
ABSTRACT
MDM2 antagonists stabilize and activate wild-type p53, and histone methyltransferase (HMT) inhibitors reduce methylation on histone lysines and arginines. Both MDM2 antagonists and HMT inhibitors are being developed as cancer therapeutics. Wild-type p53 expressing HCT116 colon cancer cells were resistant to apoptosis in response to the MDM2 antagonist Nutlin-3a. However, co-treatment with the HMT inhibitor DZNep sensitized the cells to Nutlin-3a-induced apoptosis. This sensitization resulted from reduced activity of the Bcl-2 gene promoter and a reduction in Bcl-2 mRNA and protein. Surprisingly, DZNep reduced Bcl-2 expression in other colon cancer cell lines (RKO, SW48, and LoVo) but failed to sensitize them to Nutlin-3a. We found these cell lines express elevated levels of Bcl-2 or other Bcl-2-family proteins, including Bcl-xL, Mcl-1, and Bcl-w. Knockdown of Mcl-1 and/or treatment with specific or pan Bcl-2-family inhibitors (BH3 mimetics) sensitized RKO, SW48, and LoVo cells to apoptosis by Nutlin-3a. The results demonstrate 1) DZNep represses the Bcl-2 gene promoter and affects apoptosis sensitivity by reducing Bcl-2 protein expression, and 2) elevated expression of pro-survival Bcl-2 family members protects colon cancer cells from Nutlin-3a-induced apoptosis. Targeting Bcl-2 proteins via DZNep or BH3 mimetics could increase the therapeutic potential of MDM2-antagonists like Nutlin-3a in colon cancer.
Introduction
Wild-type p53 is a tumor suppressor protein that promotes cell cycle arrest or apoptosis when activated. Approximately 50% of human cancers express wild-type p53 (p53.free.fr/Database/p53_cancer_db.html). In most p53 wild-type cancers the levels and activity of p53 are held in-check by MDM2, an E3 ubiquitin ligase that binds p53 and promotes its degradation.Citation1,Citation2 In recent years MDM2 antagonists (e.g. the Nutlins) have been developed as potential therapeutic agents against p53 wild-type cancers.Citation3,Citation4 These antagonists block MDM2-p53 binding, thus unleashing p53 to inhibit the cell cycle or induce apoptosis. Notably, some cancer cells undergo apoptosis as their primary response to MDM2 antagonists while others undergo cell cycle arrest as their primary response.Citation5-7 Cells that undergo arrest in response to MDM2 antagonists are susceptible to survival and regrowth after drug removal, which could limit the clinical effectiveness of MDM2 antagonists.Citation8,Citation9 An important goal therefore is to identify and target factors that reduce apoptosis in MDM2 antagonist treated cells.
Histone methyltransferases promote methylation of various lysine residues on histones H3 and H4.Citation10 These histone methylations constitute part of the epigenetic code that can increase or decrease gene expression.Citation11,Citation12 Aberrant histone methylation is believed to contribute to the development or maintenance of colon cancer and other cancers by increasing expression of oncogenes and inhibiting expression of tumor suppressors.Citation13 Accordingly, a number of histone methyltransferase inhibitors have been developed as potential therapeutics. 3-Deazaneplanocin A (DZNep) is a pan inhibitor of histone methyltransferases that can reduce the level of multiple histone lysine methylations.Citation14 There is some evidence that DZNep can be cytotoxic in cancer cells in a way that is dependent on wild-type p53. For example, DZNep induced arrest and apoptosis in gastric cancer cell lines that expressed wild-type p53 but not gastric cancer cell lines in which p53 was mutated.Citation15
Apoptosis is controlled in large part by the relative levels of pro- and ant-apoptotic members of the Bcl-2 protein family.Citation16,Citation17 These proteins localize at the mitochondrial membrane and control release of various apoptogenic factors from inside the mitochondria. Bcl-2 family members can interact with each other through Bcl2-homology (BH) domains. Pro-apoptotic Bcl-2 family members bax and bak interact and form pores in the outer mitochondrial membrane.Citation16,Citation17 These pores allow release of factors such as cytochrome c into the cytoplasm that activate caspases to trigger apoptosis.Citation18 BH3-only proteins such as Puma and Noxa also promote bax and bak-dependent apoptosis.Citation16,Citation17 In contrast, anti-apoptotic Bcl-2 family members such as Bcl-2, Bcl-w, Mcl-1, and Bcl-xL can bind and inhibit the pro-apoptotic family members, in this way preventing pore formation and cytochrome c release from the mitochondria and blocking apoptosis. Several studies have reported that high expression of Bcl-2 and other anti-apoptotic Bcl-2-family members is associated with poor responses to radiation or chemotherapy.Citation19 Notably, p53 can promote expression of bax, Puma, and Noxa while inhibiting expression of Bcl-2, suggesting a mechanism for p53-mediated apoptosis.Citation20 Evidence that Bcl-2 may inhibit p53-dependent apoptosis under basal conditions was first provided by Milner and colleagues.Citation21 In their study, knockdown of either Bcl-2 or Bcl-xL was sufficient to induce a low level of apoptosis in cells with wild-type p53 but not in cells where the p53 gene was deleted. Finally, it is important to note that high Bcl-2 expression is a defining characteristic and can promote the development of certain leukemias.Citation22 ABT-199 is a BH3-mimetic that specifically inhibits Bcl-2 and that was FDA approved in 2016 for treatment of patients with CLL that have a 17p deletion and have been treated with at least one prior therapy.Citation23 ABT-737 is a pan inhibitor of Bcl-2, Bcl-w, and Bcl-xL that is currently in pre-clinical development.Citation24,Citation25
In the current report we found that Nutlin-3a alone failed to induce apoptosis in 4 different colon cancer cell lines that express wild-type p53. Strikingly, however, co-treatment with DZNep sensitized HCT116 cells to Nutlin-3a-induced apoptosis. Our data indicate this effect of DZNep resulted from reduced Bcl-2 promoter activity and reduced expression of Bcl-2 protein. DZNep also reduced Bcl-2 expression in the other colon cancer cell lines (RKO, SW48, and LoVo) but failed to sensitize these cells to Nutlin-3a-induced apoptosis. We found these cell lines had elevated expression of Bcl-2 or other anti-apoptotic Bcl-2-family members, including Bcl-xL, Mcl-1, and Bcl-w. Knockdown of Mcl-1 and/or treatment with BH3 mimetics that specifically inhibit Bcl-2 (ABT-199) or Bcl-2-family proteins (ABT-737) sensitized RKO, SW48, and LoVo cells to apoptosis by Nutlin-3a. Together, the results demonstrate 1) that DZNep reduces Bcl-2 expression and affects apoptosis sensitivity by repressing Bcl-2 promoter activity, and 2) that elevated expression of pro-survival Bcl-2 family members protects colon cancer cells from Nutlin-3a-induced apoptosis. We propose targeting Bcl-2 family proteins via DZNep or BH3 mimetics could increase the therapeutic potential of MDM2-antagonists like Nutlin-3a in colon cancer.
Results
DZNep sensitizes HCT116 cells to Nutlin-induced apoptosis
MDM2 antagonists stabilize and activate wild-type p53, and histone methyltransferase inhibitors reduce methylation on histone lysine and arginine residues. Both MDM2 antagonists and histone methylation inhibitors are being developed as cancer therapeutics. We treated wild-type p53 expressing HCT116 colon cancer cells (HCT116 p53+/+) with the MDM2 antagonist Nutlin-3a (Nutlin) and the histone methyltransferase inhibitor DZNep, either alone or in combination. We then monitored apoptosis by caspase-3 cleavage and the percentage of cells with sub-G1 DNA content. As shown in and , neither Nutlin nor DZNep alone induced apoptosis in HCT116 p53+/+ cells. However, abundant apoptosis was observed in HCT116 p53+/+ cells co-treated with Nutlin and DZNep. HCT116 cells which lack the p53 gene (HCT116 p53−/−) were resistant to apoptosis when treated with Nutlin plus DZNep, indicating the apoptosis was p53-dependent.
Figure 1. DZNep sensitizes HCT116 (p53+/+) cells to Nutlin-induced apoptosis. A) HCT116 (p53+/+) and HCT116 (p53−/−) cells were treated with Nutlin (10 μM) and/or DZNep (10 μM) as indicated for 24 or 48 hrs. The percentage of cells with sub-G1 DNA content was determined by flow-cytometry and is plotted (+/− s.e.m.). Results shown are from three experiments for HCT116 (p53+/+) cells and two experiments for HCT116 (p53−/−) cells. B) HCT116 (p53+/+) and HCT116 (p53−/−) cells were treated with Nutlin (10 μM) and/or DZNep (10 μM) as indicated for 24 hrs. Protein lysates were probed with antibodies against p53 and cleaved caspase-3. Actin levels were used as a loading control. Numbers on the side indicate the position of molecular weight markers in kilodaltons.
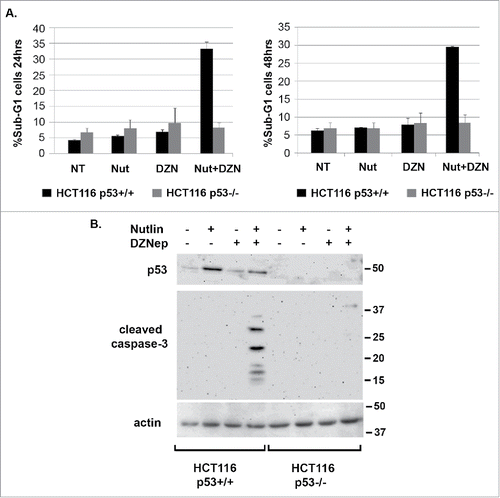
We speculated increased apoptosis in cells co-treated with Nutlin plus DZNep could result from an increase in p53 protein levels and/or an increase in p53 activity. As shown in , p53 was induced by Nutlin alone in HCT116 p53+/+ cells and induced to a slightly lower level in this experiment in cells co-treated with Nutlin plus DZNep. This indicates increased apoptosis in HCT116 cells co-treated with Nutlin plus DZNep does not result from an increase in p53 protein levels. To examine p53 activity, we monitored expression of various p53-regulated genes by qRT-PCR. This included pro-apoptotic genes that are activated by p53 (Bax, PUMA, Noxa), anti-apoptotic genes that p53 can repress (Bcl-2), and also non-apoptotic genes that are activated by p53 (P21, MDM2). As shown in , each of the genes that are activated by p53 were induced in Nutlin-treated HCT116 cells and slightly less induced in cells co-treated with Nutlin plus DZNep, consistent with the slight reduction in p53 protein levels seen in . This result suggests apoptosis in cells co-treated with Nutlin plus DZNep does not result from an increase in p53 transcriptional activity. Analysis of Bcl-2 expression was particularly interesting in these studies. Bcl-2 mRNA levels were reduced ∼40% in HCT116 cells treated with Nutlin alone (), consistent with reports p53 can repress Bcl-2 expression.Citation26-28 However, Bcl-2 mRNA was reduced ∼80% in cell treated with DZNep alone or DZNep plus Nutlin (). Immunoblotting showed a greater reduction in Bcl-2 protein levels in cells treated with DZNep or Nutlin plus DZNep compared to cells treated with Nutlin alone ().
Figure 2. DZNep reduces Bcl-2 mRNA levels in HCT116 cells. A) HCT116 (p53+/+) and HCT116 (p53−/−) cells were treated with Nutlin (10 μM) and/or DZNep (10 μM) as indicated for 24 hrs. mRNA levels for the indicated p53-responsive genes was determined by qRT-PCR. B) Protein lysates from HCT116 (p53+/+) cells were probed with antibodies against Bcl-2 and actin. Numbers on the side indicate the position of molecular weight markers in kilodaltons.
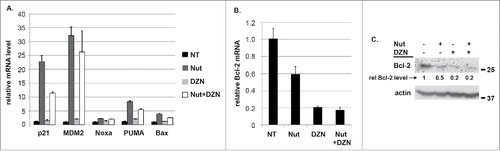
Based on the results in we speculated that DZNep may sensitize HCT116 cells to Nutlin-induced apoptosis by reducing Bcl-2 expression below a threshold needed for survival. If this were the case then we reasoned two things would be true: first, Bcl-2 overexpression would rescue cells treated with Nutlin plus DZNep from apoptosis and second, that depletion of Bcl-2 or treatment with a Bcl-2 inhibitor would have the same effect as DZNep. To test this, we first transfected HCT116 cells with control plasmid (pCDNA-3.1) or with a plasmid that expresses Bcl-2 (pCDNA-Bcl-2). Twenty four hours after transfection, the cells were either untreated or treated with Nutlin plus DZNep. The transfection process alone can stress cells and induce apoptosis. We therefore plotted the fold-increase in apoptosis (%sub-G1 cells) before and after treatment, referring to the percentage of sub-G1 cells in transfected but untreated cells as “1.0”. As shown in , in control cells there was an approximately 8-fold increase in sub-G1 (apoptotic) cells after treatment with Nutlin plus DZNep, while in Bcl-2 transfectants there was only a ∼1.5-fold increase in sub-G1 (apoptotic) cells. Immunoblotting showed overexpression of Bcl-2 in cells transfected with Bcl-2 plasmid (). The results indicate Bcl-2 overexpression can rescue HCT116 cells from apoptosis induced by Nutlin plus DZNep. This supports the idea that DZNep sensitizes the cells to Nutlin-induced apoptosis by reducing Bcl-2 expression, at least in part. Next, we transfected HCT116 cells with control siRNA or siRNA against Bcl-2. We then treated the cells with Nutlin and monitored apoptosis by the percentage of cells with sub-G1 DNA content. As shown in , control siRNA did not sensitize HCT116 cells to Nutlin-induced apoptosis. However, Bcl-2 siRNA sensitized HCT116 cells to Nutlin-induced apoptosis (). Quantitative PCR confirmed that Bcl-2 mRNA levels were reduced by Bcl-2 siRNA (). The results indicate that Bcl-2 expression protects against Nutlin-induced apoptosis, and support the idea that DZNep may sensitize HCT116 cells to apoptosis, at least in part, by reducing Bcl-2 expression. To examine this further, we made use of two different BH3 domain mimetics. ABT-199 is a BH3 mimetic that specifically inhibits Bcl-2, and ABT-737 is a BH3 mimetic that inhibits Bcl-2, Bcl-w, and Bcl-xL. As shown in , ABT-199 alone did not induce apoptosis in HCT116 cells. However, ABT-199 sensitized HCT116 cells to apoptosis by Nutlin, and to nearly the same extent as DZNep (). This finding provides further support that Bcl-2 expression protects against Nutlin-induced apoptosis in these cells. Notably, ABT-737 alone induced some apoptosis in HCT116 cells and caused a greater sensitization to Nutlin-induced apoptosis than did ABT-199. This suggests Bcl-w and/or Bcl-xL may also inhibit apoptosis in these cells, basally and in response to Nutlin.
Figure 3. Bcl-2 overexpression reduces apoptosis in Nutlin plus DZNep treated HCT116 cells. Bcl-2 knockdown or treatment with the Bcl-2-specific or pan-Bcl-2 family inhibitors sensitizes HCT116 cells to Nutlin-induced apoptosis A) HCT116 cells were transfected with either control plasmid (Ctrl; pCDNA3.1) or plasmid expressing Bcl-2. Twenty four hours after transfection the cells were treated untreated or treated with Nutlin (10 μM) plus DZNep (10 μM). The percentage of apoptotic (sub-G1) cells was determined 24 hrs later and is plotted (+/− s.e.m.). The results are from three experiments. B) Immunoblotting shows overexpression of Bcl-2 in the cells transfected with Bcl-2 expression plasmid. Numbers on the side indicate the position of molecular weight markers in kilodaltons. C) HCT116 (p53+/+) cells were transfected with control siRNA (siCtrl) or siRNA against Bcl-2 (siBcl-2). 24 hrs after transfection the cells were untreated or treated with Nutlin (10 μM) for 24 hrs and the percentage of sub-G1 cells determined. D) qRT-PCR analysis shows Bcl-2 mRNA is reduced in the cells transfected with siRNA against Bcl-2 compared to cells transfected with control siRNA. E). HCT116 (p53+/+) were treated for 24 hrs with Nutlin alone (10 μM) or in combination with increasing amounts (1, 2.5, 5, and 10 μM) of either ABT-199 or ABT-737. Apoptosis was determined by the percentage of cells with sub-G1 DNA content. For comparison, HCT116 (p53+/+) cells were treated with Nutlin (10 μM) and/or DZNep (10 μM) for 24 hrs.
DZNep reduces Bcl-2 promoter activity
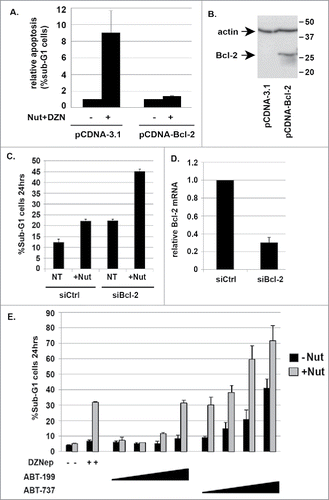
Next, we sought the mechanism how DZNep reduces Bcl-2 expression. The fact that DZNep reduces Bcl-2 mRNA suggested the reduction is at the level of transcription. To test this, we made use of two different reporter constructs in which portions of the Bcl-2 gene promoter are fused to luciferase (). The construct LB355 contains the Bcl-2 promoter fragment from position -1218 to the ATG start codon fused to luciferase, and construct LB556 contains the Bcl-2 promoter fragment from position −170 to the ATG start codon fused to luciferase. We transfected HCT116 with these reporter constructs and then monitored luciferase activity in the cells treated with Nutlin, DZNep, or both. As shown in , luciferase activity from construct LB355 was reduced ∼60% by Nutlin treatment and by ∼80-85% by DZNep or DZNep plus Nutlin. Construct LB556 had lower basal luciferase activity than did LB355 (). Notably, Nutlin treatment reduced luciferase activity from LB556 by ∼50% and this was not reduced further by Nutlin plus DZNep, and DZNep by itself did not reduce luciferase activity from LB556. The results indicate that the promoter region through which Nutlin/p53 represses Bcl-2 gene expression is located in the first 170 nucleotides upstream of the ATG, which is consistent with published results.Citation28 In contrast, the promoter region through which DZNep represses Bcl-2 gene expression is located 5′ of the ATG between positions −1218 and −170. We conclude DZNep reduces Bcl-2 expression at least in part by inhibiting Bcl-2 promoter activity and Bcl-2 gene transcription.
Figure 4. DZNep reduces activity of the Bcl-2 gene promoter. A) Schematic of reporter constructs in which different regions of the Bcl-2 gene promoter are fused upstream of luciferase. B) HCT116 cells were transfected with the indicated luciferase reporter constructs. 24 hrs after transfection the cells were either untreated or treated with Nutlin and/or DZNep as indicated. Luciferase activity in cell lysates was monitored 24 hrs later. Luciferase activity in untreated cells transfected with X construct was given the value “1.0”. Relative luciferase activity is plotted from a triplicate experiment (+/− s.e.m.).
Other colon cancer cell lines are resistant to apoptosis in response to Nutlin plus DZNep due to heightened expression of Bcl-2 family proteins
Next we asked if DZNep could sensitize other colon cancer cell lines to Nutlin-induced apoptosis. For this, we used RKO cells, LoVo cells, and SW48 cells. Each of these cell lines, like HCT116, express wild-type p53 and are mismatch repair deficient. We treated these cell lines with Nutlin and/or DZNep and monitored apoptosis by the percentage of cells with sub-G1 DNA content. As shown in , HCT116 cells again underwent apoptosis when co-treated with Nutlin plus DZNep, but the other cell lines did not. Immunoblotting showed p53 was induced to high levels in each of the cell lines treated with Nutlin or Nutlin plus DZNep (). Thus, failure to induce apoptosis in these cells was not due to a lack of p53 induction. Next we compared Bcl-2 expression in the cell lines (). The results showed that LoVo cells express much higher levels of Bcl-2 protein than HCT116, and SW48 and RKO also express higher Bcl-2 levels than HCT116 cells. Finally, we monitored Bcl-2 protein expression in each of the cell lines treated with Nutlin and/or DZNep. As shown in , Bcl-2 was again reduced in HCT116 cells treated with DZNep and further reduced in HCT116 cells treated with DZNep plus Nutlin. Nutlin alone caused little or no reduction in Bcl-2 protein in HCT116 in this experiment. Bcl-2 protein levels were also reduced by co-treatment with Nutlin plus DZNep in LoVo and RKO cells (). However, Bcl-2 protein was not reduced in LoVo cells treated with DZNep alone even though studies in showed a reduction in Bcl-2 mRNA. This suggests post-transcriptional mechanisms may be active in DZNep-treated LoVo cells that maintain Bcl-2 protein expression. In SW48 cells we observed that Bcl-2 protein was reduced by treatment with DZNep or Nutlin plus DZNep, but was not reduced by Nutlin alone ().
Figure 5. DZNep does not sensitize LoVo, RKO, and SW48 cells to Nutlin-induced apoptosis. A) LoVo, RKO, and SW48 cells were treated with Nutlin (10 μM) and/or DZNep (10 μM) as indicated for 48 hrs. Apoptosis was determined by the percentage of cells with sub-G1 DNA content. B) p53 protein levels were determined by immunoblotting in LoVo, RKO, and SW48 cells treated with Nutlin (10 μM) and/or DZNep (10 μM) for 24 hrs. Actin levels were used as a loading control. Numbers on the side indicate the position of molecular weight markers in kilodaltons. C) Bcl-2 levels were determined by immunoblotting in LoVo, RKO, and SW48 cells treated with Nutlin (10 μM) and/or DZNep (10 μM) for 24 hrs. Actin was used as a loading control. Numbers on the side indicate the position of molecular weight markers in kilodaltons. D) Basal levels of Bcl-2 were compared between the cell lines by immunoblotting. Actin was used as a loading control. Numbers on the side indicate the position of molecular weight markers in kilodaltons.
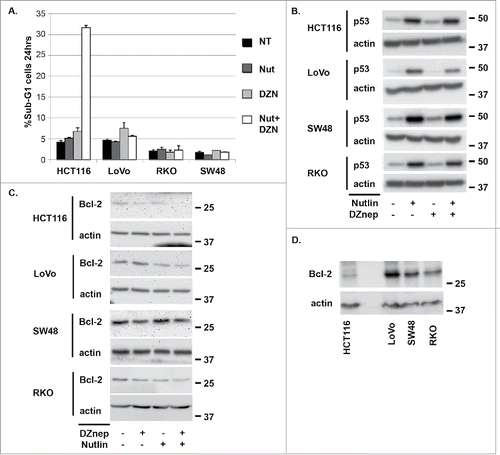
Figure 6. LoVo cells express relatively high Bcl-2 and Bcl-xL mRNA levels. ABT-199 and ABT-737 sensitize LoVo cells to Nutlin-induced apoptosis. A) mRNA levels for the different Bcl-2 family members were determined in LoVo and HCT116 cells treated with Nutlin (10 μM) and/or DZNep (10 μM) for 24 hrs. For each gene, the mRNA level in untreated HCT116 cells is given the value “1.0”, and everything else is plotted relative to that. B) LoVo cells were treated for 24 hrs with Nutlin alone (10 μM) or in combination with increasing amounts (1, 2.5, 5, and 10 μM) of either ABT-199 or ABT-737. Apoptosis was determined by the percentage of cells with sub-G1 DNA content.
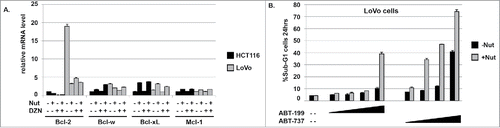
Bcl-2 was expressed basally at markedly higher levels in LoVo, SW48, and RKO cells compared to HCT116 cells (). Thus, while DZNep and/or Nutlin partially reduced Bcl-2 expression in these cells the levels of Bcl-2 still remained higher than in untreated HCT116. Based on this we speculated that continued, relatively high expression of Bcl-2 may protect LoVo, SW48, and RKO cells from apoptosis in response to Nutlin and DZNep. We compared mRNA expression of Bcl-2 as well as other anti-apoptotic Bcl-2 family members (Bcl-w, Bcl-xL, Mcl-1) in each of the cell lines treated with Nutlin and/or DZNep. As shown in , basal Bcl-2 mRNA expression was nearly 20-fold higher in LoVo cells compared to HCT116, which is consistent with Bcl-2 protein level differences between these cells. Basal Bcl-xL expression was ∼3-fold higher in LoVo compared to HCT116 cells. Bcl-2 mRNA was reduced by Nutlin and DZNep treatment in LoVo cells (); however, Bcl-2 mRNA levels remained 3–4 fold higher in Nutlin and/or DZNep treated LoVo cells compared to HCT116. We speculated this high expression of Bcl-2 might continue to protect LoVo cells from apoptosis even in the presence of DZNep. To test the role of Bcl-2 in apoptosis protection, we treated LoVo cells with the Bcl-2-specific inhibitor ABT-199 or the pan-Bcl-2-family inhibitor ABT-737. ABT-199 sensitized LoVo cells to Nutlin-induced apoptosis (), supporting the idea that high Bcl-2 expression protects these cells from apoptosis. ABT-737 caused an even greater sensitization to Nutlin-induced apoptosis than did ABT-199, suggesting elevated expression of Bcl-xL may also reduce apoptosis in LoVo cells treated with Nutlin.
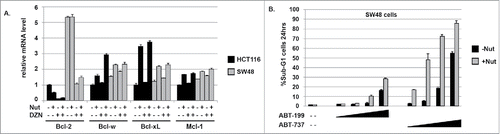
In SW48 cells, basal Bcl-2 mRNA expression was ∼5-fold higher than in HCT116 cells (), which is also consistent Bcl-2 protein level differences between these cells (). DZNep treatment reduced Bcl-2 mRNA in SW48 cells to approximately the same level as in untreated HCT116 cells (). We used ABT-199 and ABT-737 to ask if Bcl-2 or Bcl-2-family members protect the cells from Nutlin-induced apoptosis. As shown in , ABT-199 sensitized SW48 cells to Nutlin-induced apoptosis, supporting the idea that high Bcl-2 expression protects these cells from apoptosis. ABT-737 caused an even greater sensitization to Nutlin-induced apoptosis than did ABT-199, suggesting Bcl-w and/or Bcl-xL may also reduce apoptosis in SW48 cells treated with Nutlin.
Figure 7. SW48 cells express relatively high Bcl-2 and Bcl-w mRNA levels. ABT-199 and ABT-737 sensitized SW48 cells to Nutlin-induced apoptosis. A) mRNA levels for the different Bcl-2 family members were determined in SW48 and HCT116 cells treated with Nutlin (10 μM) and/or DZNep (10 μM) for 24 hrs. For each gene, the mRNA level in untreated HCT116 cells is given the value “1.0”, and everything else is plotted relative to that. B) SW48 cells were treated for 24 hrs with Nutlin alone (10 μM) or in combination with increasing amounts (1, 2.5, 5, and 10 μM) of either ABT-199 or ABT-737. Apoptosis was determined by the percentage of cells with sub-G1 DNA content.
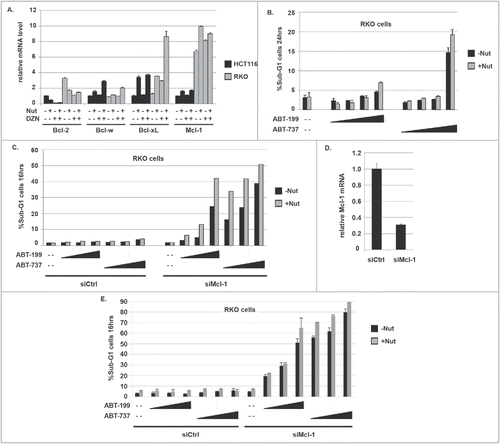
In RKO cells, basal Bcl-2 mRNA expression was ∼3-fold higher than in HCT116 cells (), consistent with differences in Bcl-2 protein expression between these cells (). Mcl-1 expression was ∼6-fold higher in RKO than in HCT116 cells (). Bcl-2 expression was reduced in RKO cells treated with Nutlin and/or DZNep, but remained at a slightly higher level than in untreated HCT116 cells. Further, Bcl-xL expression was induced ∼8-fold in RKO cells treated with Nutlin plus DZNep (). We speculated the lack of apoptosis in Nutlin plus DZNep treated RKO cells could be due to the continued expression of Bcl-2 and/or increased expression of MCL-1 or Bcl-xL. Mcl-1 is an anti-apoptotic Bcl-2 family member but is not targeted by ABT-199 or ABT-737. As shown in , neither ABT-199 nor ABT-737 could sensitize RKO cells to Nutlin-induced apoptosis. To ask if Mcl-1 was responsible for this protection from apoptosis, we inhibited Mcl-1 expression by siRNA in RKO cells and then treated the cells with Nutlin, ABT-199, and ABT-737 in different combinations. As shown in , Mcl-1 knockdown alone did not sensitize RKO cells to apoptosis by Nutlin. Importantly, however, Mcl-1 knockdown in combination with either ABT-199 or ABT-737 caused pronounced apoptosis in RKO cells treated with Nutlin. We repeated this experiment and the results again showed that Nutlin increased apoptosis in ABT-199 and ABT-737 treated cells transfected with Mcl-1 siRNA, but not in cells transfected with control siRNA (). qRT-PCR analysis showed Mcl-1 expression was reduced by Mcl-1 siRNA (). Based on these data we conclude Mcl-1 in combination with Bcl-2 (and potentially Bcl-w and/or Bcl-xL) protects RKO cells from Nutlin-induced apoptosis.
Figure 8. RKO cells express relatively high Bcl-2 and MCL-1 mRNA levels. MCL-1 knockdown sensitizes RKO cells to apoptosis in response to Nutlin and ABT-199 or Nutlin and ABT-737. A) mRNA levels for the different Bcl-2 family members were determined in RKO and HCT116 cells treated with Nutlin (10 μM) and/or DZNep (10 μM) for 24 hrs. For each gene, the mRNA level in untreated HCT116 cells is given the value “1.0”, and everything else is plotted relative to that. B) RKO cells were treated for 24 hrs with Nutlin alone (10 μM) or in combination with increasing amounts (1, 2, 5, and 10 μM) of either ABT-199 or ABT-737. Apoptosis was determined by the percentage of cells with sub-G1 DNA content. C) RKO cells were transfected with control siRNA (siCtrl) or siRNA against MCL-1 (siMCL-1). 24 hrs after transfection the cells were treated with Nutlin alone (10 μM) or in combination with increasing amounts (1, 2, and 5 μM) of either ABT-199 or ABT-737. Apoptosis was determined 16 hrs later by the percentage of cells with sub-G1 DNA content. D) qRT-PCR analysis shows MCL-1 mRNA is reduced in RKO cells from transfected with siRNA against MCL-1 compared to cells transfected with control siRNA. E) RKO cells were transfected with control siRNA (siCtrl) or siRNA against MCL-1 (siMCL-1). 24 hrs after transfection the cells were treated with Nutlin alone (10 μM) or in combination with increasing amounts (1, 2, and 5 μM) of either ABT-199 or ABT-737. Apoptosis was determined 16 hrs later by the percentage of cells with sub-G1 DNA content. Results from an experiment done in duplicate are shown (+/− s.e.m.).
Discussion
Factors that control Nutlin-induced apoptosis are not fully understood. In this report we found co-treatment with the histone methyltransferase inhibitor DZNep sensitized HCT116 colon cancer cells to Nutlin-induced apoptosis. This sensitization coincided with a pronounced decrease in Bcl-2 gene expression. Bcl-2 overexpression reduced apoptosis in Nutlin plus DZNep treated HCT116 cells, and Bcl-2 knockdown or treatment with the Bcl-2-specific inhibitor ABT-199 also sensitized HCT116 cells to Nutlin-induced apoptosis and to nearly the same extent as did DZNep. These data indicate the ability of DZNep to sensitize HCT116 cells resulted, at least in part, from reduced Bcl-2 expression. DZNep also reduced expression of Bcl-2 in other colon cancer cell lines but failed to sensitize them to Nutlin-induced apoptosis. We found that these colon cancer cell lines expressed exceedingly high levels of Bcl-2 or other anti-apoptotic Bcl-2 family members (Bcl-w, Bcl-xL, Mcl-1), either basally or in response to Nutlin and DZNep. Knockdown of individual factors and/or treatment with specific Bcl-2 family inhibitors sensitized these otherwise resistant cells to Nutlin-induced apoptosis. These results indicate anti-apoptotic Bcl-2-family members can protect cells from Nutlin-induced apoptosis, and that agents such as DZNep or Bcl-2 family inhibitors could increase the therapeutic potential of MDM2 antagonists like Nutlin and its derivatives.
Using luciferase reporter constructs, we found that DZNep represses transcription from the Bcl-2 gene promoter. We localized the DZNep responsive region between residues -170 to -1218 relative to the Bcl-2 start codon, which is different from the p53-responsive element located between position -170 and the ATG start codon. The mechanism how DZNep represses Bcl-2 promoter activity is at present unknown. DZNep was originally reported to inhibit the Polycomb repressor complex 2 and reduce histone methylations associated transcriptional repression, including histone H3 tri-methylation at lysine-27 (H3K27me3) and histone H4 tri-methylation at lysine-20 (H4K20me3).Citation29 However, later studies showed DZNep can inhibit multiple histone methylations, including methylations associated with transcriptional activation such as H3K4me3.14 This ability of DZNep to act as a global histone methylation inhibitor is most likely due to its ability to inhibit S-adenosylhomocysteine hydrolase, which leads to the indirect inhibition of S-adenosyl-methionine dependent reactions, including those carried out by many methyltransferases.Citation30,Citation31 Thus, one possibility is that DZNep reduces the level of H3K4me3 or other activating histone methylations in the Bcl-2 promoter between positions -170 and -1218, leading to inhibition of the promoter. Alternatively, DZNep could reduce Bcl-2 promoter activity indirectly by reducing expression of one or transcription factors that normally bind the Bcl-2 promoter between -170 and -1218 and promote Bcl-2 expression.
Apoptosis is controlled by the balance of pro- and anti-apoptotic Bcl-2-family protein expression. Bax and bak form pores in the outer mitochondrial membrane, allowing release of factors like cytochrome-c that trigger apoptosis. BH3-only proteins like Puma and Noxa can promote apoptosis directly by binding and activating pore formation by bax and bak, or indirectly by binding and sequestering anti-apoptotic Bcl-2-family members. Bcl-2 (as well as Bcl-xL, Bcl-w, and Mcl-1) can inhibit apoptosis by binding bax and/or bak and preventing mitochondrial pore formation, and by binding and inhibiting the activity of BH3-only proteins. High Bcl-2 expression tips the balance to favor survival whereas low or depleted Bcl-2 expression can tip the balance to favor apoptosis. Nutlin treatment increased Puma, Noxa, and bax expression in HCT116 cells and caused a partial reduction in Bcl-2, but did not cause apoptosis. In contrast, DZNep caused a pronounced decrease in Bcl-2 expression and, when combined with Nutlin, triggered apoptosis. Bcl-2 overexpression rescued HCT116 from apoptosis in response to Nutlin plus DZNep, and Bcl-2 knockdown by siRNA or treatment with the Bcl-2-specific inhibitor ABT-199 sensitized HCT116 cells to Nutlin-induced apoptosis to the same extent as did DZNep. These results indicate Bcl-2 protects HCT116 cells from Nutlin-induced apoptosis. We propose that DZNep promotes apoptosis at least in part by reducing Bcl-2 expression below a threshold needed for survival. This, in combination with increased expression of bax and/or BH3 only proteins such as Puma and Noxa in Nutlin-treated cells, triggers apoptosis. Notably, ABT-737 (which inhibits Bcl-2, Bcl-w, and Bcl-xL) caused a greater sensitization to Nutlin-induced apoptosis in HCT116 cells than did ABT-199. This suggests Bcl-w and/or Bcl-xL also contribute to the inhibition of apoptosis in HCT116 cells treated with Nutlin.
Our results demonstrate that in other colon cancer cell lines elevated expression of different Bcl-2 family members determine sensitivity to Nutlin-3a-induced apoptosis. LoVo cells expressed exceedingly high levels of Bcl-2 relative to HCT116 cells. DZNep reduced Bcl-2 expression in LoVo cells, however, the level of Bcl-2 expression in DZNep-treated LoVo cells remained at least 5-fold higher than in untreated HCT116 cells. While DZNep did not sensitize LoVo cells to apoptosis by Nutlin, the Bcl-2-specific inhibitor ABT-199 did. Based on this we conclude elevated Bcl-2 expression protects LoVo cells from Nutlin-induced apoptosis. RKO cells expressed elevated levels of Mcl-1 compared to HCT116 cells as well as elevated levels of Bcl-2 basally and in response to Nutlin. ABT-199 and ABT-737 did not sensitize RKO cells to Nutlin-induced apoptosis. Importantly, however, Mcl-1 knockdown in combination with ABT-199 or ABT-737 did sensitize RKO cells to apoptosis by Nutlin. Based on this we conclude Mcl-1 in combination with Bcl-2 (and potentially Bcl-w and Bcl-xL) protects RKO cells from Nutlin-induced apoptosis. SW48 cells expressed elevated Bcl-2 compared to HCT116 cells. ABT-199 sensitized SW48 to Nutlin-induced apoptosis and ABT-737 caused an even greater sensitization to Nutlin-induced apoptosis than did ABT-199. Based on this we conclude elevated expression of both Bcl-2 and possibly Bcl-w and/or Bcl-xL protect SW48 cells from apoptosis by Nutlin. In sum, the results indicate elevated expression of specific Bcl-2 family members protect colon cancer cells from apoptosis in response to Nutlin-3a.
MDM2 antagonists including derivatives of Nutlin-3a have entered clinical trials against p53 wild-type cancers, including cancers with relatively prevalent MDM2 gene amplifications (e.g. liposarcoma).Citation32,Citation33 While trials continue in hematologic cancers, MDM2 antagonists have not been successful against solid tumors due to dose limiting toxicities and other reasons not fully understood. Thus, identifying and then targeting factors that reduce MDM2 antagonist sensitivity could increase the therapeutic potential of MDM2 antagonists like Nutlin-3a and its derivatives. Our data indicate increased expression of various anti-apoptotic Bcl-2 family proteins block apoptosis in Nutlin-3a treated colon cancer cells, and that targeting these factors can increase apoptosis sensitivity. Based on this we envision screening tumors for relative expression of Bcl-2 family members and then using this information to direct treatments that include MDM2 antagonists and histone methyltransferase inhibitors (like DZNep) or one or more specific Bcl-2 family inhibitors.
Material and methods
Cell lines p53 wild-type SW48 cells (described in reference,Citation34 Sur et al, PNAS, 106:3964-9, 2009) and p53 wild-type and p53-null HCT116 cells (described in reference,Citation35 Bunz et al, Science 282:1497-1501, 1998) were obtained from Dr Bert Vogelstein (Johns Hopkins University). RKO and LoVo cells were purchased from ATCC. HCT116, SW48, and RKO cells were grown in McCoys 5A medium and LoVo cells were grown in DMEM medium. Both McCoys 5A and DMEM medium were supplemented with 10% fetal bovine serum (FBS), penicillin (100 U/mL) and streptomycin (100 μg/mL).
Transfection Transfections were carried out using Fugene-6 purchased from Promega according to the manufacturers instructions. HCT116 were plated in 35 mm2 dishes and transfected when they were ∼30% confluent. The cells were transfected with 2 micrograms of either pCDNA-3.1 plasmid or the Bcl-2 expression plasmid which was purchased from Addgene (Addgene catalog number 8768).
Drug treatments and siRNA knockdown Cells were plated 24 hours before being treated. Nutlin-3a and ABT-199 were obtained from Cayman Chemical (Ann Arbor, Michigan). DZNep was purchased from EMD Millipore and dissolved in DMSO. ABT-737 was purchased from Selleckchem and dissolved in DMSO. Bcl-2 (ON-TARGETplus SMARTpool), Mcl-1 (ON-TARGETplus SMARTpool), and control (ON-TARGETplus Non-targeting #1) siRNA were purchased from GE Dharmacon (Lafayette, CO). 100 nmol of each siRNA were transfected using Dharmafect I reagent following manufacturer's guidelines.
Immunoblotting For p53 and cleaved caspase-3 analysis, whole cell extracts were prepared by resuspending cell pellets in lysis buffer (150 mM NaCl, 5 mM EDTA, 0.5% Nonidet P-40, 50 mM Tris, pH 7.5), resolved by SDS-PAGE, and transferred to polyvinylidene difluoride membranes (Thermo Fisher). Cleaved Caspase-3 (#9664, 1:1,000 dilution) antibody was purchased from Cell Signaling (Danvers, MA). Antibodies to β-Actin (C4), p53 (DO-1), and Bcl-2 (C-20) were from Santa Cruz Biotechnology and were used at a 1:500 dilution. For detection of Bcl-2 we used the method described in Jiang and Milner (21). Briefly, cells were trypsinized and then washed in PBS, and an aliquot removed for cell counting. The remaining cells were lysed in 100 μl lysis buffer (150 mM NaCl, 0.5% Nonidet P-40, 50 mM Tris pH 8.0) on ice for 30 min. Samples were then diluted 1:1 in 4X Laemlli's buffer (200 mM Tris-HCl pH 6.8, 400 mM DTT, 8% SDS, 0.4% Bromophenol Blue, 50% Glycerol). Proteins from an equivalent number of cells were resolved by SDS-PAGE as described above, transferred to PVDF membranes, and probed with anti-Bcl-2 antibody. Primary antibodies were detected with goat anti-mouse (1:10,000 dilution, Thermo Fisher) or goat anti-rabbit (1:2,000 dilution, Cell Signaling) secondary antibodies conjugated to horseradish peroxidase, using Immobilon™ Western Chemiluminescent HRP Substrate (EMD Millipore).
qPCR analysis Total RNA were prepared using Total RNA mini kit (IBI Scientific, IA); the first cDNA strand was synthesized using High Capacity cDNA Reverse Transcription kit (Applied Biosystems, CA) following manufacturer's protocol. Quantitative real-time PCR (qRT-PCR) was performed to measure mRNA levels for PUMA, NOXA, BAX, P21, MDM2, BCL2, Bcl-w, Bcl-xL, MCL1 and β-Actin. The quantitative real-time PCR reaction was run in a QuantStudio 3 Real-Time PCR System (Applied Biosystems, Foster, CA) using EvaGreen qPCR master mix (Midwest Scientific) following manufacturer's instructions. Reaction was done in a final volume of 20 μL containing 2 μL of cDNA and 400 nmol/L of primers. All samples were amplified in triplicate using the following cycle scheme: 95°C for 2 minutes, 40 cycles of 95°C for 15 seconds and 60°C for 60 seconds. A single peak was obtained for targets, supporting the specificity of the reaction. Relative mRNA expression was determined using the ΔΔCT method using β-Actin to normalize. Sequences of primers used for qRT-PCR are available on request.
Luciferase Assays and transfection Reporter constructs LB355 and LB556 contain Bcl-2 promoter fragments fused upstream of luciferase and were purchased from Addgene. The Renilla Luciferase control vector FUIntroRW was obtained from Dr. Jitesh Pratap (Rush University Medical Center). HCT116 cells were plated in a 96-well plate 24 hrs prior to transfection. The cells were then transfected using FuGene transfection reagent following manufacturer's protocol. Briefly, 1.3 µg of DNA was placed in 58 µl total volume of transfection medium (McCoy's 5A w/o FBS, w/o PS) to prepare 0.020 µg/µl plasmid solution in a microfuge tube. 7.9 µl of FuGENE® HD reagent (Reagent:DNA ratio 6:1) was then added. Contents were mixed carefully by pipetting up and down 15 times, and incubated for 5 – 10 minutes at room temperature. During this incubation period, medium from the cells was aspirated and replaced with 95 µl fresh McCoy's 5A medium. 5.0 µl of the Fugene:DNA mixture was then added to each well. The following day the cells were treated with Nutlin and/or DZNep for 24 hrs as indicated in the text. Luciferase activity was determined using the Dual-Luciferase® Reporter Assay System (Promega, Madison, WI) according to manufacturer's protocol.
Flow cytometry analysis For apoptosis (% sub-G1 cells) and cell cycle analysis, cells were harvested and fixed in 25% ethanol overnight. The cells were then stained with propidium iodide (25 μg/ml; Calbiochem). Flow cytometry analysis was performed on Gallios™ flow cytometer (Beckman Coulter) and analyzed with FlowJo V10 (Treestar, Inc). For each sample, 10,000 events were collected.
Author Contributions
Y.Z. conducted the experiments for and prepared the figures.
R.E.P. conducted the experiments in and .
L.D. contributed to design of the experiments, interpretation of the data, and writing the manuscript.
C.G.M. devised the study, contributed to the design of experiments and data interpretation, and wrote the initial draft.
All authors proofread the manuscript and contributed to the final submitted manuscript.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by National Cancer Institute grant 1 R21 CA185036-01A1 (to C.G.M) and by a grant from the Swim Across America Foundation (to C.G.M).
Additional information
Funding
References
- Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387(6630):296–9. doi:10.1038/387296a0. PMID:9153395.
- Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387(6630):299–303. doi:10.1038/387299a0. PMID:9153396.
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303(5659):844–8. doi:10.1126/science.1092472. PMID:14704432.
- Selivanova, G. Wild type p53 reactivation: From lab bench to clinic. FEBS Lett. 2014;588(16):2628–38. doi:10.1016/j.febslet.2014.03.049. PMID:24726725.
- Tovar C, Rosinski J, Filipovic Z, Higgins B, Kolinsky K, Hilton H, Zhao X, Vu BT, Qing W, Packman K, et al. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: implications for therapy. Proc Natl Acad Sci. 2006;103(6):1888–93. doi:10.1073/pnas.0507493103. PMID:16443686.
- Kitagawa M, Aonuma M, Lee SH, Fukutake S, McCormick F. E2F-1 transcriptional activity is a critical determinant of Mdm2 antagonist-induced apoptosis in human tumor cell lines. Oncogene. 2008;27(40):5303–14. doi:10.1038/onc.2008.164. PMID:18521084.
- Kojima K, Konopleva M, Samudio IJ, Shikami M, Cabreira-Hansen M, McQueen T, Ruvolo V, Tsao T, Zeng Z, Vassilev LT, et al. MDM2 antagonists induce p53-dependent apoptosis in AML: implications for leukemia therapy. Blood. 2005;106(9):3150–9. doi:10.1182/blood-2005-02-0553. PMID:16014563.
- Shen H, Moran DM, Maki CG. Transient nutlin-3a treatment promotes endoreduplication and the generation of therapy-resistant tetraploid cells. Cancer Res. 2008;68(20):8260–8. doi:10.1158/0008-5472.CAN-08-1901. PMID:18922897.
- Huang B, Deo D, Xia M, Vassilev LT. Pharmacologic p53 activation blocks cell cycle progression but fails to induce senescence in epithelial cancer cells. Mol Cancer Res. 2009;7(9):1497–509. doi:10.1158/1541-7786.MCR-09-0144. PMID:19737973.
- Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nature Rev Genet. 2012;13(5):343–57. doi:10.1038/nrg3173. PMID:22473383.
- Yi X, Xiang XJ, Li XY, Jiang DS. Histone methyltransferases: novel targets for tumor and developmental defects. Am J Transl Res. 2015;7(11):2159–75. PMID:26807165.
- Black JC, Van Rechem C, Whetstine JR. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell. 2012;48(4):491–507. doi:10.1016/j.molcel.2012.11.006. PMID:23200123.
- Huang T, Lin C, Zhong LL, Zhao L, Zhang G, Lu A, Wu J, Bian Z. Targeting histone methylation for colorectal cancer. Therap Adv Gastroenterol. 2017;10(1):114–131. doi:10.1177/1756283X16671287. PMID:28286564.
- Miranda TB, Cortez CC, Yoo CB, Liang G, Abe M, Kelly TK, Marquez VE, Jones PA. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol Cancer Ther. 2009;8(6):1579–88. doi:10.1158/1535-7163.MCT-09-0013. PMID:19509260.
- Cheng LL, Itahana Y, Lei ZD, Chia NY, Wu Y, Yu Y, Zhang SL, Thike AA, Pandey A, Rozen S, et al. TP53 genomic status regulates sensitivity of gastric cancer cells to the histone methylation inhibitor 3-deazaneplanocin (DZNep). Clin Cancer Res. 2012;18(15):4201–12. doi:10.1158/1078-0432.CCR-12-0036. PMID:22675170.
- Volkmann N, Marassi FM, Newmeyer DD, Hanein D. The rheostat in the membrane: BCL-1 family proteins and apoptosis. Cell Death Diff. 2014;21(2):206–15. doi:10.1038/cdd.2013.153..
- Gillies LA, Kuwana T. Apoptosis regulation at the mitochondrial outer membrane. J Cell Biochem. 2014;115(4):632–40. doi:10.1002/jcb.24709. PMID:24453042.
- Ow Y-LP, Green DR, Hao Z, Mak TW. Cytochrome c: functions beyond respiration. Nat Rev Mol Cell Biol. 2008;9(7):532–42. doi:10.1038/nrm2434. PMID:18568041.
- Hata AN, Engelman JA, Faber AC. The BCL2 family: key mediators of the apoptotic response to targeted anticancer therapeutics. Cancer Discov. 2015;5(5):475–87. doi:10.1158/2159-8290.CD-15-0011. PMID:25895919.
- Hemann MT, Lowe SW. The p53-Bcl-2 connection. Cell Death Diff. 2006;13(8):1256–9. doi:10.1038/sj.cdd.4401962..
- Jiang M, Milner J. Bcl-2 constitutively suppresses p53-dependent apoptosis in colorectal cancer cells. Genes Dev. 2003;17(7):832–7. doi:10.1101/gad.252603. PMID:12670866.
- Kelly PN, Strasser A. The role of Bcl-2 and its pro-survival relatives in tumorigenesis and cancer therapy. Cell Death Diff. 2011;18(9):1414–24. doi:10.1038/cdd.2011.17..
- Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19(2):202–8. doi:10.1038/nm.3048. PMID:23291630.
- Chen S, Dai Y, Harada H, Dent P, Grant S. Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax tranlocation. Cancer Res. 2007;67(2):782–91. doi:10.1158/0008-5472.CAN-06-3964. PMID:17234790.
- Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, Deng X, Zhai D, Shi YX, Sneed T, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10(5):375–88. doi:10.1016/j.ccr.2006.10.006. PMID:17097560.
- Wu Y, Mehew JW, Heckman CA, Arcinas M, Boxer LM. Negative regulation of Bcl-2 expression by p53 in hematopoietic cells. Oncogene. 2001;20(2):240–51. doi:10.1038/sj.onc.1204067. PMID:11313951.
- Haldar S, Negrini M, Monne M, Sabbioni S, Croce CM. Down-regulation of Bcl-2 by p53 in breast cancer cells. Cancer Res. 1994;54(8):2095–7. PMID:8174112.
- Miyashita T, Harigai M, Hanada M, Reed JC. Identification of a p53-dependent negative response element in the Bcl-2 gene. Cancer Res. 1994;54(12):3131–5. PMID:8205530.
- Tan J, Yang X, Zhuang L, Jiang X, Chen W, Lee PL, Karuturi RK, Tan PB, Liu ET, Yu Q. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007;21(9):1050–63. doi:10.1101/gad.1524107. PMID:17437993.
- Borchardt RT, Keller BT, Patel-Thombre U, Neplanocin A. A potent inhibitor of S-adenosylhomocysteine hydrolase and of vaccinia virus multiplication in mouse L929 cells. J Biol Chem. 1984;259(7):4353–8. PMID:6707008.
- Chiang PK. Biological effects of inhibitors of S-adenosylhomocysteine hydrolase. Pharmacol Ther. 1998;77(2):115–34. doi:10.1016/S0163-7258(97)00089-2. PMID:9578320.
- Ray-Coquard I, Blay JY, Italiano A, Le Cesne A, Penel N, Zhi J, Heil F, Rueger R, Graves B, Ding M, et al. Effect of the MDM2 antagonist RG7112 on the p53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. Lancet Oncol. 2012;13(11):1133–40. doi:10.1016/S1470-2045(12)70474-6. PMID:23084521.
- Burgess A, Chia KM, Haupt S, Thomas D, Haupt Y, Lim E. Clinical overview of MDM2/X-targeted therapies. Front Oncol. 2016;6(7):1–7. PMID:26858935
- Sur S, Pagliarini R, Bunz F, Rago C, Diaz LA, Jr, Kinzler KW, Vogelstein B, Papadopoulos N. A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53. Proc Natl Acad Sci USA. 2009;106(10):3964–9. doi:10.1073/pnas.0813333106. PMID:19225112.
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogselstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282(5393):1497–501. doi:10.1126/science.282.5393.1497. PMID:9822382.