expression
We further evaluated the regulation of eIF4E by JQ1 in a H460 xenograft nude mouse model. JQ1 (oral gavage 100mg/kg/d) and the vehicle control were treated for 14 days (n = 7 for each group). The average tumor size was smaller in JQ1 treatment group than those in control vehicle group (). The statistical significance was observed from day 10. As well, the tumor weight was decreased significantly in JQ1 treatment group compared to control group (). was the photo of the tumors. Moreover, qRT-PCR assay showed that the mRNA levels of eIF4E decreased significantly compared to the control in these tumors (). In parallel, eIF4E protein levels in JQ1 treatment group were lower than those in control group (). These results suggest that JQ1 treatment inhibited A549 tumor growth and downregulated eIF4E expression in vivo.
Figure 5. JQ1 suppressed the growth of H460 tumors in parallel with decreased eIF4E expression in a xenograft mouse model. H460 cells were inoculated to the subcutaneous of nude mouse. The mice were treated with 100 mg/kg/d JQ1 or the vehicle for 15 days (n = 7 for each group). A, the size of H460 tumors. Points, means of tumor volume; bars, SD. B, the weight of the tumors. Points, the weight of each tumor; horizontal line, means of tumor weight; bars, SD. C, the photo of the tumors. D and E, the RNAs and the protein lysates of the tumors were prepared and subjected to qRT-PCR assay (D) or western blot assay (E). Relative expression of eIF4E was calculated using the 2−ΔΔCt method. Columns, means of three replicate determinations; bars, SD. *, P < 0.05.
Materials and Methods
Reagents. JQ1 (HY-13030) was purchased from Haoyuan Chemexpress Co., Ltd. I-BET151 (1300031-49-5) was purchased from Sigma-Aldrich. Reagents were dissolved in DMSO at 20mmol/L, stored at -20℃, and diluted just before use. Lipofectamine 2000 transfection reagent (11668-019) was purchased from Life Technologies Co. Invitrogen. Antibodies BRD4 (E2A7X) (13440), eIF4E (9742), cyclin D1 (2922) were purchased from Cell Signaling Technology. c-Myc (K422) and GAPDH (AP0063) were purchased from Bioworld Technology Inc.
Cell lines and cell treatment. Human NSCLC cell lines Calu-1, A549, H157, H460, and H1299 were purchased from the American Type Culture Collection (ATCC; Manassas, VA). Cells were cultured in RPMI1640 medium supplemented with 5% fetal bovine serum at 37℃ in a humidified atmosphere consisting of 5% CO2.
Sulforhodamine B assay. Cells were seeded in 96-well plates at 2,500 cells /well and treated with different concentrations of reagents or the vehicle on the second day for 3 days. Cells after transfection of siRNAs or plasmids for 24h were reseeded to 96-well plates at 1,500 cells /well and cultured for 5 days, or reseeded to 96-well plates at 2,500 cells /well and treated with reagents on the second day for another 3 days. Cell number was estimated by the sulforhodamine B (SRB) assay. The growth inhibition was calculated as we previously described.Citation19 IC50 was determined using Graphpad Prism 6.0.
RNA isolation and quantitative reverse transcription polymerase chain reaction (qRT-PCR). Total RNA extraction, reverse transcription of RNAs, and quantitative real-time PCR were conducted as we previously described.Citation20 All real-time amplifications were conducted in triplicates with the ABI Prism 7300 sequence detection system (Applied Biosystems, Carlsbad, USA). The fold-change of BRD4 and eIF4E was calculated using the 2−ΔΔCT method. Forward (F) and reverse (R) primers were used as follows: BRD4 (we designed), F: GAGCTACCCACAGAAGAAACC and R: GAGTCGATGCTTGAGTTGTGTT; eIF4E, F: 5'-CCTACAGAACAGATGGGCACTC-3' and R: 5'-GCCCAAAAGTCTTCAACAGTATCA-3', GAPDH, F, 5'-ATGGGGAAGGTGAAGGTCG-3' and R, 5'-GGGGTCATTGATGGCAACAATA-3', and synthesized by Invitrogen.Citation21,Citation22
Western blot analysis. Whole-cell protein lysates were prepared and subjected for western blotting as we described previously.Citation23 The chemiluminescent signal was collected and analyzed by Clinx 3400 Mini (Shanghai, China).
Gene knockdown by small interfering RNA. We designed three sequences of BRD4 siRNA that target: 5’-CUCCCUGAUUACUAUAAGATT-3’; 5’-GCACAAUCAAGUCUAAACUTT-3’; 5’-GGAGAUGACAUAGUCUUAATT-3’, and one eIF4E siRNA: 5’- AAGCAAACCUGCGGCUGAUCU – 3’. The other eIF4E siRNA were described previously: 5’- GGACGAUGGCUAAUUACAU – 3’.Citation24 All siRNAs and control (non-target) siRNA were synthesized by GenePharma Co. Ltd. Cells were transfected with siRNAs in a final concentration of 100 nmol/L using Lipofectamine 2000 and subjected to subsequent performances.
Plamid construction The eIF4E overexpression plasmid was a kind gift from Dr. Shi-Yong Sun at Emory University. The plasmid was established by inserting the coding sequence of eIF4E to the p3 × flag-CMV-14 plasmid as described previously.Citation25 The eIF4E promoter luciferase plasmid was also provided by Dr. Shi-Yong Sun. Briefly, the eIF4E promoter region from -1,507 to +72 was constructed to pGL3-Basic plasmid to generate pGL3-eIF4E promoter plasmid.Citation26
Luciferase reporter assay. Calu-1 and H460 were seeded in 24-well plates and transfected with vector or pGL3-eIF4E plasmid for 24 h, and then treated with JQ1 for another 24 hours. Cells were subjected to analysis following the instruction of the Dual-luciferase Reporter assay system (E1910) from Promega Co. and measured on fluorescence spectrometer (Promega GloMax 20/20 E5311, Madison, USA) for the fluorescence intensity as we previously describe.Citation20 The renilla vector was co-transfected to normalize the transfection efficiency. The relative eIF4E promoter activity was presented by the ratio of different treatment vs. control group (pGL3 vector transfection with DMSO treatment).
Chromatin immunoprecipitation assay (CHIP assay). H460 were seeded in 10cm dishes and treated with 8 μmol/L JQ1 for 24 h, then subjected to CHIP assay following the instruction of the CHIP assay kit (p2078) from Beyotime Biotechnology. Briefly, cells were first fixed with 1% formaldehyde to crosslink protein to DNA. Then DNAs in the samples were sheared to fragments of 200-100bp by sonication. Next, reserve 20 μl as input and send left samples for immunoprecipitation using BRD4 antibody. IgG antibody was used as control. The immunoprecipitates were washed with different buffers. DNAs prepared from the immunoprecipitates were subjected to quantitative real-time PCR assay. Forward (F) and reverse (R) primers were designed from -648bp to -489bp upstream of the eIF4E transcription start site as previously reported.Citation27 The sequences were as follows: F: 5'-CTCCACTTCCCAGAAGCCTCTTG-3' and R: 5'-CGGTTCCACAGTCGCCATCTTAG-3', and synthesized by Invitrogen.
Xenograft nude mouse model and treatments. Animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Nanjing Medical University. Six-week-old female anthymic (nu/nu) mice were purchased from Model Animal Research Center of Nanjing University. H460 cells were inoculated subcutaneous at 5 × 106 in serum-free medium. When tumors reached a size of about 100 mm3, the mice were randomized into two groups (n = 7 for each group), and treated with the vehicle (Beta-Cyclodextrin) or JQ1 (100mg/kg/day). Tumor volumes were measured every two days using caliper and calculated with the formula V = π (length × width2) / 6. After 14-day treatment, the mice were sacrificed. The tumors were removed, weighed, and subjected to subsequent qRT-PCR and western blot analysis.
Statistical analysis. The statistical significance of differences in cell viability between different treatments was analyzed with two-sided unpaired Student's t tests. Results were considered to be statistically significant at P < 0.05.
Discussion
In this study, we confirmed the growth inhibitory effect of BET inhibition in NSCLCs based on these findings: 1) BET inhibitors, JQ1 and I-BET151, inhibited the growth of several NSCLC cell lines; 2) Silencing the primary BET protein-BRD4 inhibited cell growth; 3) JQ1 inhibited the H460 tumors in a xenograft mouse model. Targeting BET was reported to suppress tumor progression in several types of cancers such as acute myeloid leukemia, multiple myoloma, lymphoma, castration-resistant prostate cancer, ovarian cancer, and bladder cancer.Citation4,Citation8,Citation28-32 Only in colon cancer, BRD4 functions as a tumor suppressor.Citation33 For lung cancer, Lockwood et al. screened the sensitivity of JQ1 on 17 adenocarcinoma cells lines and identified 8 cell lines, including A549, as sensitive cell lines. H460 was also evaluated and defined as non-sensitive cells in this study.Citation9 Another report by Lenhart et al. confirmed the effects of BET inhibition in small cell lung carcinoma.Citation34 We used lung adenocarcinoma cells (A549, H157, H1299), squamous cell carcinoma cells (Calu-1), as well as large cell lung carcinoma cells (H460) in this study to further confirmed the inhibitory effects of targeting BET proteins in NSCLC. And we found that all these cells were sensitive to JQ1 and I-BET151 treatment.
JQ1 and I-BET151 are small molecular inhibitors targeting all BET protein family members, including BRD2, BRD3, BRD4 and BRDT, but mainly BRD4.Citation10,Citation18 It is well established that JQ1 competitively binds to acetylated lysine recognition motifs of BRD4 and replaces it from chromatin, thus inhibits gene transcription.Citation11 It is also indicated that JQ1 may abrogate the protein-protein interaction of BRD4 with other mediators in chromatin remodeling to block gene transcription.Citation4 Thus, JQ1 is considered to block tumor progression in a context-dependent manner, for example be potency on cancers with c-Myc upregulation. Downregulation of oncogenic c-Myc transcription was proved to play important roles in targeting BRD4-induced inhibition of cancer progression in some malignant hematological diseases and some solid tumors.Citation28,Citation32,Citation35,Citation36 It is worth noting that in lung adenocarcinoma, JQ1 exerted anti-tumor effects not through downregulation of c-Myc, but through FOSL1.Citation9 In this study, we performed experiments in both adenocarcinoma cells and squamous cell carcinoma cells, and found that c-Myc and cyclin D1 were downregulated in parallel with eIF4E. Our findings are complementary to their work in lung cancer, and provide additional mechanisms for BET proteins.
In this study, we identified eIF4E as a new downstream target of BET proteins. We found that eIF4E mRNA and protein levels were downregulated by JQ1, I-BET151, and BRD4 knockdown in NSCLC cells. As well, JQ1 downregulated eIF4E expression in the xenograft mouse model. Moreover, overexpression of eIF4E partially abrogated JQ1's effects on cell growth, suggesting that downregulation of eIF4E plays a pivotal role in this process. eIF4E is the least abundant components in the eIF4F complex, thus is the speed restriction step in translation initiation.Citation12 It has been proved to be overexpressed in many types of cancers, and facilitate cancer progression, metastasis, and drug-resistance.Citation13 For the important role of eIF4E in cancer, it has been suspected as an ideal therapeutic target; however, the small molecule inhibitors are very limited, until recently 4EGI-1 and an antisense oligonucleotide of eIF4E has been developed and evaluated preclinical or in clinical studies.Citation37-41 In this study, we found that knockdown of eIF4E greatly enhanced JQ1-induced growth inhibition of NSCLCs, providing new rationales for cancer therapy by co-targeting eIF4E and BET proteins.
Currently, alternative epigenetic control for gene expression attracts great interests.Citation7 Targeting upstream regulator of eIF4E or eIF4F is emerging.Citation14 We searched 1.0 kb length of eIF4E promoter to predict the transcription factors using Alibaba 2.1 software. It showed that transcription factors NFκB, C/EBPα and β, SP-1, AP-1, RXRβ, and Oct-1 were all able to bind directly to the eIF4E promoter. Even though c-Myc is the only transcriptional factor that reported to activate eIF4E promoter by binding to E-box, Lockwood et al reported that JQ1's anti-tumor effects were not through downregulation of c-Myc in lung adenocarcinoma.Citation9,Citation16 Histone methylation was also reported to regulated eIF4E activity.Citation17 In this study, we observed a rapid decrease of eIF4E mRNA at 6h by JQ1 treatment and a significant decrease of eIF4E promoter activity by JQ1 in a luciferase reporter assay, indicating a transcriptional regulation of eIF4E. CHIP assay showed that the binding of eIF4E promoter with BRD4 was greatly decreased by JQ1, suggesting that JQ1 downregulated the eIF4E expression through inhibition the binding of BRD4 with eIF4E promoter and subsequent transcription. It should be note that we can't exclude the possibility that BRD4 may occupy super-enhancers,Citation42 interact with transcriptional factors such as Rel A of NFκB,Citation43 or recruit complex to modified chromatin,Citation7 to facilitate eIF4E transcription. For BRD4 mediated epigenetic regulation of eIF4E, such as DNA methylation, histone acetylation, or methylation, in lung cancer are worthy of further study.
In summary, the currently study reveals that targeting BRD4 by JQ1, I-BET151, or BRD4 silencing suppresses the growth of non-small cell lung carcinoma through releasing the binding of eIF4E promoter with BRD4 and decreasing subsequent eIF4E mRNA and protein expression. Our findings not only reveal a new mechanism of BRD4-regulated eIF4E in lung cancer, but also indicate a novel strategy of co-targeting eIF4E to enhance BET-targeted cancer therapy.
Acknowledgments
This work is supported by the National Natural Science Foundation of China under Grant No. 81102458, 81473241, 81172004 for X, Wang; No. 81372395 for Z, Cheng; Key Laboratory of Human Functional Genomics of Jiangsu Province, Nanjing Medical University, Nanjing, Jiangsu, China, 210029 (X, Wang); and Six Talents Peak Project of Jiangsu Province (No.2013-WSN-048) (Z, Cheng). We thank Dr. Shi-Yong Sun in Emory University for providing p3 × flag-eIF4E overexpression plasmid and pGL3-eIF4E promoter plasmid. We thank Dr. Jialiang Wang in Vanderbilt University for discussion.
Disclosure statement
No conflicts of interest.
Additional information
Funding
References
- Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA: a cancer journal for clinicians. 2015;65(2):87–108. doi:10.3322/caac.21262
- Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowland JH, Stein KD, Alteri R, Jemal A. Cancer treatment and survivorship statistics, 2016. CA: a cancer journal for clinicians. 2016;66(4):271–89. doi:10.3322/caac.21349
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA: a cancer journal for clinicians. 2016;66(1):7–30. doi:10.3322/caac.21332
- Wu X, Liu D, Tao D, Xiang W, Xiao X, Wang M, Wang L, Luo G, Li Y, Zeng F, et al. BRD4 Regulates EZH2 Transcription through Upregulation of C-MYC and Represents a Novel Therapeutic Target in Bladder Cancer. Mol Cancer Ther. 2016;15(5):1029–42. doi:10.1158/1535-7163.MCT-15-0750
- Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, Zhou Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell. 2005;19(4):535–45. doi:10.1016/j.molcel.2005.06.029
- Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell. 2005;19(4):523–34. doi:10.1016/j.molcel.2005.06.027
- Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell. 2014;54(5):728–36. doi:10.1016/j.molcel.2014.05.016
- Ott CJ, Kopp N, Bird L, Paranal RM, Qi J, Bowman T, Rodig SJ, Kung AL, Bradner JE, Weinstock DM. BET bromodomain inhibition targets both c-Myc and IL7R in high-risk acute lymphoblastic leukemia. Blood. 2012;120(14):2843–52. doi:10.1182/blood-2012-02-413021
- Lockwood WW, Zejnullahu K, Bradner JE, Varmus H. Sensitivity of human lung adenocarcinoma cell lines to targeted inhibition of BET epigenetic signaling proteins. Proc Natl Acad Sci U S A. 2012;109(47):19408–13. doi:10.1073/pnas.1216363109
- Sanchez R, Meslamani J, Zhou MM. The bromodomain: from epigenome reader to druggable target. Biochim Biophys Acta. 2014;1839(8):676–85. doi:10.1016/j.bbagrm.2014.03.011
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. Selective inhibition of BET bromodomains. Nature. 2010;468(7327):1067–73. doi:10.1038/nature09504
- Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136(4):731–45. doi:10.1016/j.cell.2009.01.042
- Wang J, Ye Q, She QB. New insights into 4E-BP1-regulated translation in cancer progression and metastasis. Cancer Cell Microenviron. 2014;1(5):e331. doi:10.14800/ccm.331.
- Pelletier J, Graff J, Ruggero D, Sonenberg N. Targeting the eIF4F translation initiation complex: a critical nexus for cancer development. Cancer Res. 2015;75(2):250–63. doi:10.1158/0008-5472.CAN-14-2789
- Robichaud N, del Rincon SV, Huor B, Alain T, Petruccelli LA, Hearnden J, Goncalves C, Grotegut S, Spruck CH, Furic L, et al. Phosphorylation of eIF4E promotes EMT and metastasis via translational control of SNAIL and MMP-3. Oncogene. 2015;34(16):2032–42. doi:10.1038/onc.2014.146
- Jones RM, Branda J, Johnston KA, Polymenis M, Gadd M, Rustgi A, Callanan L, Schmidt EV. An essential E box in the promoter of the gene encoding the mRNA cap-binding protein (eukaryotic initiation factor 4E) is a target for activation by c-myc. Mol Cell Biol. 1996;16(9):4754–64. doi:10.1128/MCB.16.9.4754
- Zhang B, Dong S, Zhu R, Hu C, Hou J, Li Y, Zhao Q, Shao X, Bu Q, Li H, et al. Targeting protein arginine methyltransferase 5 inhibits colorectal cancer growth by decreasing arginine methylation of eIF4E and FGFR3. Oncotarget. 2015;6(26):22799–811. doi:10.18632/oncotarget.4332
- Seal J, Lamotte Y, Donche F, Bouillot A, Mirguet O, Gellibert F, Nicodeme E, Krysa G, Kirilovsky J, Beinke S, et al. Identification of a novel series of BET family bromodomain inhibitors: binding mode and profile of I-BET151 (GSK1210151A). Bioorg Med Chem Lett. 2012;22(8):2968–72. doi:10.1016/j.bmcl.2012.02.041
- Ma Z, Zhu L, Luo X, Zhai S, Li P, Wang X. Perifosine enhances mTORC1-targeted cancer therapy by activation of GSK3beta in NSCLC cells. Cancer Biol Ther. 2012;13(11):1009–17. doi:10.4161/cbt.20989
- Chen X, Zhu L, Ma Z, Sun G, Luo X, Li M, Zhai S, Li P, Wang X. Oncogenic miR-9 is a target of erlotinib in NSCLCs. Sci Rep. 2015;5:17031. doi:10.1038/srep17031
- Mathis JM, Williams BJ, Sibley DA, Carroll JL, Li J, Odaka Y, Barlow S, Nathan CO, Li BD, DeBenedetti A. Cancer-specific targeting of an adenovirus-delivered herpes simplex virus thymidine kinase suicide gene using translational control. J Gene Med. 2006;8(9):1105–20. doi:10.1002/jgm.935
- Liang S, Guo R, Zhang Z, Liu D, Xu H, Xu Z, Wang X, Yang L. Upregulation of the eIF4E signaling pathway contributes to the progression of gastric cancer, and targeting eIF4E by perifosine inhibits cell growth. Oncol Rep. 2013;29(6):2422–30. doi:10.3892/or.2013.2397
- Huang W, Yang L, Liang S, Liu D, Chen X, Ma Z, Zhai S, Li P, Wang X. AEG-1 is a target of perifosine and is over-expressed in gastric dysplasia and cancers. Dig Dis Sci. 2013;58(10):2873–80. doi:10.1007/s10620-013-2735-5
- Svitkin YV, Herdy B, Costa-Mattioli M, Gingras AC, Raught B, Sonenberg N. Eukaryotic translation initiation factor 4E availability controls the switch between cap-dependent and internal ribosomal entry site-mediated translation. Mol Cell Biol. 2005;25(23):10556–65. doi doi:10.1128/MCB.25.23.10556-10565.2005
- Li Y, Yue P, Deng X, Ueda T, Fukunaga R, Khuri FR, Sun SY. Protein phosphatase 2A negatively regulates eukaryotic initiation factor 4E phosphorylation and eIF4F assembly through direct dephosphorylation of Mnk and eIF4E. Neoplasia. 2010;12(10):848–55. doi:10.1593/neo.10704
- Li Y, Fan S, Koo J, Yue P, Chen ZG, Owonikoko TK, Ramalingam SS, Khuri FR, Sun SY. Elevated expression of eukaryotic translation initiation factor 4E is associated with proliferation, invasion and acquired resistance to erlotinib in lung cancer. Cancer Biol Ther. 2012;13(5):272–80. doi:10.4161/cbt.18923
- Lynch M, Chen L, Ravitz MJ, Mehtani S, Korenblat K, Pazin MJ, Schmidt EV. hnRNP K binds a core polypyrimidine element in the eukaryotic translation initiation factor 4E (eIF4E) promoter, and its regulation of eIF4E contributes to neoplastic transformation. Mol Cell Biol. 2005;25(15):6436–53. doi:10.1128/MCB.25.15.6436-6453.2005
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146(6):904–17. doi:10.1016/j.cell.2011.08.017
- Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, Nekritz EA, Zeid R, Gustafson WC, Greninger P, et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Dis. 2013;3(3):308–23. doi:10.1158/2159-8290.CD-12-0418
- Sengupta S, Biarnes MC, Clarke R, Jordan VC. Inhibition of BET proteins impairs estrogen-mediated growth and transcription in breast cancers by pausing RNA polymerase advancement. Breast Cancer Res Treat. 2015;150(2):265–78. doi:10.1007/s10549-015-3319-1
- Baratta MG, Schinzel AC, Zwang Y, Bandopadhayay P, Bowman-Colin C, Kutt J, Curtis J, Piao H, Wong LC, Kung AL, et al. An in-tumor genetic screen reveals that the BET bromodomain protein, BRD4, is a potential therapeutic target in ovarian carcinoma. Proc Natl Acad Sci U S A. 2015;112(1):232–7. doi:10.1073/pnas.1422165112
- Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478(7370):524–8. doi:10.1038/nature10334
- Rodriguez RM, Huidobro C, Urdinguio RG, Mangas C, Soldevilla B, Dominguez G, Bonilla F, Fernandez AF, Fraga MF. Aberrant epigenetic regulation of bromodomain BRD4 in human colon cancer. J Mol Med. 2012;90(5):587–95. doi:10.1007/s00109-011-0837-0
- Lenhart R, Kirov S, Desilva H, Cao J, Lei M, Johnston K, Peterson R, Schweizer L, Purandare A, Ross-Macdonald P, et al. Sensitivity of Small Cell Lung Cancer to BET Inhibition Is Mediated by Regulation of ASCL1 Gene Expression. Mol Cancer Ther. 2015;14(10):2167–74. doi:10.1158/1535-7163.MCT-15-0037
- Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, Robson SC, Chung CW, Hopf C, Savitski MM, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478(7370):529–33. doi:10.1038/nature10509
- Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, Bergeron L, Sims RJ, 3rd. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci U S A. 2011;108(40):16669–74. doi:10.1073/pnas.1108190108
- Moerke NJ, Aktas H, Chen H, Cantel S, Reibarkh MY, Fahmy A, Gross JD, Degterev A, Yuan J, Chorev M, et al. Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell. 2007;128(2):257–67. doi:10.1016/j.cell.2006.11.046
- Hong DS, Kurzrock R, Oh Y, Wheler J, Naing A, Brail L, Callies S, Andre V, Kadam SK, Nasir A, et al. A phase 1 dose escalation, pharmacokinetic, and pharmacodynamic evaluation of eIF-4E antisense oligonucleotide LY2275796 in patients with advanced cancer. Clin Cancer Res: an official journal of the American Association for Cancer Research. 2011;17(20):6582–91. doi:10.1158/1078-0432.CCR-11-0430
- Jacobson BA, Thumma SC, Jay-Dixon J, Patel MR, Dubear Kroening K, Kratzke MG, Etchison RG, Konicek BW, Graff JR, Kratzke RA. Targeting eukaryotic translation in mesothelioma cells with an eIF4E-specific antisense oligonucleotide. PloS One. 2013;8(11):e81669. doi 10.1371/journal.pone.0081669. doi:10.1371/journal.pone.0081669
- Thumma SC, Jacobson BA, Patel MR, Konicek BW, Franklin MJ, Jay-Dixon J, Sadiq A, De A, Graff JR, Kratzke RA. Antisense oligonucleotide targeting eukaryotic translation initiation factor 4E reduces growth and enhances chemosensitivity of non-small-cell lung cancer cells. Cancer Gene Ther. 2015;22(8):396–401. doi:10.1038/cgt.2015.34
- Lu C, Makala L, Wu D, Cai Y. Targeting translation: eIF4E as an emerging anticancer drug target. Expert Rev Mol Med. 2016;18:e2. doi:10.1017/erm.2015.20
- Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153(2):320–34. doi:10.1016/j.cell.2013.03.036
- Huang B, Yang XD, Zhou MM, Ozato K, Chen LF. Brd4 coactivates transcriptional activation of NF-kappaB via specific binding to acetylated RelA. Mol Cell Biol. 2009;29(5):1375–87. doi:10.1128/MCB.01365-08