
ABSTRACT
The irreversible ERBB1/2/4 inhibitor neratinib has been shown to rapidly down-regulate the expression of ERBB1/2/4 as well as the levels of c-MET, PDGFRα and mutant RAS proteins via autophagic degradation. Neratinib interacted in an additive to synergistic fashion with the approved PARP1 inhibitor niraparib to kill ovarian cancer cells. Neratinib and niraparib caused the ATM-dependent activation of AMPK which in turn was required to cause mTOR inactivation, ULK-1 activation and ATG13 phosphorylation. The drug combination initially increased autophagosome levels followed later by autolysosome levels. Preventing autophagosome formation by expressing activated mTOR or knocking down of Beclin1, or knock down of the autolysosome protein cathepsin B, reduced drug combination lethality. The drug combination caused an endoplasmic reticulum stress response as judged by enhanced eIF2α phosphorylation that was responsible for reducing MCL-1 and BCL-XL levels and increasing ATG5 and Beclin1 expression. Knock down of BIM, but not of BAX or BAK, reduced cell killing. Expression of activated MEK1 prevented the drug combination increasing BIM expression and reduced cell killing. Downstream of the mitochondrion, drug lethality was partially reduced by knock down of AIF, but expression of dominant negative caspase 9 was not protective. Our data demonstrate that neratinib and niraparib interact to kill ovarian cancer cells through convergent DNA damage and endoplasmic reticulum stress signaling. Cell killing required the induction of autophagy and was cathepsin B and AIF -dependent, and effector caspase independent.
Introduction
The epidermal growth factor receptor (EGFR, HER1, ERBB1) and HER2 (ERBB2) are widley known to promote the growth and malignancy of tumor cells; such cells also generally are more resistant to anti-cancer drugs [Citation1, and references therein]. ERBB1 is frequently expressed in triple negative breast cancer (TNBC). ERBB2, ERBB3 and ERBB4 are also known to facilitate tumor growth through multiple downstream pathways.Citation2-4 We recently discovered that the ERBB1/2/4 inhibitor neratinib, could down-regulate the levels of ERBB1/2/3/4, c-MET, PDGFRα and mutant K-/N-RAS proteins.Citation5-7 Neratinib-treated cells contained many vesicles of different sizes inside the cell, both close to the plasma membrane and within the cytoplasm. These vesicles contained ERBB family receptors and the mutant RAS proteins, and which co-stained for Beclin1, phosphorylated ATG13 S318 and/or cathepsin B and LAMP2. ERBB2 and cathepsin B co-localization has been noted by others.Citation8 The precise biology by which neratinib rapidly causes protein degradation are unknown.
Poly(ADP-ribose) polymerase (PARP) is required for repairing DNA damage through the base excision repair pathway.Citation9 Upon DNA damage, PARP1 associates with the DNA where its catalytic activity is enhanced. PARP then catalyzes the synthesis of branched, protein-conjugated poly(ADP-ribose) to itself and other proteins that are complexed together in the execution of base excision repair. BRCA1/2 mutant cells, that are deficient in homologous repair, are particularly sensitive to PARP inhibitors.Citation10-14 Multiple inhibitors of Poly (ADP ribose) polymerase 1 (PARP1) have been developed over the past decade with olaparib, rucaparib and niraparib now approved for use in ovarian cancer. Olaparib is only approved for ovarian cancers that have BRCA1/2 mutations whereas niraparib has been shown to have activity in this disease regardless of BRCA1/2 status, and is an approved second line therapy for all ovarian cancer patients.Citation15
One observation we made several y ago was that PARP1 inhibitors could activate the kinase ataxia telangiectasia mutated (ATM), causing γH2AX phosphorylation.Citation16 More recently we have demonstrated that ATM signaling can activate the AMPK which in turn promotes a toxic form of autophagosome formation.Citation16-19 From our prior studies with neratinib, we were also aware that in addition to inhibiting ERBB1/2/4 and down-regulating other signaling proteins, neratinib too could enhance ATM-AMPK signaling. Collectively, based on this knowledge, we hypothesized that neratinib and niraparib would interact to kill tumor cells; as niraparib is approved in ovarian cancer for all patients, we chose this system for our studies. Our conclusions are that neratinib and niraparib synergize to kill ovarian cancer cells through coordinated DNA damage and endoplasmic reticulum stress signaling resulting in the formation of toxic autophagosomes.
Results
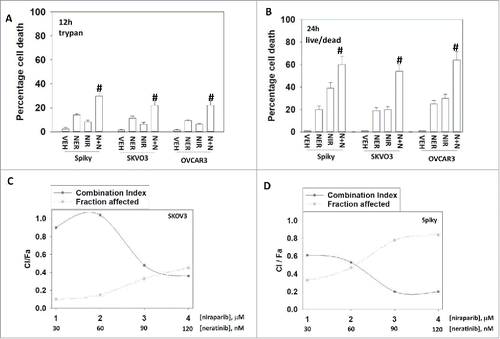
The irreversible ERBB1/2/4 inhibitor neratinib and the PARP1 inhibitor niraparib interacted using short-term viability assays in an additive to greater than additive fashion to kill ovarian cancer cells ( and ).Citation20-25 In median dose effect isobologram colony formation assays neratinib and niraparib interacted in a synergistic fashion, with combination index values of less than 1.0 to kill ovarian cancer cells ( and ). The drug combination did not kill peripheral blood mononuclear cells (negative control; data not shown)
Figure 1. Neratinib and niraparib combine to kill ovarian cancer cells. A. Ovarian cancer cells were treated with vehicle control, neratinib (50 nM), niraparib (2.0 μM) or the drugs in combination for 12 h. Cell viability was determined by trypan blu exclusion assay (n = 3 +/− SEM) # p < 0.05 greater than neratinib alone. B. Ovarian cancer cells were treated with vehicle control, neratinib (50 nM), niraparib (2.0 μM) or the drugs in combination for 24 h. Cell viability was determined by live / dead assay (n = 3 +/− SEM). # p < 0.05 greater than neratinib alone. C. SKOV3 cells and D. Spiky cells were plated in 6-well plates in sextuplicate as individual cells (500 cells per well). After 12 h the cells were treated with vehicle control, neratinib, niraparib or the drugs combined, at the indicated concentrations in the figure, at a fixed ratio. After 24 h, the media is removed, the cells washed with warm drug-free media, and fresh drug-free media placed on the cells. After 10 d, colonies of > 50 cells have formed and the cells are fixed in place and stained with crystal violet. The plating efficiency under each treatment condition is determined and the fraction affected determined. Synergy was determined using the Calcusyn for Windows program using the method of Cho and Tallalay (n = 2 independent studies in sextuplicate). A combination index of less than 1.00 indicates a synergy of drug interaction.
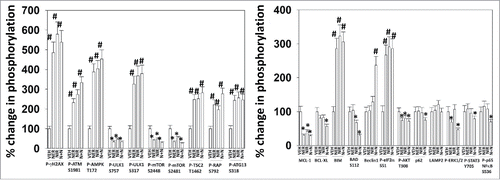
We performed descriptive studies to define changes in cell signaling caused by the drugs as single agents and in combination after 6 hours of exposure. The drugs, both alone or in combination activated ATM, AMPK and ULK-1 (decreased S757, increased S317) which was associated with increased ATG13 S318 phosphorylation (). The drugs, both alone or in combination inactivated mTOR (decreased S2448 and decreased S2481; increased TSC2 T1462 and increased Raptor S792) which was also associated with increased ATG13 S318 phosphorylation. The drugs both increased eIF2α phosphorylation; increased expression of BIM; decreased expression of MCL-1, BCL-XL and p62; and decreased phosphorylation of BAD S112 and AKT T308. The drugs, but only in combination, increased the expression of Beclin1, and decreased the phosphorylation of STAT3 Y705 and NFκB S536. Neratinib as a single agent reduced ERK1/2 phosphorylation, that correlated with the ability of this drug to reduce mutant N-RAS expression (Figure S1). These findings, in a broad perspective, suggest that we are inactivating protective signaling pathways, decreasing expression of protective proteins, increasing expression/activity of toxic proteins and enhancing autophagosome formation.
Figure 2. Neratinib and niraparib interact to activate ATM-AMPK-ULK1 signaling and in parallel activate eIF2α signaling that correlates with reduced expression of MCL-1 and BCL-XL. Spiky ovarian cancer cells were treated with vehicle control, neratinib (50 nM), niraparib (2.0 μM) or the drugs in combination for 6 h. The cells were fixed in place and immunostaining performed to determine the expression and phosphorylation of the indicated proteins at 10X magnification (data from multiple separate images & treatments +/− SEM) *p < 0.05 less than vehicle control; #p < 0.05 greater than vehicle control.
Based on the data in , we performed additional mechanistic studies to address the role of autophagy and other survival-regulatory pathways in cells treated with [neratinib + niraparib]. The drug combination increased autophagosome formation within 4 hours, an effect that was partially blocked by knock down of eIF2α, ATM or AMPKα (). No increase in autolysosome formation was observed at this time point. After 8 hours, the numbers of autophagosomes had declined and the number of autolysosomes had increased. Again, autolysosome formation was reduced by knock down of eIF2α, ATM or AMPKα. Using siRNA knock down or protein over-expression approaches we then interrogated our cells to determine the role of autophagy and other survival-regulatory pathways in the cell death response to the drug combination.
Figure 3. Neratinib and niraparib kill via toxic autophagy and necroptotic processes. A. Spiky ovarian cancer cells were transfected with: a scrambled siRNA molecules or siRNA molecules to knock down the expression of eIF2α, ATM or AMPKα; a plasmid to express LC3-GFP-RFP. Twenty-four h after transfection cells were treated with vehicle control or [neratinib (50 nM) + niraparib (2.0 μM)] in combination for 4 h and 8 h. The cells were imaged at 60X magnification and the mean number of intense fluorescent GFP+ and RFP+ foci in the cells determined (from 40 cells per condition in triplicate). *p < 0.05 less than corresponding value in siSCR cells. B. Ovarian cancer cells were transfected with: a scrambled siRNA molecules or siRNA molecules to knock down the expression of CD95, AIF, AMPKα, ATG5, ATM, BAD, BAX, BAK, Beclin1, BIM, cathepsin B, eIF2α, FADD and ULK-1. Twenty-four h after transfection cells were treated with vehicle control or with [neratinib (50 nM) + niraparib (2.0 μM)] in combination for 24 h. Cell viability was determined by live / dead assay (n = 3 +/− SEM). *p < 0.05 less than corresponding value in siSCR cells; **p < 0.01 less than corresponding value in siSCR cells. C. Ovarian cancer cells were transfected with an empty vector plasmid (CMV) or with plasmids to express c-FLIP-s, BCL-XL or dominant negative caspase 9. Twenty-four h after transfection cells were treated with vehicle control or with [neratinib (50 nM) + niraparib (2.0 μM)] in combination for 24 h. Cell viability was determined by live / dead assay (n = 3 +/− SEM). *p < 0.05 less than corresponding value in CMV transfected cells.
![Figure 3. Neratinib and niraparib kill via toxic autophagy and necroptotic processes. A. Spiky ovarian cancer cells were transfected with: a scrambled siRNA molecules or siRNA molecules to knock down the expression of eIF2α, ATM or AMPKα; a plasmid to express LC3-GFP-RFP. Twenty-four h after transfection cells were treated with vehicle control or [neratinib (50 nM) + niraparib (2.0 μM)] in combination for 4 h and 8 h. The cells were imaged at 60X magnification and the mean number of intense fluorescent GFP+ and RFP+ foci in the cells determined (from 40 cells per condition in triplicate). *p < 0.05 less than corresponding value in siSCR cells. B. Ovarian cancer cells were transfected with: a scrambled siRNA molecules or siRNA molecules to knock down the expression of CD95, AIF, AMPKα, ATG5, ATM, BAD, BAX, BAK, Beclin1, BIM, cathepsin B, eIF2α, FADD and ULK-1. Twenty-four h after transfection cells were treated with vehicle control or with [neratinib (50 nM) + niraparib (2.0 μM)] in combination for 24 h. Cell viability was determined by live / dead assay (n = 3 +/− SEM). *p < 0.05 less than corresponding value in siSCR cells; **p < 0.01 less than corresponding value in siSCR cells. C. Ovarian cancer cells were transfected with an empty vector plasmid (CMV) or with plasmids to express c-FLIP-s, BCL-XL or dominant negative caspase 9. Twenty-four h after transfection cells were treated with vehicle control or with [neratinib (50 nM) + niraparib (2.0 μM)] in combination for 24 h. Cell viability was determined by live / dead assay (n = 3 +/− SEM). *p < 0.05 less than corresponding value in CMV transfected cells.](/cms/asset/ad60db00-5cd4-4f8b-9a7b-90056250f3c2/kcbt_a_1436024_f0003_b.gif)
In all three lines tested knock down of ATM, AMPKα, ULK-1, ATG5, Beclin1, cathepsin B or eIF2α, reduced the killing of cells by [neratinib + niraparib] (). This suggests, based on our prior studies, that an ATM – AMPK – ULK-1 – ATG13 – cathepsin B autophagy pathway plays a role in cell killing and that endoplasmic reticulum stress signaling is also required for tumor cell death. Knock down of death receptor signaling via CD95 or FADD was weakly / not protective in any ovarian cancer line whereas in two of the three lines, over-expression of the caspase 8 inhibitor c-FLIP-s was protective, and, when combined with our knock down data for CD95 and FADD, this argues that a non-receptor dependent activation of caspases 8/10 can play a role in the killing process (). Downstream of death receptors and of autolysosomes at the level of the mitochondrion, knock down of BAD weakly altered the cell death response, as did, surprisingly knock down of the toxic BH3 domain proteins BAX and BAK, whereas knock down of BIM exhibited protection. Over-expression of BCL-XL, which protects both mitochondrial and endoplasmic reticulum function, protected cells from [neratinib + niraparib] exposure. Downstream of the mitochondrion, expression of dominant negative caspase 9 had no protective effect whereas knock down of apoptosis inducing factor (AIF) was partially protective ( and ). Collectively, with our knock down data with AIF and cathepsin B, and with the lack of caspase 9 / 3 influencing cell killing, we argue that [neratinib + niraparib]-stimulated autophagy causes tumor cell death via lysosomal dysfunction (cathepsin B) and through a mitochondrial-dependent pathway (AIF).
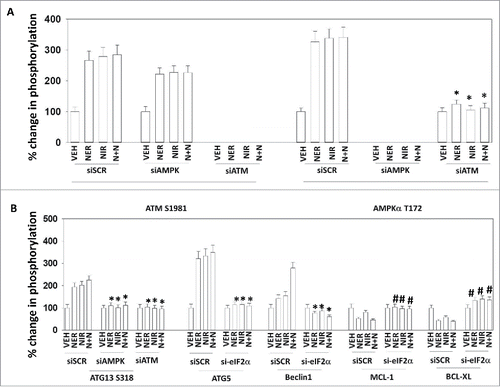
In prior studies using the thymidylate synthase inhibitor pemetrexed or the histone deacetylase inhibitors sodium valproate and AR42, we determined that DNA damage signaling and ATM activation played key roles in regulating autophagosome formation and tumor cell killing.Citation17,Citation18 The drugs alone or in combination activated ATM (). The drugs, alone or in combination, increased AMPKα T172 phosphorylation, an effect blocked by knock down of ATM. The drugs alone or in combination enhanced the phosphorylation of ATG13 S318 in an ATM-AMPK -dependent fashion (). In an eIF2α (endoplasmic reticulum stress)-dependent fashion the drugs alone or in combination increased the expression of ATG5 and Beclin1 and decreased the expression of MCL-1 and BCL-XL. Thus, ER stress signaling facilitates autophagosome formation and lowers the ability of the ER and mitochondria to defend itself from toxic pore-forming BH3 domain proteins.
Figure 4. Neratinib and niraparib activate the AMPK and cause ATG13 S318 phosphorylation via activation of ATM. A. and B. Spiky ovarian cancer cells were transfected with a scrambled siRNA control or with siRNA molecules to knock down the expression of eIF2α, AMPKα or ATM. Twenty-four h after transfection, cells were treated with vehicle control, neratinib (50 nM), niraparib (2.0 μM) or the drugs in combination for 6 h. The cells were fixed in place and immunostaining performed to determine the expression and phosphorylation of the indicated proteins at 10X magnification. (data from multiple separate images & treatments +/− SEM) *p < 0.05 less than corresponding value in siSCR cells; #p < 0.05 greater than corresponding value in siSCR cells.
In agreement with our ATG13 S318 phosphorylation data, knock down of ATM or AMPKα prevented the drugs alone or in combination from enhancing ULK-1 S317 phosphorylation and from reducing ULK-1 S757 phosphorylation (). Knock down of ATM or AMPKα prevented the drugs alone or in combination from reducing the phosphorylation of mTORC1 S2448 and mTORC2 S2481 (). Collectively, this data demonstrates that the drug-dependent regulation of two key gate-keeper kinases who control ATG13 S318 phosphorylation / autophagosome formation, mTOR and ULK1, requires elevated ATM-AMPK signaling. Three other important survival-regulatory pathways are the NFκB pathway, the PI3K-AKT pathway and the ERK1/2 pathway. In an ATM-AMPK dependent fashion, the drugs alone or in combination reduced AKT T308 phosphorylation (). In contrast, but also in an ATM-AMPK -dependent manner, neratinib, but not niraparib, reduced ERK1/2 phosphorylation. The drug combination via ATM-AMPK, inactivated NFκB signaling. Thus, [neratinib + niraparib] combine, via ATM-AMPK signaling, to reduce the activities of AKT, mTOR and ERK1/2.
Figure 5. The inactivation of mTOR, AKT and ERK1/2 requires ATM-AMPK signaling. A. and B. Spiky cells were transfected with a scrambled siRNA control or with siRNA molecules to knock down the expression of AMPKα or ATM. Twenty-four h after transfection, cells were treated with vehicle control, neratinib (50 nM), niraparib (2.0 μM) or the drugs in combination for 6 h. The cells were fixed in place and immunostaining performed to determine the expression and phosphorylation of mTOR, ULK-1 and mTOR at 10X magnification. (data from multiple separate images & treatments +/− SEM) *p < 0.05 less than corresponding value in siSCR cells; #p < 0.05 greater than corresponding value in siSCR cells.
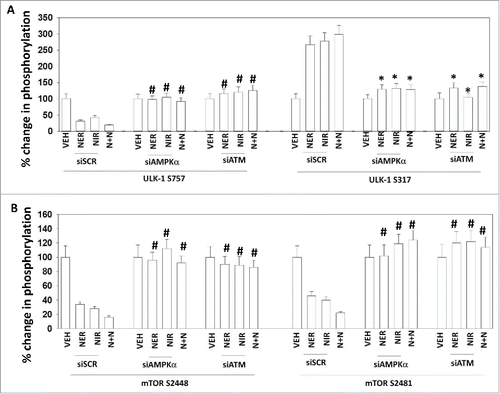
Figure 6. Loss of mTOR activity facilitates toxic autophagy and loss of ERK1/2 activity facilitates BIM expression. A. Spiky cells were transfected with a scrambled siRNA control or with siRNA molecules to knock down the expression of AMPKα or ATM. Twenty-four h after transfection, cells were treated with vehicle control, neratinib (50 nM), niraparib (2.0 μM) or the drugs in combination for 6 h. The cells were fixed in place and immunostaining performed to determine the expression and phosphorylation of AKT, ERK1/2 and p65 NFκB. (data from multiple separate images & treatments +/− SEM) *p < 0.05 less than vehicle control; **p < 0.05 less than neratinib alone value. B. Ovarian cancer cells were transfected with an empty plasmid control (CMV) or with plasmids to express activated MEK1 or activated mTOR. Twenty-four h after transfection cells were treated with vehicle control or with [neratinib (50 nM) + niraparib (2.0 μM)] in combination for 24 h. Cell viability was determined by trypan blue exclusion assay (n = 3 +/− SEM). *p < 0.05 less than corresponding value in CMV cells; **p < 0.05 less than corresponding value in caMEK1 cells. C. Ovarian cancer cells were transfected with an empty plasmid control (CMV) or with a plasmid to express activated MEK1. Twenty-four h after transfection cells were treated with vehicle control or with [neratinib (50 nM) + niraparib (2.0 μM)] in combination for 6 h. Immunostaining was performed to determine the expression of c-FLIP-s and BIM. (data from multiple separate images & treatments +/− SEM) *p < 0.05 less than corresponding value in CMV control; #p < 0.05 greater than corresponding value in CMV control.
![Figure 6. Loss of mTOR activity facilitates toxic autophagy and loss of ERK1/2 activity facilitates BIM expression. A. Spiky cells were transfected with a scrambled siRNA control or with siRNA molecules to knock down the expression of AMPKα or ATM. Twenty-four h after transfection, cells were treated with vehicle control, neratinib (50 nM), niraparib (2.0 μM) or the drugs in combination for 6 h. The cells were fixed in place and immunostaining performed to determine the expression and phosphorylation of AKT, ERK1/2 and p65 NFκB. (data from multiple separate images & treatments +/− SEM) *p < 0.05 less than vehicle control; **p < 0.05 less than neratinib alone value. B. Ovarian cancer cells were transfected with an empty plasmid control (CMV) or with plasmids to express activated MEK1 or activated mTOR. Twenty-four h after transfection cells were treated with vehicle control or with [neratinib (50 nM) + niraparib (2.0 μM)] in combination for 24 h. Cell viability was determined by trypan blue exclusion assay (n = 3 +/− SEM). *p < 0.05 less than corresponding value in CMV cells; **p < 0.05 less than corresponding value in caMEK1 cells. C. Ovarian cancer cells were transfected with an empty plasmid control (CMV) or with a plasmid to express activated MEK1. Twenty-four h after transfection cells were treated with vehicle control or with [neratinib (50 nM) + niraparib (2.0 μM)] in combination for 6 h. Immunostaining was performed to determine the expression of c-FLIP-s and BIM. (data from multiple separate images & treatments +/− SEM) *p < 0.05 less than corresponding value in CMV control; #p < 0.05 greater than corresponding value in CMV control.](/cms/asset/09acdae5-fc4c-424a-bca6-dece58e20625/kcbt_a_1436024_f0006_b.gif)
Finally, as [neratinib + niraparib] exposure was reducing the activities of AKT, ERK1/2 and mTOR via ATM-AMPK signaling, we determined whether expression of activated forms of AKT, MEK1 or of mTOR could protect tumor cells from the drug combination. Expression of an activated form of MEK1 or an activated form of AKT partially reduced the lethality of [neratinib + niraparib] exposure (). Expression of an activated form of mTOR significantly reduced the lethality of [neratinib + niraparib] exposure below that observed for activated MEK1 and activated AKT; activated mTOR also significantly suppressed the induction of autophagy by [neratinib + niraparib].Citation5 The drug combination increased the expression of BIM and reduced the expression of c-FLIP-s, effects that were prevented by expression of activated MEK1 (). Thus, inactivation of mTOR and of ERK1/2 coordinate to enhance caspase 8 activity and mitochondrial dysfunction in parallel with promoting autophagosome formation.
Discussion
The present studies were performed to determine whether the approved ovarian cancer therapeutic niraparib interacted with the recently approved breast cancer therapeutic neratinib at clinically relevant concentrations to kill cisplatin-resistant / multi-drug resistant ovarian cancer cells. Neratinib and niraparib interacted in an additive to synergistic fashion to kill ovarian cancer cells in vitro.
Neratinib is an irreversible inhibitor of ERBB1/2/4 and niraparib is an inhibitor of PARP1. Both drugs reduced the activities of mTORC1 and mTORC2 that was associated with increased autophagosome and subsequently autolysosome levels, and knock down of Beclin1 or ATG5 protected the cells from the lethal effects of the drug combination (Figure S2). Inactivation of the mTOR complexes and ULK-1 was mediated by ATM-AMPK signaling, as was the inactivation of AKT, ERK1/2 and NFκB. The drug combination activated eIF2α which was responsible for enhancing ATG5 and Beclin1 expression and decreasing MCL-1 and BCL-XL expression. The drug combination also rapidly reduced, in a Beclin1 / ATG5 -dependent fashion, the expression of mutant N-RAS. We recently presented evidence that neratinib in combination with the HDAC inhibitor sodium valproate had the potential to cause the rapid degradation of mutant K-RAS and mutant N-RAS proteins that was associated with reduced tumor growth in vivo.Citation5-7 Collectively, our data argues that [neratinib + niraparib] through the regulation of multiple pathways ultimately generates a toxic form of autophagy that leads to necrotic / necroptotic tumor cell killing.
Based on our findings, we argue that the induction of a DNA damage response, i.e. γH2AX phosphorylation and ATM activation, plays a critical role in the initiation of tumor cell killing. Neratinib, niraparib and the drug combination all caused similar amounts of γH2AX phosphorylation and ATM activation, and downstream AMPK activation, when compared to each other. Neratinib, niraparib and the drug combination also all caused similar amounts of mTORC1 / mTORC2 inactivation, and downstream ULK-1 activation, when compared to each other. This was reflected in similar amounts of ATG13 phosphorylation being induced by the drugs alone or in combination. Thus, activation of the ATM-AMPK-mTOR / ULK1-ATG13 pathway does not appear to be the pathway by which the two drugs interact to cause an additive to synergistic induction of tumor cell killing.
Neratinib, but not niraparib, reduced ERK1/2 phosphorylation that was enhanced by niraparib. In other systems ERK1/2 phosphorylation has been proposed to down-regulate the expression of the toxic BH3 domain protein BIM. And, in ovarian cancer cells, we discovered that BIM expression played a vital role in cell killing caused by [neratinib + niraparib]. In other systems ERK1/2 phosphorylation has been proposed to enhance the expression of the caspase 8 inhibitor c-FLIP-s. And, in ovarian cancer cells, we discovered that c-FLIP-s expression played a vital role in the prevention of cell killing caused by [neratinib + niraparib]. Expression of activated MEK1 partially suppressed [neratinib + niraparib] lethality that was associated with activated MEK1 preventing the down-regulation of c-FLIP-s and the up-regulation of BIM. As [neratinib + niraparib] was capable of rapidly reducing the expression of mutant N-RAS in Spiky cells we conclude that reduced signaling by RAS proteins and enhanced ATM-AMPK signaling results in lower ERK1/2 activity that facilitates caspase activation and mitochondrial stability.
In ovarian cancer cells, neratinib, niraparib or the drugs in combination enhanced the expression of ATG5, whereas the expression of Beclin1 was only strongly enhanced by the drug combination. Only the drug combination caused an obvious reduction in the expression of p62, indictive of its proteolytic degradation in autolysosomes. Neratinib, niraparib or the drugs in combination all enhanced ER stress signaling as judged by S51 phosphorylation of eIF2α. Activation of eIF2α was causal in the enhanced expression of ATG5 and Beclin1, and reduced expression of the mitochondrial protective proteins MCL-1 and BCL-XL. Hence eIF2α-dependent down-regulation of BCL-XL and MCL-1 will positively interact with the de-repression of BIM expression due to ERK1/2 inactivation, to cause mitochondrial dysfunction. Thus, as a point of signaling convergence for the two drugs, how does eIF2α regulate the protein levels of Beclin1?.Citation26-31 Allelic loss of Beclin1 is common in ovarian cancer patients however elevated Beclin1 expression is associated with a poorer prognosis. Some studies suggest that STAT3 can inhibit Beclin1 expression, and the drug combination but not the individual drugs inhibit STAT3 Y705 phosphorylation. Other data argues that under certain circumstances inactivation of NFκB can facilitate Beclin1 transcription, and only the [neratinib + niraparib] combination inactivated NFκB. Other findings argue that reduced activity through the ERK1/2 pathway can facilitate other stimuli to increase Beclin1 expression. Studies beyond the scope of the present manuscript will be required to understand how signaling by eIF2α, STAT3, NFκB and ERK2 converge to enhance Beclin1 protein levels.
In conclusion, we have presented evidence that the ERBB1/2/4 inhibitor neratinib and the PARP1 inhibitor niraparib interact to kill ovarian cancer cells, but not normal PMBCs. At present, due to financial constraints for the corresponding author, we have been unable to perform animal studies combining neratinib and niraparib, which somewhat limits the impact of our studies; studies that would more readily facilitate clinical translation. Both agents are FDA approved, and have non-overlapping toxicities in patients and following additional in vivo work it is hoped that clinical translation of this drug combination can eventually be performed.
Materials and methods
Materials
Niraparib was purchased from Selleckchem (Houston, TX). Neratinib was supplied by Puma Biotechnology Inc. (Los Angeles, CA). Trypsin-EDTA, DMEM, RPMI, penicillin-streptomycin were purchased from GIBCOBRL (GIBCOBRL Life Technologies, Grand Island, NY). SKOV3 and OVCAR3 cells were purchased from the ATCC and were not further validated beyond that claimed by ATCC.Citation20-22 Cells were re-purchased every ∼6 months. Spiky ovarian cancer cells, an established PDX model, were kindly provided by Dr. Karen Paz (Champions Oncology, NJ).Citation23 Commercially available validated short hairpin RNA molecules to knock down RNA / protein levels were from Qiagen (Valencia, CA) (Figures S3 and S4, and see below). Reagents / performance of experimental procedures were described in refs: 5–7, 16–19. Antibodies used in the manuscript were: (a) from Cell Signaling Technology: AIF (5318), BAX (5023), BAK (12105), BAD (9239), BIM (2933), BAK1 (12105), Beclin1 (3495), cathepsin B (31718), CD95 (8023), FADD (2782), eIF2α (5324), P-eIF2α S51 (3398), ULK-1 (8054), P-ULK-1 S757 (14202), P-AMPK S51 (2535), AMPKα (2532), P-ATM S1981 (13050), ATM (2873), and ATG5 (12994). TSC2 (4308), P-TSC2 T1462 (3617), Raptor (2280), P-Raptor S792 (2083), mTOR (2983), P-mTOR S2448 (5536), P-mTOR S2481 (2974), ATG13 (13468), MCL-1 (94296), BCL-XL (2764), P-AKT T308 (13038), P-ERK1/2 (5726), P-STAT3 Y705 (9145), P-p65 S536 (3033), p62 (23214), LAMP2 (49067); (b) from Abgent: P-ULK-1 S317 (3803a); (c) from Novus Biologicals: P-ATG13 S318 (19127). The pre-validated smart-pool siRNA molecules used in the manuscript were from Qiagen: siSCR (SI03650318), ATM (SI00604737), cathepsin B (1027416), BAX (GS581), BAK (GS578); AMPKα (GS5562), BIM (GS10018), BAD (GS572), Beclin1 (GS8678), ATG5 (GS9474), CD95 (GS355), AIF (GS9131), eIF2α (GS83939), FADD (GS8772), ULK-1 (GS8408), ATG13 (GS9776).
Methods
Culture and in vitro exposure of cells to drugs
All cell lines were cultured at 37°C (5% (v/v CO2) in vitro using RPMI supplemented with dialyzed 5% (v/v) fetal calf serum and 1% (v/v) Non-essential amino acids. For short term cell killing assays, immune-staining studies, cells were plated at a density of 3 × 103 per cm2 and 24 h after plating treated with various drugs, as indicated. In vitro drug treatments were generally from a 100 mM stock solution of each drug and the maximal concentration of Vehicle carrier (VEH; DMSO) in media was 0.02% (v/v). Cells were not cultured in reduced serum media during any study in this manuscript. The safe achievable plasma Cmax for neratinib is ∼150 nM; for niraparib it is ∼3.5 μM.Citation24,Citation25
Transfection of cells with siRNA or with plasmids
For Plasmids
Cells were plated and 24 h after plating, transfected. Plasmids expressing a specific mRNA (or siRNA) or appropriate vector control plasmid DNA was diluted in 50 μl serum-free and antibiotic-free medium (1 portion for each sample). Concurrently, 2 μl Lipofectamine 2000 (Invitrogen), was diluted into 50 μl of serum-free and antibiotic-free medium (1 portion for each sample). Diluted DNA was added to the diluted Lipofectamine 2000 for each sample and incubated at room temperature for 30 min. This mixture was added to each well / dish of cells containing 200 μl serum-free and antibiotic-free medium for a total volume of 300 μl, and the cells were incubated for 4 h at 37 °C. An equal volume of 2x medium was then added to each well. Cells were incubated for 24 h, then treated with drugs.
Transfection for siRNA
Cells were transfected with control and with specific Smartpool siRNAs (Qiagen) using Lipofectamine 2000 (Thermo Fisher Scientific, 11668027) following the supplier's guidelines. Briefly, cells from a fresh culture growing in log phase as described above, and 24 h after plating transfected. Prior to transfection, the medium was aspirated and serum-free medium was added to each plate. For transfection, 10 nM of the annealed siRNA, the positive sense control doubled stranded siRNA targeting GAPDH or the negative control (a “scrambled” sequence with no significant homology to any known gene sequences from mouse, rat or human cell lines) were used. Ten nM siRNA (scrambled or experimental) was diluted in serum-free media. Four μl Hiperfect (Qiagen) was added to this mixture and the solution was mixed by pipetting up and down several times. This solution was incubated at room temp for 10 min, then added drop-wise to each dish. The medium in each dish was swirled gently to mix, then incubated at 37°C for 2 h. Serum-containing medium was added to each plate, and cells were incubated at 37°C for 24 h before then treated with drugs (0–24 h). Additional immuno-fluorescence / live-dead analyses were performed at the indicated time points.
Colony formation / isobologram assays
Single cells were plated in 6-well plates in sextuplicate as individual cells (500 cells per well). After 12 h the cells were treated with vehicle control, neratinib, niraparib or the drugs combined, at the indicated concentrations in the figure, at a fixed ratio. After 24 h, the media is removed, the cells washed with warm drug-free media, and fresh drug-free media placed on the cells. After 10 d, colonies of > 50 cells have formed, and the cells are fixed in place and stained with crystal violet. The plating efficiency under each treatment condition is determined and the fraction affected determined (any group of cells > 50 cells is considered a colony). Synergy was determined using the Calcusyn for Windows program using the method of Cho and Talalay (n = 2 independent studies in sextuplicate). A combination index of less than 1.00 indicates a synergy of drug interaction.
Detection of cell viability, protein expression and protein phosphorylation by immuno-fluorescence using a Hermes WiScan wide-field microscope. http://www.idea-bio.com/, Cells (4 × 103) are plated into each well of a 96 well plate, and cells permitted to attach and grow for the next 18 h. Based on the experiment, after 18 h, cells are then either genetically manipulated, or are treated with drugs. For genetic manipulation, cells are transfected with plasmids or siRNA molecules and incubated for an additional 24 h. Cells are treated with vehicle control or with drugs at the indicated final concentrations, alone or in combination. Cells are then isolated for processing at various times following drug exposure. The 96 well plate is centrifuged / cyto-spun to associate dead cells (for live-dead assays) with the base of each well. For live dead assays, after centrifugation, the media is removed and cells treated with live-dead reagent (Thermo Fisher Scientific, Waltham MA) and after 10 min this is removed and the cells in each well are visualized in the Hermes instrument at 10X magnification. Green cells = viable; yellow/red cells = dying/dead. The numbers of viable and dead cells were counted manually from three images taken from each well combined with data from another two wells of separately treated cells (i.e. the data is the mean cell dead from 9 independent data points from three separate exposures).
For immuno-fluorescence studies, after centrifugation, the media is removed and cells are fixed in place and permeabilized using ice cold PBS containing 0.4% paraformaldehyde and 0.5% Triton X-100. After 30 min the cells are washed three times with ice cold PBS and cells are pre-blocked with rat serum for 3 h. Cells are then incubated with a primary antibody to detect the expression / phosphorylation of a protein (usually at 1:100 dilution from a commercial vendor) overnight at 37°C. Cells are washed three times with PBS followed by application of the secondary antibody containing an associated fluorescent red, green or blue chemical tag. After 3 h of incubation the antibody is removed, and the cells washed again. A secondary alone negative control was included to assess background staining in the absence of primary antibody; at present, using mouse, rabbit and goat secondary antibodies the background staining effect in our system is essentially zero. The cells are visualized at either 10X or 60X in the Hermes microscope for imaging assessments of fluorescence intensity. All immunofluorescent images for each individual protein / phospho-protein are taken using the identical machine settings so that the levels of signal in each image can be directly compared to the level of signal in the cells treated with drugs. Imaging for fluorescence intensity is made using software integral to the Hermes microscope. Selections of groups of cells are made (∼40 in each well) from each treatment in triplicate and the mean intensity determined. The software automatically assesses and removes background fluorescence from each data set so that comparisons are made examining actual cell staining intensity between conditions / treatments.
For presentation, the enhancement of image brightness/contrast is processed at 9999 dpi using Adobe Photoshop CS6 and is simultaneously performed for each individual set of protein/phospho-protein to permit direct comparison of the image intensity between treatments. Images are labeled, and figures generated in Microsoft PowerPoint.
Detection of cell death by Trypan Blue assay
Cells were harvested by trypsinization with Trypsin/EDTA for ∼10 min at 37°C. Harvested cells were combined with the culture media containing unattached cells and the mixture centrifuged (800 rpm, 5 min). Cell pellets were resuspended in PBS and mixed with trypan blue agent. Viability was determined microscopically using a hemocytometer. Five hundred cells from randomly chosen fields were counted and the number of dead cells was counted and expressed as a percentage of the total number of cells counted.
Assessment of autophagy
Cells were transfected with a plasmid to express a green fluorescent protein (GFP) and red fluorescent protein (RFP) tagged form of LC3 (ATG8). For analysis of cells transfected with the GFP‐RFP‐LC3 construct, the GFP/RFP‐positive vesicularized cells were examined under the × 40 objective of a Zeiss Axiovert fluorescent microscope.
Data analysis
Comparison of the effects of various treatments (performed independently in triplicate and then three times) was using one-way analysis of variance (ANOVA) and a two tailed Student's t-test. Differences with a p-value of < 0.05 were considered statistically significant. Experiments shown are the means of multiple individual points from multiple experiments (± SEM).
Abbreviations
| ca | = | constitutively active |
| CMV | = | empty vector plasmid or virus |
| dn | = | dominant negative |
| ER | = | endoplasmic reticulum |
| ERK | = | extracellular regulated kinase |
| HDAC | = | histone deacetylase. |
| mTOR | = | mammalian target of rapamycin |
| MAPK | = | mitogen activated protein kinase |
| PI3K | = | phosphatidyl inositol 3 kinase |
| PTEN | = | phosphatase and tensin homologue on chromosome ten |
| ROS | = | reactive oxygen species |
| si | = | small interfering |
| SCR | = | scrambled |
| VEH | = | vehicle |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Sup_mat_2018CBT11103R1-f07-z-4c.pdf
Download PDF (564.4 KB)Acknowledgments
Support for the present study was funded from philanthropic funding from Massey Cancer Center, the Universal Inc. Chair in Signal Transduction Research and PHS R01-CA192613. Thanks to Dr. H.F. Young and the Betts family fund for support in the purchase of the Hermes Wiscan instrument. The authors have no conflicts of interest to report.
Additional information
Funding
References
- Booth L, Roberts JL, Tavallai M, Webb T, Leon D, Chen J, McGuire WP, Poklepovic A, Dent P. The afatinib resistance of in vivo generated H1975 lung cancer cell clones is mediated by SRC/ERBB3/c-KIT/c-MET compensatory survival signaling. Oncotarget. 2016;7:19620–30. doi:10.18632/oncotarget.7746. PMID:26934000.
- Henson E, Chen Y, Gibson S. EGFR family members' regulation of autophagy is at a crossroads of cell survival and death in cancer. Cancers (Basel). 2017;9(4). pii: E27. doi:10.3390/cancers9040027.
- Appert-Collin A, Hubert P, Crémel G, Bennasroune A. Role of ErbB receptors in cancer cell migration and invasion. Front Pharmacol. 2015;6:283 doi:10.3389/fphar.2015.00283. PMID:26635612.
- Arteaga CL, Engelman JA. ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell. 2014;25:282–303 doi:10.1016/j.ccr.2014.02.025. PMID:24651011.
- Booth L, Roberts JL, Poklepovic A, Avogadri-Connors F, Cutler RE, Lalani AS, Dent P. HDAC inhibitors enhance neratinib activity and when combined enhance the actions of an anti-PD-1 immunomodulatory antibody in vivo. Oncotarget. 2017 IN PRESS;8(52):90262–90277 doi:10.18632/oncotarget.21660. PMID:29163826.
- Booth L, Roberts JL, Poklepovic A, Kirkwood J, Sander C, Avogadri-Connors F, Cutler RE, Lalani AS, Dent P. The levels of mutant K-RAS and mutant N-RAS are rapidly reduced by the irreversible ERBB1/2/4 inhibitor neratinib. Cancer Biol Ther. 2017.
- Booth L, Roberts JL, Kirkwood J, Avogadri-Connors F, Cutler RE, Lalani AS, Poklepovic A, Dent P. [Neratinib + Valproate] exposure permanently reduces ERBB1 and RAS expression in 4T1 mammary tumors and enhances M1 macrophage infiltration. Oncotarget. 2017. doi:10.18632/oncotarget.23681.
- Zhang Y, Zhang J, Liu C, Du S, Feng L, Luan X, Zhang Y, Shi Y, Wang T, Wu Y, Cheng W, Meng S, Li M, Liu H. Neratinib induces ErbB2 ubiquitylation and endocytic degradation via HSP90 dissociation in breast cancer cells. Cancer Lett. 2016;382:176–185. doi:10.1016/j.canlet.2016.08.026. PMID:27597738.
- Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010;10:293–301. doi:10.1038/nrc2812. PMID:20200537.
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917 doi:10.1038/nature03443. PMID:15829966.
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921 doi:10.1038/nature03445. PMID:15829967.
- Martin SA, Lord CJ, Ashworth A. DNA repair deficiency as a therapeutic target in cancer. Curr Opin Genet Dev. 2008;18:80–86 doi:10.1016/j.gde.2008.01.016. PMID:18343102.
- Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, Friedlander M, Arun B, Loman N, Schmutzler RK, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376:235–244. doi:10.1016/S0140-6736(10)60892-6. PMID:20609467.
- Jenner ZB, Sood AK, Coleman RL. Evaluation of rucaparib and companion diagnostics in the PARP inhibitor landscape for recurrent ovarian cancer therapy. Future Oncol. 2016;12:1439–56. doi:10.2217/fon-2016-0002. PMID:27087632.
- McLachlan J, George A, Banerjee S. The current status of PARP inhibitors in ovarian cancer. Tumori. 2016;102:433–440 doi:10.5301/tj.5000558. PMID:27716873.
- Tang Y, Hamed HA, Poklepovic A, Dai Y, Grant S, Dent P. Poly(ADP-ribose) polymerase 1 modulates the lethality of CHK1 inhibitors in mammary tumors. Mol Pharmacol. 2012;82:322–32. doi:10.1124/mol.112.078907. PMID:22596349.
- Booth L, Roberts JL, Poklepovic A, Dent P. [pemetrexed + sildenafil], via autophagy-dependent HDAC down-regulation, enhances the immunotherapy response of NSCLC cells. Cancer Biol Ther. 2017. doi:10.1080/15384047.2017.1362511.
- Booth L, Roberts JL, Sander C, Lee J, Kirkwood JM, Poklepovic A, Dent P. The HDAC inhibitor AR42 interacts with pazopanib to kill trametinib/dabrafenib-resistant melanoma cells in vitro and in vivo. Oncotarget. 2017;8:16367–16386. doi:10.18632/oncotarget.14829. PMID:28146421.
- Booth L, Roberts JL, Poklepovic A, Gordon S, Dent P. PDE5 inhibitors enhance the lethality of pemetrexed through inhibition of multiple chaperone proteins and via the actions of cyclic GMP and nitric oxide. Oncotarget. 2017;8:1449–1468. doi:10.18632/oncotarget.13640. PMID:27903966.
- Hamilton TC, Young RC, McKoy WM, Grotzinger KR, Green JA, Chu EW, Whang-Peng J, Rogan AM, Green WR, Ozols RF. Characterization of a human ovarian carcinoma cell line (NIH:OVCAR-3) with androgen and estrogen receptors. Cancer Res. 1983;43:5379–89. PMID:6604576.
- Morimoto H, Safrit JT, Bonavida B. Synergistic effect of tumor necrosis factor-alpha- and diphtheria toxin-mediated cytotoxicity in sensitive and resistant human ovarian tumor cell lines. J Immunol. 1991;147:2609–16. PMID:1918981.
- Liu JR, Opipari AW, Tan L, Jiang Y, Zhang Y, Tang H, Nuñez G. Dysfunctional apoptosome activation in ovarian cancer: implications for chemoresistance. Cancer Res. 2002;62:924–31. PMID:11830553.
- Teng PN, Bateman NW, Wang G, Litzi T, Blanton BE, Hood BL, Conrads KA, Ao W, Oliver KE, Darcy KM, McGuire WP, Paz K, Sidransky D, Hamilton CA, Maxwell GL, Conrads TP. Establishment and characterization of a platinum- and paclitaxel-resistant high grade serous ovarian carcinoma cell line. Hum Cell. 2017;30:226–236. doi:10.1007/s13577-017-0162-1. PMID:28251557.
- Sandhu SK, Schelman WR, Wilding G, Moreno V, Baird RD, Miranda S, Hylands L, Riisnaes R, Forster M, Omlin A, Kreischer N, Thway K, Gevensleben H, Sun L, Loughney J, Chatterjee M, Toniatti C, Carpenter CL, Iannone R, Kaye SB, de Bono JS, Wenham RM. The poly (ADP-ribose) polymerase inhibitor niraparinib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2013;14:882–92. doi:10.1016/S1470-2045(13)70240-7. PMID:23810788.
- Kourie HR, Chaix M, Gombos A, Aftimos P, Awada A. Pharmacodynamics, pharmacokinetics and clinical efficacy of neratinib in HER2-positive breast cancer and breast cancer with HER2 mutations. Expert Opin Drug Metab Toxicol. 2016;12:947–57. doi:10.1080/17425255.2016.1198317. PMID:27284682.
- Yu L, Wang Y, Yao Y, Li W, Lai Q, Li J, Zhou Y, Kang T, Xie Y, Wu Y, Chen X, Yi C, Gou L, Yang J. Eradication of growth of HER2-Positive ovarian cancer with Trastuzumab-DM1, an antibody-cytotoxic drug conjugate in mouse Xenograft Model. Int J Gynecol Cancer 2014;24:1158Y1164. doi:10.1097/IGC.0000000000000179.
- Canonici A, Gijsen M, Mullooly M, Bennett R, Bouguern N, Pedersen K, O'Brien NA, Roxanis I, Li JL, Bridge E, Finn R, Siamon D, McGowan P, Duffy MJ, O'Donovan N, Crown J, Kong A. Neratinib overcomes trastuzumab resistance in HER2 amplified breast cancer. Oncotarget. 2013;4:1592–605. doi:10.18632/oncotarget.1148. PMID:24009064.
- Miao LJ, Huang FX, Sun ZT, Zhang RX, Huang SF, Wang J. Stat3 inhibits Beclin 1 expression through recruitment of HDAC3 in non-small cell lung cancer cells. Tumour Biol. 2014;35:7097–103. doi:10.1007/s13277-014-1961-6. PMID:24760274.
- Jiang Q, Wang Y, Li T, Shi K, Li Z, Ma Y, Li F, Luo H, Yang Y, Xu C. Heat shock protein 90-mediated inactivation of nuclear factor-κB switches autophagy to apoptosis through becn1 transcriptional inhibition in selenite-induced NB4 cells. Mol Biol Cell. 2011;22:1167–80 doi:10.1091/mbc.E10-10-0860. PMID:21346199.
- Zhao Y, Chen S, Gou WF, Xiao LJ, Takano Y, Zheng HC. Aberrant Beclin 1 expression is closely linked to carcinogenesis, differentiation, progression, and prognosis of ovarian epithelial carcinoma. Tumour Biol. 2014;35:1955–64. doi:10.1007/s13277-013-1261-6. PMID:24132590.
- Ji D, Zhang Z, Cheng L, Chang J, Wang S, Zheng B, Zheng R, Sun Z, Wang C, Zhang Z, Liu R, Zhang X, Liu X, Wang X, Li J. The combination of RAD001 and MK-2206 exerts synergistic cytotoxic effects against PTEN mutant gastric cancer cells: involvement of MAPK-dependent autophagic, but not apoptotic cell death pathway. PLoS One. 2014;9:e85116. doi:10.1371/journal.pone.0085116. PMID:24416349.