
ABSTRACT
Background: Anaplastic lymphoma kinase (ALK) is a validated molecular target in non–small-cell lung cancer (NSCLC). However, the clinical benefits of ALK inhibitors are almost universally limited by the emergence of drug resistance.
Methods: We monitored the plasma circulating tumor DNA (ctDNA) using captured-based ultra-deep sequencing analysis of one patient with metastatic ALK-positive NSCLC who had received therapies including first-, second- and third-generation ALK inhibitors. Functional in vitro studies were further undertaken to elucidate the mechanism of resistance.
Results: ALK T1151Sins mutation was detected when the patient developed resistance to ceritinib, and undetectable when she responded to lorlatinib. MET amplification was present when the tumor developed resistance to lorlatinib, and reduced when the patient received combination therapy of lorlatinib with crizotinib, which corresponded to clinical radiologic responses. In addition, further functional in vitro studies demonstrated that ALK harboring the T1151Sins mutation, while conferring resistance to ceritinib, was inhibited by lorlatinib.
Conclusions: Clinical evidence and in vitro validation revealed the clinical usefulness of captured-base ultra-deep sequencing on longitudinal plasma ctDNA in revealing the underlying resistance mechanism and guiding the precise administration of ALK inhibitors in patients with advanced ALK-positive NSCLC.
Introduction
Anaplastic lymphoma kinase (ALK) rearrangements have been reported in 3–7% of non-small-cell lung cancer (NSCLC) cases.Citation1 The therapeutic landscape for advanced ALK-positive NSCLC has been transformed recently by the development of increasingly potent and selective ALK inhibitors. However, the clinical benefits of these ALK inhibitors are almost universally limited by the emergence of drug resistance.Citation2,Citation3
To identify the resistance mechanism, repeat biopsies are always needed. However, because of unreachable sites of disease, not many patients can undergo repeat biopsies. The clinical application of circulating tumor DNA (ctDNA) as a “liquid biopsy” has been investigated in recent years. The results have showed that ctDNA could represent an unbiased way to assess tumor genetic profile noninvasively and monitor genetic resistance mechanisms expediently.Citation4,Citation5
In this study, we report a patient with metastatic ALK-positive NSCLC who had received multiple prior therapies, including first-, second- and third-generation ALK inhibitors. The disease eventually relapsed while the patient was receiving the third-generation inhibitor, but the patient again had a response to the first-generation inhibitor ( and ). We monitored the serially collected plasma ctDNA using capture-based ultra-deep sequencing analysis and described the molecular basis for the resistance in this patient.
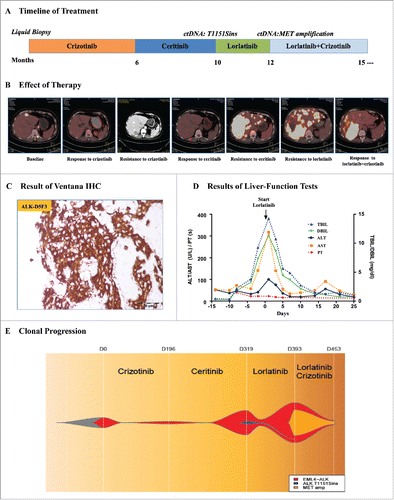
Figure 1. Plasma ctDNA revealing the resistance mechanism of ALK inhibitors. A. The multiple treatments the patient received for metastatic anaplastic lymphoma kinase (ALK)-positive lung adenocarcinoma and the duration of each treatment. B. Computed tomography (CT) or positron emission tomography (PET)-CT images of the patient's metastatic liver lesions chronicling the response to treatment with various ALK inhibitors. C. Ventana (D5F3) ALK Immunohistochemical (IHC) assay analysis of the left lung biopsy tissue. D. Serial monitoring of liver-function before and after treatment with lorlatinib. E. Clonal evolution of resistance to ALK inhibitors in the patient. This model was according to captured-based ultra deep sequencing analysis of pretreatment and resistant plasma circulating tumor DNA (ctDNA). TBIL, total bilirubin; DBIL, direct bilirubin; ALT, alanine aminotransferase; AST, Aspertate Aminotransferase; PT, prothrombin time.
Case report
A 59-year-old female with no history of smoking was diagnosed with metastatic ALK-positive lung adenocarcinoma (; Supplementary Fig. 1A and B). She received first-line crizotinib treatment, a first-generation ALK inhibitor, and had a nearly complete response for 6 months. A computed tomography (CT) scan with contrast at the time revealed new metastatic liver lesions (; Supplementary Fig. 1B). The patient refused to have a needle biopsy of the liver lesions; thus, we conducted the first liquid biopsy assessing plasma ctDNA coupled with captured-based targeted ultra-deep sequencing and the results only showed EML4-ALK fusion (abundance 0.09%). The patient discontinued crizotinib and started to receive ceritinib, a second-generation ALK inhibitor, at a dose of 750 mg once per day. The positron emission computed tomography (PET)-CT scan at 6 weeks showed acceptable radiologic responses (; Supplementary Fig. 2A).
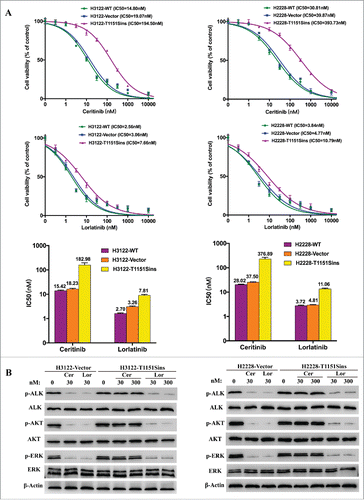
Figure 2. Inhibitory effects of ALK inhibitors on ALK T1151Sins mutation. A. Cell viability assays upon H3122 or H2228 cells harboring ALKWT and ALKT1151S constructs, respectively. H3122 or H2228 cells were treated with the indicated doses of ceritinib or lorlatinib for 48 hours. After the incubation, the cell survival was assayed using CCK8 assay. As depicted by cell viability curves and IC50 values, lorlatinib displayed superior inhibitory activity against T1151Sins mutation compared with ceritinib. Data are representative of three independent biological replicates. B. Immunoblot analyses show differential activity of ceritinib and lorlatinib upon intracellular signaling inhibition in H3122 or H2228 cells harboring ALKWT and ALKT1151S constructs. H3122 or H2228 cells were treated with the indicated concentrations of ceritinib or lorlatinib for 2 hours. Lysates were probed with antibodies directed against the indicated proteins. Inhibition of ALK and intracellular intermediates of MAPK and PI3K-AKT pathways was achieved at lower doses for lorlatinib compared with ceritinib in ALKWT and ALKT1151S cells. The protein levels of total ERK, total AKT and β-actin were as loading controls. Experiments were repeated three times.
The patient was admitted to the hospital with progressive right upper quadrant pain, nausea, and fatigue after 4 months of ceritinib treatment. The laboratory studies were notable for worsening hepatic dysfunction with an elevated total bilirubin level of 14.2 mg per deciliter (). The PET-CT scan at that time showed markedly worsening disease with nearly confluent metastatic liver lesions (; Supplementary Fig. 2A). We performed another captured-based sequencing on plasma ctDNA, and the results revealed EML4-ALK fusion (abundance 19.21%) with an acquired ALK T1151Sins mutation (abundance 2.25%) (). In addition, two gene aberrances associated with cell cycle have been identified at this time. TP53 variation (p.H214fs, c.642_643delTA), occurred on exon 6 which caused frameshift, was detected with the abundance of 11.22%. Another gene frameshift, CDKN2A (p.Met53fs, c158_171delTGATGGGCAGCGCC) also appeared on exon 2 with the abundance of 4.54%. It had been reported that the same site mutation T1151Tins led to resistance to ceritinib or alectinib but sensitivity to lorlatinib, a third-generation ALK inhibitor.Citation6 The patient chose to receive HY-12215 containing the active pharmaceutical ingredient (API) of lorlatinib at a dose of 100 mg once per day on her own volition and experienced a rapid and dramatic clinical improvement with resolution of her liver failure ().
A restaging PET-CT scan showed progressive disease with new bilateral multiple pulmonary nodules, enlarged lymph nodes and bone metastases after 2 months of HY-12215 API treatment (Supplementary Fig. 2B). The third sequencing on plasma ctDNA showed EML4-ALK fusion (abundance increased to 31.42%) with a new MET amplification (copy number, CN = 3.59) (). Besides, the sequencing results also reported the existence of the same TP53 and CDKN2A frameshift mutations (abundance increased to 20.43% and 12.42%, respectively) and the absence of the acquired ALK T1151Sins mutation. Crizotinib is also recommended for the therapy of high-level MET amplification or MET exon 14 skipping mutation NSCLC.Citation7,Citation8 The patient restarted full-dose crizotinib and continued HY-12215 API, due to the increased level of EML4-ALK abundance in plasma ctDNA ().
Unexpectedly, a restaging PET-CT scan showed significant radiologic responses after 2 months. The results indicated the pulmonary nodules disappeared and there were decreases of the liver, lymph node, and bone metastases (; Supplementary Fig. 2B). The fourth sequencing on plasma ctDNA indicated decreased levels of EML4-ALK abundance and MET amplification (abundance to 4.07% and CN to 2.38) (). Meanwhile, the abundance of the same TP53 and CDKN2A frameshift mutations also decreased to 1.93% and 1.45%, respectively. The patient achieved partial response in her multiple lesions and continued the combination therapy of HY-12215 API and crizotinib with no significant side effects. She remained on therapy for more than six months with stabilization of her disease. Her response to the therapy of HY-12215 API combined with crizotinib was her longest response to any agent. After progression, her performance status deteriorated and she elected hospice care.
Functional characterization of novel ALK T1151Sins mutation
In order to investigate the effect of ALK T1151Sins mutation on ALK resistance to ceritinib and sensitivity to lorlatinib, we generated H3122 and H2228 cells expressing ALK T1151Sins mutation. The cells were treated with increasing concentrations of ceritinib or lorlatinib (). The results of cell viability assays confirmed the superiority of lorlatinib (6–10 fold lower IC50) against ALKWT-expressing cells compared with ceritinib. As we expected, ALK T1151Sins mutation led to marked resistance to ceritinib and lorlatinib maintained its strong inhibitory activity against this mutant (25–36 fold lower IC50) compared with ceritinib (). Immunoblot analyses of ALK and downstream pathway activation corroborated the cell viability assays. Lower doses of lorlatinib were required to decrease ALK phosphorylation, whereas only a slightly high concentration of lorlatinib appeared to have effects on ALKT1151S phosphorylation (). Ceritinib had no effect on ALK phosphorylation in cells harboring T1151Sins mutation, which led to persistent ERK and AKT phosphorylation. However, lorlatinib was able to completely switch off ALK, ERK, and AKT signaling in the presence of T1151Sins mutation (). Taken together, these in vitro results revealed ceritinib resistance conferred by ALK T1151Sins mutation, against which lorlatinib maintained its inhibitory potency.
Discussion
ALK is a validated molecular target in NSCLC and the therapies of ALK inhibitors can be highly effective.Citation9,Citation10 However, the resistance often develops.Citation2,Citation3 Recently, liquid biopsy has emerged to becoming a routine diagnostic test and numerous studies have shown that plasma ctDNA can be used as a surrogate for patient stratification, diagnosis, disease monitoring and identification of resistant mechanisms.Citation4,Citation5 In this study, we collected serial plasma ctDNA samples and applied to capture-based sequencing using the LungPlasma panel (Burning Rock Biotech Ltd, Guangzhou China.), consisting of critical exons and introns of 168 genes and 160KB of human genomic regions.
This patient received multiple prior treatments and eventually developed resistance to lorlatinib. Unexpectedly, the patient responded to crizotinib after lorlatinib failure. A recent study described the acquired L1198F mutation confers resistance to lorlatinib but restores sensitivity to crizotinib.Citation3 However, in our patient, we monitored plasma ctDNA and the results revealed MET amplification was present when the tumor developed resistance to lorlatinib. The tumor showed reduced EML4-ALK abundance and MET amplification when the patient received combination therapy of lorlatinib with crizotinib, which corresponded to clinical radiologic responses. Therefore, our results highlight MET amplification as a novel mechanism of resistance to lorlatinib and this combination therapy may be a promising strategy to overcome lorlatinib resistance.
Our results also indicate the acquired ALK T1151Sins mutation may be a novel resistance mechanism to ceritinib. The dynamic monitoring of plasma ctDNA showed an ALK T1151Sins mutation was detectable when the patient developed a resistance to ceritinib, and undetectable when she responded to lorlatinib. Using H3122 or H2228 cells expressing native or mutated ALK fusion, we functionally demonstrated that T1151Sins mutation conferred ceritinib resistance. Lorlatinib, more potent against WT ALK compared with ceritinib, maintained strong growth inhibition of ALKT1151S cells (). Immunoblot analyses confirmed that lorlatinib was able to completely switch off ALK and downstream signaling phosphorylation in engineered H3122 or H2228 cells ().
In summary, this is the first report of MET amplification as the major mechanism mediating lorlatinib resistance and restoring crizotinib sensitivity. Besides, we also find a novel ALK T1151Sins mutation confers resistance to ceritinib and lorlatinib has the superior potency against this mutant.. At last, we highlight the clinical usefulness of captured-base ultra-deep sequencing on longitudinal plasma ctDNA in revealing the underlying resistance mechanism and guiding the precise administration of ALK inhibitors in patients with advanced ALK-positive NSCLC.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Suppl_KCBT_1433496.pdf
Download PDF (2.5 MB)Acknowledgments
We thank Dr. Yongjian Deng for help with histopathological analysis.
Additional information
Funding
References
- Shaw AT, Yeap BY, Mino-Kenudson M, Digumarthy SR, Costa DB, Heist RS, Solomon B, Stubbs H, Admane S, McDermott U, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol. 2009;27:4247–53. doi:10.1200/JCO.2009.22.6993. PMID:19667264.
- Gainor JF, Dardaei L, Yoda S, Friboulet L, Leshchiner I, Katayama R, Dagogo-Jack I, Gadgeel S, Schultz K, Singh M, et al. Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK-rearranged lung cancer. Cancer Discov. 2016;6:1118–33. doi:10.1158/2159-8290.CD-16-0596. PMID:27432227.
- Shaw AT, Friboulet L, Leshchiner I, Gainor JF, Bergqvist S, Brooun A, Burke BJ, Deng Y-L, Liu W, Dardaei L, et al. Resensitization to crizotinib by the lorlatinib ALK resistance mutation L1198F. N Engl J Med. 2016;374:54–61. doi:10.1056/NEJMoa1508887. PMID:26698910.
- Jiang T, Ren S, Zhou C. Role of circulating-tumor DNA analysis in non-small cell lung cancer. Lung Cancer. 2015;90:128–34. doi:10.1016/j.lungcan.2015.09.013. PMID:26415994.
- Alix-Panabieres C, Pantel K. Clinical applications of circulating tumor cells and circulating tumor DNA as liquid biopsy. Cancer Discov. 2016;6:479–91. doi:10.1158/2159-8290.CD-15-1483. PMID:26969689.
- Zou HY, Friboulet L, Kodack DP, Engstrom LD, Li Q, West M, Tang RW, Wang H, Tsaparikos K, Wang J, et al. PF-06463922, an ALK/ROS1 inhibitor, overcomes resistance to first and second generation ALK inhibitors in preclinical models. Cancer Cell. 2015;28:70–81. doi:10.1016/j.ccell.2015.05.010. PMID:26144315.
- Ou S-HI, Kwak EL, Siwak-Tapp C, Dy J, Bergethon K, Clark JW, Camidge DR, Solomon BJ, Maki RG, Bang Y-J, et al. Activity of crizotinib (PF02341066), a dual mesenchymal-epithelial transition (MET) and anaplastic lymphoma kinase (ALK) inhibitor, in a non-small cell lung cancer patient with de novo MET amplification. J Thorac Oncol. 2011;6:942–6. doi:10.1097/JTO.0b013e31821528d3. PMID:21623265.
- Paik PK, Drilon A, Fan P-D, Yu H, Rekhtman N, Ginsberg MS, Borsu L, Schultz N, Berger MF, Rudin CM, et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 2015;5:842–9. doi:10.1158/2159-8290.CD-14-1467. PMID:25971939.
- Solomon BJ, Mok T, Kim D-W, Wu Y-L, Nakagawa K, Mekhail T, Felip E, Cappuzzo F, Paolini J, Usari T, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371:2167–77. doi:10.1056/NEJMoa1408440. PMID:25470694.
- Shaw AT, Kim TM, Crino L, Gridelli C, Kiura K, Liu G, Novello S, Bearz A, Gautschi O, Mok T, et al. Ceritinib versus chemotherapy in patients with ALK-rearranged non-small-cell lung cancer previously given chemotherapy and crizotinib (ASCEND-5): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2017;18:874–86. doi:10.1016/S1470-2045(17)30339-X. PMID:28602779.