
ABSTRACT
mTOR is an important therapeutic target in many types of cancers. In melanoma, the mTOR nonsynonymous mutation rate is up to 10.4%. However, mTOR inhibitors have shown limited effects in clinical trials of melanoma. Because mTOR mutations are distributed, not selecting patients with specific mTOR mutations may be the main reason for therapeutic failures. Our previous research found that mutations in the mTOR P2213S and S2215Y kinase domains resulted in gain-of-function and were sensitive to specific inhibitors. The purpose of this study was to test the effect of heterozygous/homozygous H2189Y mutations on downstream pathways and sensitivity to inhibitors. mTOR kinase activity was analyzed by western blot and a K-LISA™ mTOR activity kit. The sensitivity of melanoma cells to inhibitors was tested by a proliferation assay. The expression of downstream pathway proteins was also analyzed by western blot. The results showed that heterozygous/homozygous H2189Y mutations were gain-of-function. The heterozygous H2189Y mutation was sensitive to the AKT inhibitor, AZD5363, and the phosphoinositide 3-kinase inhibitor, LY294002. The homozygous H2189Y mutation was sensitive to the mTOR inhibitor, everolimus, and the AKT inhibitors AZD5363 and MK-2206 2HCL,and the phosphoinositide 3-kinase inhibitor, LY294002. These results indicated that homozygous mutations in the kinase domain have a greater effect on protein function than heterozygous mutations. The mTOR kinase domain may play an important role in mTOR kinase activity and may be the target of selective inhibitors. Our study can facilitate the selection of appropriate inhibitors for patients in clinical trials.
Introduction
Melanoma is an aggressive cancer with a low five-year survival rate. Globally, the incidence of melanoma is increasing.Citation1,Citation2 In recent years, targeted therapy has shown promising clinical efficacy in melanoma treatment. For example, BRAF, KIT, and MEK inhibitors performed better than dacarbazine in enhancing progression-free and overall survival for patients harboring specific mutations.Citation3-6 However, these validated targets cannot benefit all melanoma patients, and resistance to these inhibitors is common. This prompted an increasing number of preclinical studies and clinical trials seeking new targets for pathway inhibitors.
Mammalian target of rapamycin (mTOR) is an important serine/threonine protein kinase involved in transcriptional regulation, protein synthesis, metabolism, and apoptosis.Citation7-10 Therefore, mTOR has been considered as a key target for treating many cancers. mTOR inhibitors include rapamycin and a new generation of derivatives such as RAD001, temsirolimus, and ridaforolimus. Combined with FK-506-binding protein 12, mTOR inhibitors can inhibit mTOR activity and arrest tumor cells in the G1 phase, thus inhibiting tumor cell growth and causing apoptosis. However, mTOR inhibitors showed limited effects in clinical trials in melanoma.Citation11-13 Considering the complex structure of mTOR, not selecting patients with specific mTOR mutations may be the main reason for these therapeutic failures.
Multiple sequencing studies have shown that there are few driver genes in the tumor genome, and hot-spot mutations such as BRAF V600E are even less frequent.Citation14 The majority of driver gene mutations are distributed. For example, the incidence of KIT somatic mutations was 10.8% in Asia, of which 50% were distributed.Citation3 Although the coding sequence of the c-Kit gene is covered by 21 exons, patients with mutations in exons 11 and 13 have a better response to imatinib. This suggests that finding the functional mutation is meaningful in selecting appropriate therapy.
Our previous study showed that 10.4% of Asia melanoma patients harbored mTOR missense mutations. mTOR missense mutations in acral and mucosal melanoma are 11.0 and 14.3%, respectively. mTOR H1968Y and P2213S mutations are functional acquired mutations that are sensitive to specific inhibitors.Citation15 In the current study, we examined another mTOR mutation, H2189Y, and constructed stable heterozygous/homozygous HEK293T cells using transcription activator-like effector nucleases (TALENs). Downstream signaling pathway proteins expressing kinase activities in these cells, as well as their sensitivity to phosphoinositide 3-kinase (PI3K)-AKT-mTOR pathway inhibitors, were evaluated.
Results
Expression levels of mTOR pathway proteins
Western blotting was used to investigate the effects of mTOR H2189Y mutants on the expression of PI3K/AKT/mTOR pathway proteins. Wild-type HEK293T cells were used as the negative control. mTOR S2215Y and P2213S cells were used as heterozygous and homozygous positive controls.Citation15, Citation16
Heterozygous/homozygous H2189Y mutant cells had a significantly higher expression level of phospho-mTOR, phospho-p70 ribosomal protein S6 kinase (p70S6K), phospho-AKT, and phospho-4EBP1 than those in wild-type HEK293T cells (). Differences ranged from 4- to 16-fold. Compared with mTOR S2215Y and P2213S cells, the expression levels of phospho-mTOR, phospho-p70S6K, phospho-AKT, and phospho-4EBP1 were slightly lower in heterozygous/homozygous H2189Y mTOR mutants (). These results indicate that mTOR heterozygous/homozygous H2189Y mutations may be new gain-of-function mutations.
Figure 1. Protein Expression Levels of mTOR Mutations.HEK293T cells stably expressing mTOR mutants (H2189Y, P2213S and S2215Y) were constructed by TALEN. After nutrient starvation, wild type or mutated HEK293T cells were lysed.The phosphorylation levels of mTOR, p70S6K, AKT and 4EBP1 were examined by Western blot (A).Results in (B-E) were quantified by measuring the relative intensity of phosphorylated protein bands to corresponding total protein bands with the method of mean ± SD of 3 scans.
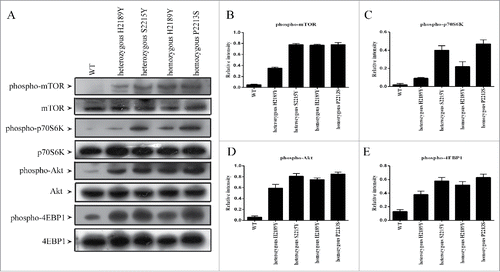
mTOR kinase activity of mTOR mutations
mTOR kinase analyses were performed in these cells using the K-LISA method. As illustrated in , heterozygous and homozygous H2189Y mutant cells all showed significantly higher mTOR kinase activities in comparison with wild-type cells, and lower mTOR kinase levels compared with their respective positive controls (). These results were consistent with the western blotting findings ().
Figure 2. mTOR kinase activity of mTOR mutants.HEK293T cells stably expressing mTOR mutants (H2189Y, P2213S and S2215Y) were constructed by TALEN. After nutrient starvation, wild type or mutated HEK293T cells were lysed, and the activation of mTOR kinase were examined by K-LISA™ mTOR (Recombinant) Activity Kit.
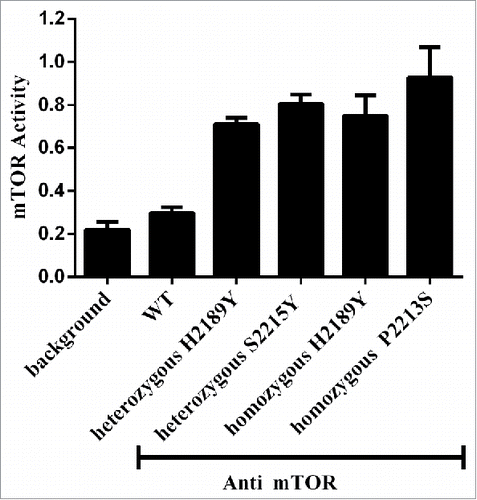
Sensitivity of mTOR mutants to PI3K-AKT-mTOR pathway inhibitors
To analyze the sensitivity of mTOR mutants to PI3K-AKT-mTOR pathway inhibitors (everolimus, AZD5363, MK-2206, LY294002, and Wortmannin), cell proliferation assays were performed. Heterozygous H2189Y mutant cells were sensitive to AKT (AZD5363, P = 0.015) and PI3K (LY294002, P = 0.047) inhibitors, compared with the negative control. Everolimus, MK-2206, and Wortmannin had no significant effect on proliferation.
Proliferation of homozygous H2189Y mutant cells was significantly inhibited by the mTOR inhibitor everolimus (P = 0.018), the PI3K inhibitor LY294002 (P = 0.016), and the AKT inhibitors AZD5363 (P = 0.009) and MK-2206 (P = 0.003). The PI3K inhibitor Wortmannin had no significant effect on proliferation in these five cell lines ().
Figure 3. Protein Expression Levels of mTOR mutations after PI3K-AKT-mTOR inhibitors treatment. HEK293T cells stably expressing mTOR mutants (H2189Y, P2213S and S2215Y) were constructed by TALEN. After nutrient starvation, wild type or mutated HEK293T cells were treated with indicated inhibitors or vehicle for 24 hours. The activation of indicated molecules was examined by Western blot.
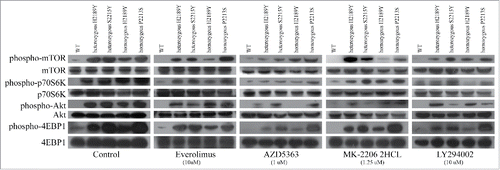
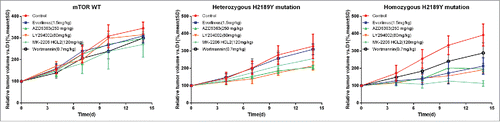
Phosphorylation levels of mTOR, p70S6K, AKT, and 4EBP1 were examined by western blot after drug treatments for 24 hours. For everolimus and MK-2206, phosphorylation levels of mTOR, p70S6K, and 4EBP1 induced by the homozygous H2189Y mutant were inhibited remarkably more than the other four cell lines (). For AZD5363, expression of phospho-mTOR, phospho-p70S6K, phospho-AKT, and phospho-4EBP1 in heterozygous H2189Y and P2213S cells, and homozygous H2189Y and S2215Y cells, was significantly decreased compared with the control group. For LY294002, phosphorylation levels of p70S6K, AKT, and 4EBP1 in homozygous H2189Y cells were significantly inhibited compared with the other groups. These expression levels were consistent with the cell proliferation results ().In order to verify the reliability of the experimental results in vitro, we conducted experiments in the nude mouse xenograft model.As shown in ,this results were consistent with cell experiments.
Figure 4. Sensitivity of gain-of-function mTOR mutations to PI3K-AKT-mTOR inhibitors. HEK293T cells stably expressing mTOR mutants (H2189Y, P2213S and S2215Y) were constructed by TALEN. After nutrient starvation, wild type or mutated HEK293T cells were treated with indicated inhibitors or vehicle.The proliferation of HEK293T cells was evaluated by CellTiter-Glo® Luminescent Cell Viability Assay, and the results were presented as mean ± SD of 3 independent experiments.
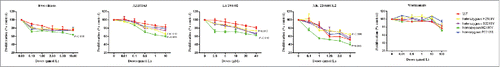
Figure 5. Sensitivity of xenograft models to inhibitors in vivo. When the tumor size reached approximately 200 mm3, mice were treated with buffer control or inhibitors daily. Tumor volume was evaluated as % of the tumor volume on day 0 and presented as mean ± SD. The data are representative of these independent experiments.
Discussion
In the era of precision medicine, finding and targeting the driver gene is an important approach to treat cancer. As a key molecule downstream of the PI3K/AKT pathway, mTOR makes an important impact on cell transcriptional regulation, protein synthesis, cell proliferation, and apoptosis.Citation8-10 In melanoma, the mTOR somatic nonsynonymous mutation rate is up to 10.4%.Citation15 Therefore, mTOR may be an important therapeutic target for melanoma.
To obtain stable phenotypes, the present study used TALENs technology to construct model cells carrying heterozygous/homozygous mTOR mutations. TALENs is a genomic editing technique that can introduce a stable single base mutation by changing the genetic code artificially.Citation17 Compared with transient transfection, TALENs operates at the DNA level which can better simulate the impact of mutations on the functions of signaling pathways, and the responses to different inhibitors. Thus, the current study differs from others in the choice of gene editing technique.Citation18
mTOR consists of several independent and conserved domains: HEAT, FAT, FRB, kinase, and FATCs.Citation19 The kinase domain is an important mTOR structure that mediates serine/threonine phosphorylation. mTOR mutants activated in the kinase domain contribute to apoptotic resistance, and might contribute to cellular transformation.Citation20 Construction of the S2215Y point mutation in the wild-type mTOR kinase domain could increase mTOR kinase activity and activate the p70S6K pathway.Citation16 Our previous research found that both P2213S and S2215Y mutations in the mTOR kinase domain resulted in gain-of-function and were sensitive to selective inhibitors. These findings indicated that the mTOR kinase domain may be a key target for melanoma treatment.
H2189Y is a new repeated missense mutation found in Chinese melanoma patients that is located in the mTOR kinase domain. In our study, mTOR kinase activity was significantly increased in H2189Y cells, indicating that H2189Y mutations located in the kinase domain result in gain-of-function. Cell proliferation and western blot assays showed that the effect of a homozygous H2189Y mutation on downstream pathways was more significant than that of the heterozygous H2189Y mutation. The heterozygous H2189Y mutation was only sensitive to AZD5363 and LY294002, which was similar to P2213S and S2215Y mutations.Citation15 However, homozygous H2189Y cells were sensitive to multiple inhibitors, including the mTOR inhibitor everolimus and the AKT inhibitor MK-2206. In contrast, previous mutations in the mTOR kinase domain were insensitive to everolimus. This is the first mutation in the kinase domain resulting in sensitivity to everolimus, pointing out the necessity of screening for specific mutations in preclinical studies.
mTOR has two distinct complexes, mTORC1 (mTOR-Raptor) and mTORC2 (mTOR-Rictor). mTORC2 plays an important role in the phosphorylation of AKT.Citation21 When PI3K or AKT are activated, mTORC1 expression is elevated. Activation of mTORC1 may phosphorylate the downstream molecule, p70S6K1, and eukaryotic initiation factor 4EBP1, which affect mRNA translation and protein synthesis. Current mTOR inhibitors affect mainly mTORC1 rather than mTORC2. Clinical trials of mTOR inhibitors might be limited by a lack of mTORC2 inhibition and selecting for sensitive mTOR mutations.Citation22 By integrating our findings with previous research, we found that mTOR H2189Y, P2213S, and S2215Y cells were all sensitive to the AKT inhibitor, AZD5363, and the PI3K inhibitor, LY294002. Homozygous H2189Y cells were sensitive to multiple inhibitors, including the mTOR inhibitor, everolimus. Thus, for some mTOR mutation patients, PI3K-AKT inhibitors may have a stronger effect than mTOR inhibitors. However, the use of mTOR inhibitors requires screening for specific mutations such as in the phase II clinical trial, NCT01960829.Citation23
In summary, our study provides evidence that the mTOR kinase domain may play an important role in mTOR kinase activity, and may be the target of selective inhibitors. The results of the study can facilitate the development of clinical trials by directing the selection of appropriate inhibitor(s) for each patient.
Materials and methods
Construction of single base mutation HEK293T cell lines
We selected the mTOR missense mutation, H2189Y, from DNA sequencing analysis results of Chinese melanoma patients.Citation15 Single-base mutation HEK293T cell lines were constructed using a single base substitution method mediated by TALENs. Briefly, a TALEN binding pair, TALEN-L and TALEN-R , were designed on both sides of the 6565 locus in the mTOR gene. A single-base mutation single-stranded oligodeoxynucleotide (from C to T) in locus 6565 was also designed. All three mutations were co-transfected into HEK293T cells using FuGENE HD and positive clones were chosen by culturing with puromycin. The positive clones were then proliferated and purified. After DNA sequencing analysis, heterozygous/homozygous single-base mutation 293T cell lines on the mTOR gene 6565 locus (from C to T, H2189Y) were obtained.
Cell culture and cell lysate preparation
HEK293T cells (catalog no. ACC-635) obtained from Leibniz Institute DSMZ were used as the negative control. Mutant S2215Y (mTOR c6644a)Citation16 and P2213S (mTOR c6637t) HEK293T cellsCitation15 were used as positive heterozygous and homozygous controls. All cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Invitrogen) containing 10% fetal bovine serum (FBS, Hyclone) and 1% penicillin/streptomycin (Invitrogen). When the proportion of adherent cells reached 70 to 80%, cells were partially serum-starved in DMEM containing 0.1% FBS and 1% penicillin/streptomycin for 15 h, and then completely serum-starved in Dulbecco's phosphate-buffered saline (Thermo Fisher Scientific) for 4 h. After that, cells were incubated with inhibitors or DMSO for 24 h.Cells were then collected and lysates prepared by adding pre-chilled PhosphoSafe Extraction Reagent (EMD Millipore). The cell lysates were used for western blot and mTOR kinase activity analyses.
Western blotting
Western blotting was performed with the prepared cell lysates and the following antibodies: anti-mTOR (Cat #ab134903, Abcam), anti-phospho-mTOR (Cat#ab109268, Abcam), anti-p70 S6 kinase (49D7) (Cat #2708, Cell Signaling Technology), anti-S6K1 (phospho T421+S424) (Cat #ab32525, Abcam), anti-AKT1 [Y89] (Cat #ab32505, Abcam), anti-AKT1 (phospho Ser473) (Cat #ab66134, Abcam), anti-4E (eIF4E) binding protein-1 (4EBP1) [Y330] (Cat #ab32130, Abcam), and anti-phospho-4EBP1 (Thr37/46) (Cat #2855, Cell Signaling Technology).
mTOR kinase activity analyses
The K-LISA™ mTOR (recombinant) activity kit (Cat #CBA104) was purchased from EMD Millipore. Cell lysates were prepared from mTOR wild-type and mutant HEK293T cells as described in the previous section. Cell lysates were used immediately to immunoprecipitate mTOR. mTOR in vitro kinase analyses were performed according to the protocol recommended by the manufacturer. Absorbance was measured at 450 nm.
PI3K-AKT-mTOR pathway inhibitors and cell proliferation assays
Everolimus (Cat #S1120), AZD5363 (Cat #S8019), MK-2206-2HCl (Cat #S1078), LY294002 (Cat #S1105), and Wortmannin Cat (#S2758) were purchased from Selleck Chemicals. Everolimus is an mTOR inhibitor. AZD5363 and MK-2206 are AKT inhibitors. LY294002 and Wortmannin are PI3K inhibitors. All inhibitors were dissolved in dimethyl sulfoxide and diluted with DMEM containing 0.1% FBS (Hyclone) to reach specific concentrations. Corresponding concentrations of dimethyl sulfoxide in DMEM supplemented with 0.1% FBS were used as controls.
mTOR wild-type or mutant HEK293T cells were seeded into 96-well plates at a density of 3 × 103 cells per well. Cells were cultured in DMEM containing 10% FBS and 1% penicillin/streptomycin for 24 h. Cells were then partially serum-starved in DMEM containing 0.1% FBS and 1% penicillin/streptomycin for 15 h, and then completely serum-starved in Dulbecco's phosphate-buffered saline (Thermo Fisher Scientific) for 4 h.
After that, cells were incubated with varying concentrations of inhibitors or DMSO for 24 h. Cell proliferation was evaluated using the CellTiter-Glo® Luminescent Cell Viability Assay (Promega) according to the manufacturer's instructions. All cell proliferation experiments were repeated three times.
Xenograft models and treatment
mTOR wild-type(HEK293T cells) or mutant HEK293T cells (heterozygous/homozygous H2189Y) were digested into single cell suspensions with more than 90% living cells.The cells were subcutaneouslys injected under the skin of the nude mice to establish the xenograft model.
When the tumor size reached approximately 200 mm3, mice were randomized (treatment arm versus control arm) and treated with control buffer or different inhibitors . For Everolimus treatment, mice received Everolimus (1.5 mg/kg) via oral gavage daily.Citation23 For AZD5363 treatment, mice received AZD5363 (250 mg/kg) via oral gavage daily.Citation24 For LY294002 treatment, mice received LY294002 (80 mg/kg) via intraperitoneal injection daily.Citation25 For MK-2206 HCL2 treatment, mice received MK-2206 HCL2 (120 mg/kg) via oral gavage daily.Citation26 For Wortmanmia treatment, mice received Wortmanmia (0.7 mg/kg) via intraperitoneal injection daily.Citation27 Tumor sizes were measured every 3 days and tumor volume calculated using the formula: volume = length*width2/2. The treatment lasted for 14 days. The above experiments were repeated twice.
Statistical analysis
Statistical analyses were performed using SPSS 20.0 software. All statistical analyses were two-sided, and P <0 .05 was considered as statistically significant.The differences in protein expression were analyzed by T test.The results of cell proliferation were analyzed by repeated measurements of variance.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by grants from the Major State Basic Research Development Program of China (2013CB911004), National Natural Science Foundation of China (81672696; 81772912), Beijing Municipal Natural Science Foundation (7152033), Beijing Baiqianwan Talents Project, Beijing Municipal Administration of Hospitals Clinical medicine Development of special funding support (ZYLX201603) and Beijing Municipal Science & Technology Commission (Z151100003915074).
Additional information
Funding
References
- Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J.Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–32. doi:10.3322/caac.21338. PMID:26808342.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi:10.3322/caac.21332. PMID:26742998.
- Guo J, Si L, Kong Y, Flaherty KT, Xu X, Zhu Y, Corless CL, Li L, Li H, Sheng X, et al. .Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol. 2011;29:2904–9. doi:10.1200/JCO.2010.33.9275. PMID:21690468.
- Carvajal RD, Antonescu CR, Wolchok JD, Chapman PB, Roman RA, Teitcher J, Panageas KS, Busam KJ, Chmielowski B, Lutzky J, et al. .KIT as a therapeutic target in metastatic melanoma. JAMA. 2011;305:2327–34. doi:10.1001/jama.2011.746. PMID:21642685.
- Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O'Dwyer PJ, Lee RJ, Grippo JF, Nolop K, et al. .Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–19. doi:10.1056/NEJMoa1002011. PMID:20818844.
- Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, Demidov LV, Hassel JC, Rutkowski P, Mohr P, et al. .Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367:107–14. doi:10.1056/NEJMoa1203421. PMID:22663011.
- Hay N, Sonenberg N .Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–45. doi:10.1101/gad.1212704. PMID:15314020.
- Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, Schreiber SL. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 1994;369:756–8. doi:10.1038/369756a0. PMID:8008069.
- Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell. 2000;103:253–62. doi:10.1016/S0092-8674(00)00117-3. PMID:11057898.
- Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441:424–30. doi:10.1038/nature04869. PMID:16724053.
- Dronca RS, Allred JB, Perez DG, Nevala WK, Lieser EA, Thompson M, Maples WJ, Creagan ET, Pockaj BA, Kaur JS, et al. .Phase II study of temozolomide (TMZ) and everolimus (RAD001) therapy for metastatic melanoma: a North Central Cancer Treatment Group study, N0675. Am J Clin Oncol. 2014;37:369–76. doi:10.1097/COC.0b013e31827b45d4. PMID:23357973.
- Margolin K, Longmate J, Baratta T, Synold T, Christensen S, Weber J, Gajewski T, Quirt I, Doroshow JH .CCI-779 in metastatic melanoma: a phase II trial of the California Cancer Consortium. Cancer. 2005;104:1045–8. doi:10.1002/cncr.21265. PMID:16007689.
- Rao RD WHE, Allred JB LVJ, Maples WJ GMK. Phase II trial of the mTOR inhibitor everolimus (RAD-001) in metastatic melanoma. J Clin Oncol. 2006;24(18_suppl):8043–43, Abs8043.
- Niu B, Scott AD, Sengupta S, Bailey MH, Batra P, Ning J, Wyczalkowski MA, Liang WW, Zhang Q, McLellan MD, et al. .Protein-structure-guided discovery of functional mutations across 19 cancer types. Nat Genet. 2016;48:827–37. doi:10.1038/ng.3586. PMID:27294619.
- Kong Y, Si L, Li Y, Wu X, Xu X, Dai J, Tang H, Ma M, Chi Z, Sheng X, et al. .Analysis of mTOR Gene Aberrations in Melanoma Patients and Evaluation of Their Sensitivity to PI3K-AKT-mTOR Pathway Inhibitors. Clin Cancer Res. 2016;22:1018–27. doi:10.1158/1078-0432.CCR-15-1110. PMID:26490311.
- Sato T, Nakashima A, Guo L, Coffman K, Tamanoi F. Single amino-acid changes that confer constitutive activation of mTOR are discovered in human cancer. Oncogene. 2010;29:2746–52. doi:10.1038/onc.2010.28. PMID:20190810.
- Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, Baller JA, Somia NV, Bogdanove AJ, Voytas DF. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011;39:e82. doi:10.1093/nar/gkr218. PMID:21493687.
- Murugan AK, Alzahrani A, Xing M. Mutations in critical domains confer the human mTOR gene strong tumorigenicity. J Biol Chem. 2013;288:6511–21. doi:10.1074/jbc.M112.399485. PMID:23322780.
- Tee AR, Blenis J, Proud CG. Analysis of mTOR signaling by the small G-proteins, Rheb and RhebL1. FEBS Lett. 2005;579:4763–8. doi:10.1016/j.febslet.2005.07.054. PMID:16098514.
- Sekulić A, Hudson CC, Homme JL, Yin P, Otterness DM, Karnitz LM, Abraham RT. A direct linkage between the phosphoinositide 3-kinase-AKT signaling pathway and the mammalian target of rapamycin in mitogen-stimulated and transformed cells. Cancer Res. 2000;60:3504–13. PMID:10910062.
- Bellacosa A, Chan TO, Ahmed NN, Datta K, Malstrom S, Stokoe D, McCormick F, Feng J, Tsichlis P. Akt activation by growth factors is a multiple-step process: the role of the PH domain. Oncogene 1998;17:313–25. doi:10.1038/sj.onc.1201947. PMID:9690513.
- Strickland LR, Pal HC, Elmets CA, Afaq F. Targeting drivers of melanoma with synthetic small molecules and phytochemicals. Cancer Lett. 2015;359:20–35. doi:10.1016/j.canlet.2015.01.016. PMID:25597784.
- https://clinicaltrials.gov/ct2/show/NCT01960829?term=NCT01960829&rank=1.
- Kurdi A, De Doncker M, Leloup A, Neels H, Timmermans JP, Lemmens K, Apers S, De Meyer GRY, Martinet W. Continuous administration of the mTORC1 inhibitor everolimus induces tolerance and decreases autophagy in mice. Br J Pharmacol. 2016;173:3359–71. doi:10.1111/bph.13626. PMID:27638766.
- Davies BR, Greenwood H, Dudley P, Crafter C, Yu DH, Zhang J, Li J, Gao B, Ji Q, Maynard J, et al. .Preclinical pharmacology of AZD5363, an inhibitor of AKT: pharmacodynamics, antitumor activity, and correlation of monotherapy activity with genetic background. Mol Cancer Ther. 2012;11:873–87. doi:10.1158/1535-7163.MCT-11-0824-T. PMID:22294718.
- Hu L, Zaloudek C, Mills GB, Gray J, Jaffe RB. In vivo and in vitro ovarian carcinoma growth inhibition by a phosphatidylinositol 3-kinase inhibitor (LY294002). Clin Cancer Res. 2000;6:880–6. PMID:10741711.
- Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, Ueno Y, Hatch H, Majumder PK, Pan BS, et al. .MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther. 2010;9:1956–67. doi:10.1158/1535-7163.MCT-09-1012. PMID:20571069.
- Ng SS, Tsao MS, Nicklee T, Hedley DW. Wortmannin inhibits pkb/akt phosphorylation and promotes gemcitabine antitumor activity in orthotopic human pancreatic cancer xenografts in immunodeficient mice. Clin Cancer Res. 2001;7:3269–75. PMID:11595724.