
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Non-small cell lung cancer (NSCLC) is the leading cause of cancer-related death worldwide, and novel effective drugs against NSCLC are urgently needed. Isodon species are rich in ent-kaurane diterpenoids that have been reported to have antitumor bioactivity. Acetyl-macrocalin B (A-macB) is a novel ent-kaurane diterpenoid isolated from Isodon silvatica, and its antitumor efficacy against NSCLC and the underlying mechanisms were scrutinized in depth. The viability of cells treated with A-macB was detected by CCK-8 and colony formation assays. Apoptosis and cell cycle distribution were analyzed by flow cytometry. The mechanisms were investigated by detecting ROS and performing western blotting and verification experiments with specific inhibitors. The in vivo effect of A-macB was explored in a nude mouse xenograft model. A-macB effectively inhibited H1299 and A549 cell viability, triggered apoptosis and delayed cells in the G2/M phase. A-macB induced cellular ROS production and then activated the p38 MAPK-mediated, caspase 9-dependent apoptotic pathway. Both the ROS scavenger NAC and the specific p38 inhibitor SB203580 inactivated the function of p38 induced by A-macB, thus preventing cells from apoptosis. A-macB activated the Chk1/2-Cdc25C-Cdc2/cyclin B1 axis to induce G2/M phase arrest. AZD7762 abrogated the function of Chk1/2, abolished the G2/M delay and enhanced the cytotoxicity of A-macB. Moreover, A-macB efficiently suppressed tumor growth in a mouse xenograft model without noticeable toxicity to normal tissues. Having both efficacy and relative safety, A-macB is a potential lead compound that is worthy of further exploration for development as an anticancer agent.
Introduction
Non-small cell lung cancer (NSCLC) accounts for 80–85% of lung cancers and is a leading cause of cancer-related death worldwide.Citation1–3 Among patients with early-stage resectable or locally advanced disease who receive definitive chemoradiotherapy, up to 90% patients eventually relapse.Citation4 Considering all stages of lung cancer, the 5-year survival rate of NSCLC is less than 20%; the 5-year survival rate for those with metastatic disease is only 2%.Citation2,Citation3,Citation5 Despite recent advances in the treatment of NSCLC with targeted therapies and immunotherapy, patients usually develop resistance to such targeted drugs.Citation1,Citation6 Thus, cytotoxic chemotherapy remains of vital importance for the majority of patients with advanced disease.Citation1,Citation6 Therefore, there is a constant need to develop new and effective drugs against NSCLC.
Natural products have attracted considerable attention due to their broad-spectrum biological activities and have become a prolific source of novel drug discovery over past decades.Citation7,Citation8 The widely used Chinese popular folk medicine genus Isodon is rich in natural ent-kaurane diterpenoids, which are reported to have antitumor bioactivity.Citation9,Citation10 We purified a novel natural ent-kaurane diterpenoid, acetyl-macrocalin B (A-macB, ), from Isodon silvatica by high-performance liquid chromatography. Macrocalin B (macB) was first isolated from Isodon macrocalyx and was shown to have cytotoxicity against Hela, K562, HL-60, MKN-28, A549, HCT and CA tumor cells in vitro.Citation11–13 A-macB is the natural acetylate form of macB, which was extracted from Isodon silvatica, and the antitumor activity of A-macB against NSCLC haven't been explored before.
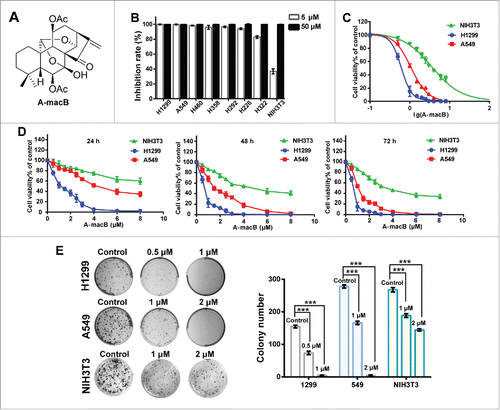
. A-macB inhibits the viability of NSCLC cell lines. (A) The structure of A-macB. (B) Growth inhibition effects of A-macB on seven NSCLC cell lines and the mouse embryo fibroblast cell line NIH3T3. (C) IC50 curves for H1299, A549 and NIH3T3 treated with A-macB for 72 h. (D) Proliferation inhibition curves of H1299, A549 and NIH3T3 incubated with A-macB for different times and at different concentrations. (E) Colony formation of NSCLC and the normal NIH3T3 cells treated with A-macB. Data are presented as the mean ± SEM (*p < 0.05, **p < 0.01, ***p < 0.001).
Chemotherapeutic agents usually cause oxidative DNA damage.Citation14 Reactive oxygen species (ROS) are a source of oxidative stress involved in DNA damage, cell proliferation, apoptosis and senescence.Citation15 In particular, ROS are important mediators of the activity of many chemotherapeutics, including numerous natural products extracted from Isodon species. Previous studies have shown that intracellular ROS accumulation induced by ent-kaurane diterpenoids extracted from Isodon results in cancer cell apoptosis; for example, Jaridonin induces the apoptosis of esophageal cancer cells,Citation16 and Longikaurin ACitation17 and Isoforretin ACitation18 evoke hepatocellular carcinoma cell apoptosis. Excess cellular ROS levels are cytotoxic and serve as an early signal that regulates apoptosis.Citation19,Citation20 The p38 pathway is an important stress response pathway that can be activated by increased ROS production.Citation21 By inducing apoptotic cascades, the ROS–p38 axis participates in regulating apoptosis and inducing cell death.Citation22
Ent-kaurane diterpenoids have been implicated in mediating cell cycle arrest in tumor cells. In particular, ent-kaurane diterpenoids were reported to participate in the regulation of G2/M cell cycle delay.Citation17,Citation23,Citation24 An essential step in the cell cycle transition from G2 to mitosis includes the activation of the cell division cycle protein 2 (Cdc2)/cyclin B complex.Citation25 In response to DNA damage, checkpoint kinases 1 and 2 (Chk1 and Chk2) are activated to promote G2/M arrest via destruction or sequestration of Cdc25C phosphatases, preventing the reversal of the inhibitory phosphorylation of Cdc2 at Thr14 and Tyr15.Citation26 Therefore, Cdc2/cyclin B activity was inhibited and cells were arrested in G2/M phase.Citation25
In this study, we show that A-macB significantly induced NSCLC cell apoptosis through the ROS/p38 signaling pathway and delayed cells in G2/M phase by regulating the Chk1/2-Cdc25-Cdc2/cyclin B axis. The cytotoxic activity of A-macB was enhanced by the Chk1/2 inhibitor AZD7762. Moreover, A-macB inhibited the growth of tumor xenografts in vivo without notable injury to the mice.
Results
A-macB inhibited NSCLC cell viability and colony formation
Seven NSCLC cell lines and a mouse embryo fibroblast cell line NIH3T3 were screened to detect the inhibitory activity of A-macB. As shown in , the viability of all tumor cell lines was significantly inhibited by treatment with 5 μM or 50 μM A-macB for 72 h, while NIH3T3 were more resistant to A-macB treatment at 5 μM. Then, H1299 and A549 were selected for further study and NIH3T3 was used as the normal cell line control. The IC50 value of A-macB toward H1299 and A549 cells at 72 h was 0.61 μM and 2.20 μM, respectively (). Cell inhibition curves showed that A-macB inhibited both cell lines in a dose-and time-dependent manner (). What's more, colony generation assays revealed that cells treated with A-macB formed fewer and smaller colonies compared with control-treated cells (). In contrast, the IC50 value of the normal NIH3T3 was 4.57 μM, which was much higher than the tumor cell lines. Moreover, A-macB displayed only moderate cytotoxicity against NIH3T3, as manifested by the inhibition ability of cell proliferation and clone generation (, ).
A-macB induced apoptosis through the p38 MAPK/caspase 9-mediated apoptotic pathway
The effect of A-macB on the induction of apoptosis in H1299 and A549 cells was evaluated by flow cytometry. Treatment with A-macB markedly induced apoptosis after 24 h. The percentage of apoptotic H1299 cells increased from 2.00% to 14.20% (3 μM) and 89.58% (6 μM), and that of apoptotic A549 cells increased from 2.04% to 8.16% (4 μM) and 19.17% (8 μM) ( and ).
Figure 2. A-macB induces NSCLC apoptosis through the p38 MAPK-caspase 9-mediated apoptosis pathway. (A and B) Flow cytometry analyses of NSCLC cells after A-macB treatment for 24 h. Cells that were Annexin V (+) were considered as apoptotic cell population. H1299 were incubated with 3 μM or 6 μM and A549 were incubated with 4 μM or 8 μM of A-macB for 24 h, and apoptosis in these cell lines was examined by flow cytometry. (C) Western blot analysis showed that the p38 MAPK-caspase 9-mediated apoptosis pathway was activated by A-macB treatment. (D and E) Differential effects of caspase inhibitors on A-macB-induced apoptosis. For pretreated groups, H1299 and A549 cells were incubated with 20 μM of Z-IETD-FMK (caspase-8 inhibitor), Z-LEHD-FMK (caspase-9 inhibitot) or Z-VAD-FMK (pan-caspase inhibitor) for 2 h and then incubated with A-macB (6 μM for H1299 and 8μM for A549) for another 24 h; for non-pretreated groups, H1299 and A549 cells were incubated with A-macB alone for 24 h. Then, the apoptotic statuses of the cells were examined by flow cytometry. (F) WB analysis was performed to verify that protein expression and activation were correlated with cell apoptosis regulation in response to A-macB with or without caspase inhibitor pretreatment. Data are presented as the mean ± SEM. (*p < 0.05, **p < 0.01, ***p < 0.001). (c-Cas9: cleaved caspase-9, c-Cas3: cleaved caspase-3, c-Cas8: cleaved caspase-8, Z-IETD: Z-IETD-FMK, Z-LEHD: Z-LEHD-FMK, Z-VAD: Z-VAD-FMK).
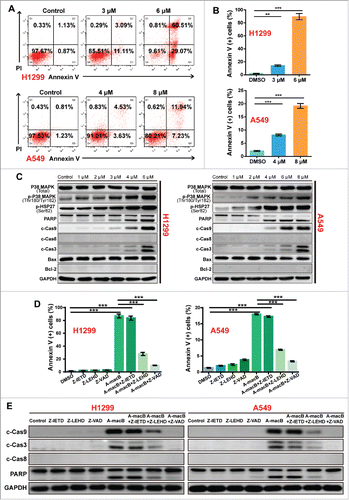
To explore the underlying mechanism by which A-macB induces apoptosis, we used a mini stress and apoptotic array that includes the main signaling pathways involved in regulating the stress response and apoptosis. The results indicated that p38 MAPK was consistently activated by A-macB treatment in both cell lines, and this activation may regulate the caspase 9-dependent apoptotic pathway (Supplementary ). The western blot (WB) results confirmed this hypothesis. As shown in , p-p38 (Thr180/Tyr182) levels significantly increased after A-macB treatment, and p-HSP27 (Ser82) levels increased in line with p38 activation, although total p38 expression was not affected by A-macB. Accordingly, caspase 9, caspase 3 and PARP were cleaved, but caspase 8, Bax and Bcl-2 remained unchanged ( and Supplementary ). The apoptosis induced by A-macB could be partially reversed by a caspase-9-specific inhibitor (Z-LEHD-FMK) and a pan-caspase inhibitor (Z-VAD-FMK) but was not reversed by a caspase-8 inhibitor (Z-IETD-FMK) ( and Supplementary ). In addition, the caspase-9 and pan-caspase inhibitors at least partially protected procaspase-9, procaspase-3 and PARP from cleavage (). These results confirmed that A-macB induced apoptosis mainly via a caspase-9-dependent intrinsic apoptotic pathway.
Figure 3. (A and B) SB203580 co-administration partially reversed A-macB-induced apoptosis. Cells were divided into four groups, three single-agent groups and the co-administration group. The co-administraion group were pretreated with 10 μM SB203580, a specific p38 MAPK inhibitor, for 2 h and then co-treated with A-macB for 24 h; and the single-agent groups were treated with 0.1% DMSO, 10 μM SB203580, 6 μM A-macB for H1299 or 8 μM A-macB for A549, respectively. SB203580 co-administration significantly reversed A-macB-induced apoptosis. (C) SB203580 co-treatment inhibited the function of p38 MAPK, as indicated by a decrease in p-HSP27 (Ser82) levels. As a result, the caspase 9/caspase 3/PARP signaling pathway was suppressed. Data are presented as the mean ± SEM (*p < 0.05, **p < 0.01, ***p < 0.001). (c-Cas9: cleaved caspase 9; c-Cas3: cleaved caspase 3; c-Cas8: cleaved caspase 8).
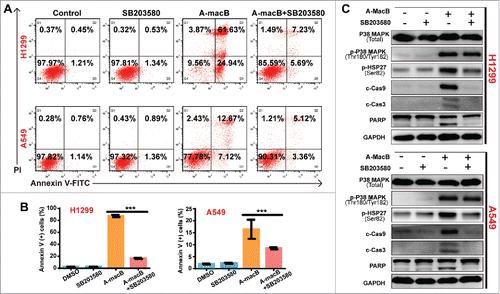
To further validate the results, we performed the reverse experiment using SB203580, a specific inhibitor of p38. As shown in and , SB203580 co-administration significantly reversed A-macB-induced apoptosis. The mechanism involved the inhibition of the downstream activation function of p38 MAPK since SB203580 significantly suppressed the phosphorylation of HSP27, the downstream target, instead of p38 itself (). When the p38 signaling pathway was blocked by SB203580, signaling through the caspase 9-dependent apoptotic pathway was restricted accordingly (). As a result, caspases 9 and 3 were not activated, and cells were prevented from undergoing apoptosis.
A-macB increased intracellular ROS and activated the p38 MAPK pathway
ROS plays an important role in apoptotic induction, and natural products have been shown to exert antitumor activity by increasing intracellular ROS generation.Citation15–18 Therefore, we determined whether A-macB stimulates ROS production. As shown in and , A-macB significantly increased ROS levels in H1299 and A549 cells, and this effect was abolished by the ROS scavenger N-acetylcysteine (NAC). The percentage of apoptotic cells was obviously increased, corresponding with the high ROS levels induced by A-macB. Furthermore, A-macB induced apoptosis could be at least partially reverted by NAC. As shown in and , NAC reduced apoptosis by approximately 86.70% in H1299 and 56.81% in A549. The results indicated that A-macB increased intracellular ROS levels in NSCLC cells, thus causing apoptosis.
Figure 4. A-macB enhances intracellular ROS production and induced p38/caspase 9 mediated apoptotic pathway. (A and B) A-macB treatment markedly increased ROS levels in A549 and H1299 cells. The antioxidant N-acetylcysteine (NAC, a precursor of glutathione, 10 mM) effectively prevented A-macB-induced ROS generation. (C and D) The population of H1299 and A549 cells undergoing apoptosis increased significantly in response to increasing ROS levels. NAC pretreatment prevented H1299 and A549 apoptosis. (E) Western blot analysis showed that NAC pretreatment protected cells from DNA damage and inhibited the p38 MAPK signaling pathway, thus suppressing the caspase 9-caspase 3-PARP signaling pathway. Data are presented as the mean ± SEM (*p < 0.05, **p < 0.01, ***p < 0.001).
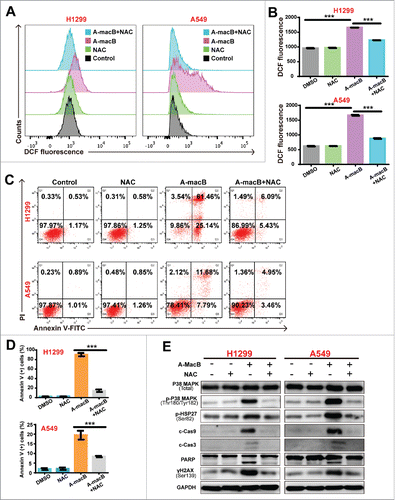
Previous studies have shown that p38 and its downstream signaling pathway are critical responders to ROS stress,Citation27,Citation28 and both ROS and p38 regulate apoptosis.Citation20,Citation29 In the present study, we found that along with ROS increasing, the p38 pathway was activated by A-macB treatment, as evidenced by the increased expression of p-p38 (Thr180/Tyr182) and its downstream target p-HSP27 (Ser82) (). Consequently, caspase 9 dependent-apoptosis was initiated. NAC co-treatment partially reversed apoptosis and abrogated the phosphorylation of both p38 and HSP27 (), which indicated that A-macB-induced ROS generation can initiate p38/caspase 9-mediated apoptosis. As expected, WB analysis confirmed that NAC co-administration alleviated DNA damage and blocked the activation of caspases 9 and 3, thereby protecting cells from undergoing apoptosis ().
A-macB induced G2/M arrest via the Chk1/2-Cdc25C-Cdc2/cyclin B axis
Many cytotoxic agents influence cell cycle progression, so we explored the effect of A-macB on the cell cycle distribution of H1299 and A549 cells. As shown in , A-macB delayed H1299 and A549 cells in G2/M phase. We further analyzed crucial regulatory proteins involved in the G2/M phase by WB. A-macB caused severe DNA damage, indicated by the elevated expression of γH2AX (). In addition, p-Chk1 (Ser345) and p-Chk2 (Thr68) levels increased to delay the cell cycle in response to DNA damage. In response to Chk1/2 mediation, Cdc25C, the phosphatase responsible for the dephosphorylation and activation of Cdc2, is usually regulated by degradation, phosphorylation and subcellular localization.Citation30 In this study, Cdc25C expression levels were noticeably decreased along with Chk1/2 activation, suggesting the marked degradation of Cdc25C. Consistent with the decreased Cdc25C levels, Cdc2 was phosphorylated at the inhibitory site, Tyr15, which resulted in the inactivation of the Cdc2/cyclin B1 complex that promotes G2/M progression (). Moreover, cyclin B1 protein levels were suppressed simultaneously, which enhanced the inhibition of the Cdc2/cyclin B1 complex (). Taken together, these results suggested the possible engagement of the Chk1/2-Cdc25C-Cdc2/cyclin B1 axis, which prompted the delay of cells in G2/M phase.
Figure 5. A-macB induces G2/M arrest via the Chk1/2-Cdc25C-Cdc2/cyclin B1 axis. (A) Cell cycle distribution of NSCLC cells after A-macB treatment. H1299 were incubated with 2 μM or 4 μM A-macB and A549 were incubated with 3 μM or 6 μM A-macB for 24 h. Cell cycle distribution was analyzed by flow cytometry. The histograms show the percentages of cells in each phase of the cell cycle. (B) Western blot analysis showed that A-macB induces G2/M arrest through the Chk1/2-Cdc25C-Cdc2/cyclin B1 signaling pathway. Data are presented as the mean ± SEM (*p < 0.05, **p < 0.01, ***p < 0.001).
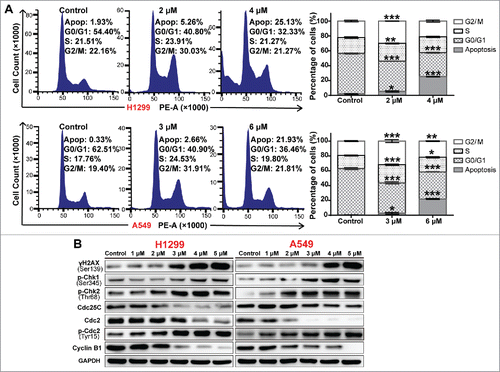
The Chk1/2 inhibitor AZD7762 abrogated A-macB-induced G2/M arrest and enhanced cell cytotoxicity
To confirm the involvement of the Chk1/2 pathway in G2/M regulation, we performed the reverse experiment using AZD7762, a specific Chk1/2 inhibitor. Cell cycle analysis revealed that AZD7762 partially reversed the A-macB-induced G2/M arrest and enhanced cell cytotoxicity (-). Furthermore, AZD7762 co-administration with A-macB abrogated the function of Chk1/2, as indicated by the stabilization of Cdc25A expression (). DNA damage-induced Chk1/2 activation usually stimulates the ubiquitylation and subsequent proteolysis of Cdc25A;Citation31 thus, stable Cdc25A expression indicates the functional inhibition of Chk1/2. Cdc25C expression was partially restored when Chk1/2 function was blocked by AZD7762. Consistent with the recovered function of Cdc25C, p-Cdc2 (Tyr15) levels were decreased, and analysis of cyclin B1 suggested the activation of the Cdc2/cyclin B complex. As a result, the population of cells in G2/M decreased, and cells were promoted into the mitotic phase, as evidenced by the increased levels of p-histone H3 (Ser10) (). However, cells co-treated with A-macB and AZD7762 experienced more severe DNA injury, evidenced by the increased γH2AX signal. The cells with damaged DNA were forced into mitosis and underwent mitotic catastrophe, manifested as the positive expression of γH2AX, p-histone H3 (Ser10) and cleaved caspase 3 (). Apoptosis analyses revealed significantly increased apoptosis of cells co-treated with A-macB and AZD7762, indicating the enhanced cytotoxic effect of AZD7762 to A-macB ( and ).
Figure 6. AZD7762 abrogates A-macB-induced G2/M arrest and enhances apoptosis triggered by A-macB. (A-C) The Chk1/2-specific inhibitor AZD7762 abrogated the G2/M arrest induced by A-macB. NSCLC cells were treated with AZD7762 (100 nM) or DMSO (vehicle) for 2 h prior to exposure to 1 μM A-macB. The cell cycle distribution was detected by flow cytometry. (D) The function of Chk1/2 was suppressed by AZD7762, leading to Cdc25C hyperactivity and decreased Cdc2/cyclin B dephosphorylation; this enabled the cells to enter mitosis with DNA damage, resulting in mitotic catastrophe, a phenomenon reflected by the levels of γH2AX, p-hH3 and cleaved caspase 3. (E and F) Enhanced cytotoxic effect of AZD7762 to A-macB. H1299 and A549 cells were pretreated with or without 100 nM AZD7762 for 2 h. Then the cells were incubated with 0.1% DMSO, 100 nM AZD7762, A-macB, or A-macB+AZD7762 for 24 h, respectively. A-macB was administered at a non-cytotoxic concentration (1 μM). The apoptotic status of the cells was examined by flow cytometry. Data are presented as the mean ± SEM (*p < 0.05, **p < 0.01, ***p < 0.001).
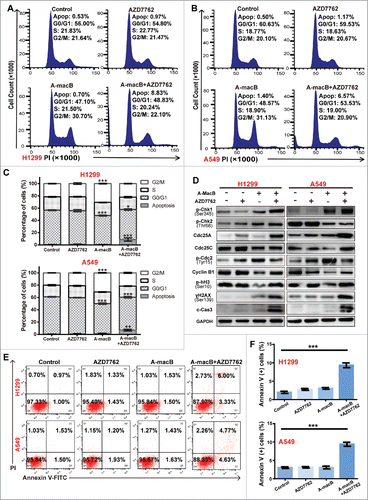
A-macB suppressed tumor growth in vivo
H1299 xenograft models were established to evaluate the role of A-macB in vivo. Mice were randomly divided into three groups and injected intraperitoneally every other day with 12 mg/kg A-macB (test group), 2 mg/kg cisplatin (positive control), or 1% Pluronic F68 (solvent negative control). Two mouse tumors in the negative control group reached 2000 mm3 at day 27 after transplantation; at this time point, all mice were sacrificed, and the tumors were harvested. As shown in and B, A-macB markedly inhibited tumor growth in mice with an inhibition rate of 63.71%, which was similar to the effect of cisplatin (61.79%). Tumors were obviously lighter in the A-macB and cisplatin treatment groups than in the negative control group (). Mouse weight did not decrease in any of the three groups (). A-macB did not cause significant histological injury to the liver or kidneys, as evidenced by hematoxylin and eosin (HE) staining ().
Figure 7. A-macB has efficient antitumor activity in vivo without inducing significant cytotoxicity. (A) A-macB and cisplatin inhibited H1299 xenograft growth. (B) Growth curves showed that tumor volume was significantly smaller in the A-macB- and cisplatin-treated groups than in the vehicle control-treated group. (C) Xenograft tissue weight was substantially lower in the A-macB- and cisplatin-treated groups than in the vehicle control-treated group. (D) Mouse weight did not decline in any of the three groups. (E) No significant tissue injury occurred after A-macB or cisplatin administration compared with vehicle injection, as shown by H&E staining. (F) Histochemical analyses of the tumors. Sections of paraffin-embedded tumor tissue from each group of H1299 xenograft mice were stained with H&E (upper panel). Ki67 (middle panel) and cleaved caspase 3 (lower panel) was examined by IHC. (G) TUNEL assays confirmed that apoptosis was induced by A-macB or cisplatin. Data are presented as the mean ± SD (*p < 0.05, **p < 0.01, ***p < 0.001). (DDP: cisplatin, Fluo-12-dUTP: fluorescein-12-dUTP).
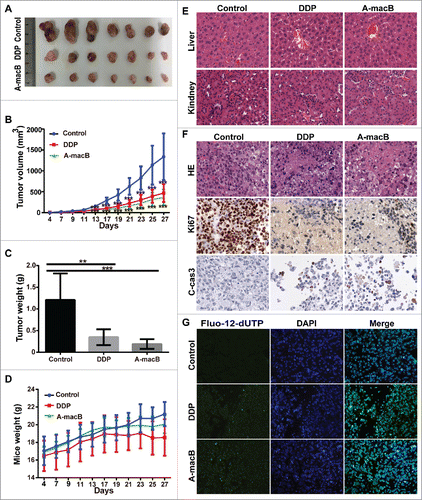
Growth inhibition by A-macB was further verified by histological staining. As shown in , HE staining clearly showed widespread histopathological disorder, such as swollen cells and necrosis in the A-macB- and cisplatin-treated groups, while viable tumor cell nests were pervasive in the negative control group. Moreover, tumor tissue in the groups treated with A-macB or cisplatin displayed rare Ki67 expression, indicating decreased proliferative ability, while Ki-67 expression was widespread in the negative control group (). Moreover, significant cleaved caspase-3 was expressed in the A-macB or DDP treated xenografts, while the control group didn't show such signal (). Terminal deoxynucleotide transferase-mediated dUTP end labeling (TUNEL) assay assays further confirmed the cytotoxic effect of A-macB. As shown in , A-macB or cisplatin treatment substantially increased the number of TUNEL-positive cells in H1299 tumor tissue, whereas there was no appreciable apoptosis in the negative control group.
Discussion
In past decades, approximately 75% of compounds approved for cancer therapy have been natural products or their derivatives.Citation32 Isodon diterpenoids are natural compounds extracted from Isodon species, and these compounds have attracted considerable attention for their strong antitumor activity and limited toxicity.Citation33,Citation34 The current treatments for advanced NSCLC are far from desirable, and novel strategies to enhance treatment efficacy are being pursued by researchers. A-macB is an ent-kaurane diterpenoid isolated from Isodon silvatica, and the antitumor activity of this compound against NSCLC was evaluated in this study. A-macB significantly inhibited NSCLC cells in vitro, with limited cytotoxicity to normal cells. Furthermore, A-macB showed efficacious antitumor activity in vivo with little cytotoxicity to normal tissues, making it a potential candidate for further clinical application.
A-macB suppressed cell viability and inhibited tumor growth mainly due to the induction of apoptosis, which was confirmed by flow cytometry-based apoptotic analyses in NSCLC cells and TUNEL assays in tumor xenograft tissues. Apoptosis can be triggered when cells suffer overwhelming damage. Caspases 9 and 8 are important initiator caspases that commit cells to the intrinsic or extrinsic apoptotic pathway, respectively.Citation35 Increased intracellular ROS cause cell damage and are tightly linked to the activation of the intrinsic apoptotic pathway.Citation19,Citation35 In this study, we discovered that A-macB induced significant apoptosis of H1299 and A549 cells and a marked increase in cellular ROS levels. NAC, a ROS scavenger, appreciably decreased cellular ROS levels and abolished A-macB-evoked apoptosis, verifying the link between ROS and apoptosis stimulated by A-macB.
P38 MAPK has been classically known as a stimulus-activated stress kinase that is associated with the promotion of apoptosis.Citation36 Apoptotic stimuli usually activate the p38 pathway by secondary routes, such as excessive ROS generation.Citation21,Citation36 In the present study, activation of the p38 MAPK pathway was abrogated by NAC and SB203580. Meanwhile, the induction of apoptosis and caspase 9-mediated intrinsic signals by A-macB were attenuated by co-treatment with NAC or SB203580. Taken together, these results evinced that A-macB increased intracellular ROS, activated the p38-MAPK pathway, and initiated cell death via the caspase 9-mediated apoptotic pathway.
Cytotoxic agents usually cause DNA damage, and γH2AX serves as a marker of DNA double-strand breaks (DSBs).Citation37 Checkpoint signaling is activated in response to DNA damage to prevent further progression through the cell cycle.Citation38 In this study, A-macB induced significant DSBs, as indicated by increased γH2AX levels, and arrested cells in G2/M via Chk1/2 activation. Chk1/2 targets and inactivates the Cdc25 family, in which Cdc25C assists in the G2/M transition, and Cdc25A plays a role in G1/S progression.Citation38,Citation39 Cdc25C regulates both Cdc2 and cyclin B1 to activate the Cdc2/cyclin B complex to facilitate mitosis.Citation40,Citation41 Activated Cdc25C dephosphorylates Cdc2 at Thr14 and Tyr15 and promotes entry into mitosis.Citation39 Cdc25C also regulates cyclin B1. Chou et al. reported that decreased Cdc25C could reduce cyclin B1 expression.Citation42 Concurrent treatment with AZD7762 and A-macB impeded the function of Chk1/2 and partially restored Cdc25C expression, resulting in the activation of Cdc2/cyclin B.Citation43 Moreover, AZD7762 and A-macB co-treatment increased DNA damage, as evidenced by high γH2AX expression. Consequently, cells were forced into mitosis despite severe DNA damage evoked by AZD7762 and A-macB, thus attenuating the G2/M delay. As the cells entered mitosis with damaged DNA, they experienced mitotic catastrophe, and the apoptosis pathway was activated to eliminate cells harboring DNA damage. Mitotic catastrophe is indicated by the co-expression of p-pH3, γH2AX and cleaved caspase 3.Citation44 These results were consistent with the flow cytometry analyses of apoptosis and cell cycle distribution. What's more, AZD7762 promoted mitotic catastrophe and apoptosis at relatively low concentrations of A-macB, while single-agent A-macB could not induce significant apoptosis at these concentrations. These results indicated that AZD7762 has the potential to sensitize cells to A-macB, making it possible to reduce the dose and side effects of A-macB.
In our research, we found that A-macB could induce apoptosis and G2/M arrest in H1299 at lower concentrations than in A549 cells. Previous studies have reported that A549 harbored wide type p53 and mutated KRAS, while H1299 was a p53-null-and-KRAS-wide type cell line.Citation45,Citation46 The genetic differences may associated with the different sensitivity of H1299 and A549 and the underlying mechanisms will be future studied by our team. What's more, the direct target of A-macB and the ability of AZD7762 to sensitize cells to A-macB will be investigated in-depth, and the efficacy of the combination treatment should be evaluated in cell line-xenograft and patient-derived xenogragft (PDX) models.
In conclusion, this study is the first to show that A-macB inhibits the growth of NSCLC cells in vitro and in vivo. A-macB increased intracellular ROS generation, which activated the p38-mediated, caspase 9/caspase 3/PARP signaling pathway, leading to extensive cell death. A-macB treatment arrested H1299 and A549 cells in the G2/M phase via the Chk1/2-Cdc25C-Cdc2/cyclin B axis. Additionally, A-macB showed less cytotoxicity against the normal NIH3T3 cell line than NSCLC cells in vitro, and A-macB did not exhibit any noticeable toxicity to normal tissues in nude mice. These results suggest that A-macB possesses selectivity between normal and cancer cells, thus making it a relatively safe candidate treatment for malignancy. These properties of A-macB render it a potential lead compound worthy of further exploration in the development of anticancer agents.
Materials and methods
Cell lines and animals
Seven NSCLC cell lines, A549, H1299, H226, H460, H358, H322 and H292, were maintained in RPMI 1640 culture medium (Corning, Logan, UT, USA) supplemented with 10% fetal bovine serum (FBS; Corning, Mediatech Inc., Manassas, VA, USA) at 37°C in a Thermo Scientific CO2 Incubator (Thermo, Rockford, IL, USA). NIH3T3, a mouse embryo fibroblast cell line, were cultured in Dulbecco's Modified Eagle's Medium (Corning). All the cell lines were authenticated via short tandem repeat (STR) analysis.
Female BALB/c nude mice were used to establish the xenograft model. The mice purchased from Huafukang Bioscience (Beijing, China) were 3–4 weeks old and weighed 13–17 g, and they were maintained in a laminar airflow cabinet under specific pathogen-free conditions.Citation47 Mice were cared for in accordance with institutional guidelines for animal welfare.
Chemicals and reagents
A-macB, a natural ent-kaurane diterpenoid extracted from Isodon silvatica, was initially dissolved in DMSO (D2650, Sigma, St. Louis, MO, USA) at 500 mM and stored at −20°C. Cisplatin (P4394, Sigma) was dissolved in 0.9% w/v NaCl solution at 1 mg/mL and stored at −20°C. Pluronic F68 (10%; #24040032) was purchased from Life Technologies Corporation and used as surfactant to facilitate A-macB delivery in vivo.
The Reactive Oxygen Species Assay Kit (S0033) and N-acetylcysteine (S0077) were obtained from Beyotime (Jiangsu, China). SB203580 (S1076) and AZD7762 (S1532) was purchased from SelleckChem (Houston, TX, USA). Caspase-9 inhibitor (Z-LEHD-FMK, B3233), caspase-8 inhibitor (Z-IETD-FMK, B3232) and pan-caspase inhibitor (Z-VAD-FMK, A1902) were purchased from APExBIO (Houston, TX, USA). Cell proliferation was detected with the Cell Counting Kit-8 (CCK8) from Dojindo (Kumamoto, Japan). A BD Pharmingen FITC Annexin V Apoptosis Detection Kit (#556547) was purchased from BD Biosciences (NJ, USA). Cell cycle distribution was detected using a Cell Cycle Detection Kit (KGA512, KeygenTec., Jiangsu, China). The DeadEnd™ Fluorometric TUNEL System (Promega, Madison, WI, USA) was used to detect and quantitate apoptotic signals in tissues. Cell colonies were stained with crystal violet (Amresco, Solon, OH, USA). RIPA buffer (89900), protease and phosphatase inhibitor cocktail (78442) and the BCA protein assay kit (23227) were purchased from Thermo. All the antibodies used in this study and their dilutions are listed in Supplementary Table 1.
Cell viability assay
Cell viability was measured using the CCK8 assay according to the manufacturer's instruction. Briefly, cells were seeded at 2000 cells/well into 96-well plates, allowed to attach overnight, and then treated with different doses of A-macB. The optical density was measured at 450 nm using a spectrophotometer (SpectraMax® 190, Molecular Devices, Sunnyvale, CA, USA).
Plate colony formation assay
Cells (500 per well) were seeded in 6-well plates and allowed to attach overnight before being treated with different concentrations of A-macB for 24 h. The cells were then cultured in RPMI 1640 medium supplemented with 10% FBS for another 10 days. The surviving colonies were fixed and stained with 1% crystal violet.
Cell cycle analysis by flow cytometry
Cells were plated in 6-cm dishes at 2 × 105 cells/dish, incubated overnight, synchronized in serum-free RPMI 1640 medium for 12 h and exposed to A-macB for 24 h. Thereafter, cells were collected, fixed in 70% ethanol overnight, digested with RNase A and stained with propidium iodide (PI) according to the manufacturer's protocol. Cell distribution in each phase of the cell cycle was determined using a BD Flow Cytometer (Becton Dickinson FACSCanto II).Citation48
Annexin V/propidium iodide staining assay for apoptosis analysis
The percentage of apoptotic cells was detected via Annexin V-FITC staining; Annexin V binds to cells with surface-exposed phosphatidylserine (PS). PI is used to distinguish viable cells from nonviable cells. Apoptotic cells were defined as Annexin V-FITC-positive cells. After treatment, cells were collected, washed twice with PBS, and labeled with Annexin V-FITC and PI according to the manufacturer's instruction. The percentage of apoptotic cells was evaluated using a BD Flow Cytometer.
Detection of intracellular ROS
Intracellular ROS levels were determined with the DCFH-DA fluorescent dye according to the instructions of the Reactive Oxygen Species Assay Kit. Briefly, cells were harvested after treatment and incubated with 10 μM DCFH-DA for 30 min in the dark. The fluorescence intensity in cells was analyzed using a BD Flow Cytometer with excitation and emission wavelengths of 488 nm and 525 nm, respectively.
Western blotting
Protein was isolated from cells using RIPA buffer containing a protease and phosphatase inhibitor cocktail and quantified with the BCA protein assay kit. Equal amounts of protein from each sample were separated by SDS-PAGE and transferred to polyvinylidene fluoride (PVDF) membranes. The membranes were blocked in 5% nonfat milk for 1 h at room temperature, washed and then incubated with specific primary antibodies at 4°C overnight. Thereafter, the blots were washed and incubated with secondary antibody for 1 h at room temperature. Signals were visualized by chemiluminescence detection.
Immunohistochemistry
Sections were deparaffinized 3 times in xylene, rinsed 2 times in 100% ethanol and rehydrated in ethanol (95%, 90%, 80%, and 70%) and then water. Endogenous peroxidase was inactivated by 0.3% H2O2. After antigen retrieval, nonspecific antigens were blocked with goat serum, and specific antigens were reacted with a primary antibody against Ki-67 at 4°C overnight. Then, the slides were incubated with the secondary antibody and visualized with 3, 3′-diaminobenzidine.
TUNEL assay
Apoptotic signals in tissues were detected with the DeadEnd™ Fluorometric TUNEL System according to the manufacturer's instruction. In brief, tissue sections were deparaffinized, rehydrated, and permeabilized with proteinase K; DNA strand breaks were labeled with fluorescein-12-dUTP. After stopping the reaction, all cells were stained with DAPI in mounting medium and analyzed. Apoptotic cells appear with green fluorescence (fluorescein-12-dUTP) on a blue background (nuclear DAPI stain) by fluorescence microscopy.
In vivo animal experiments
For the xenograft model, a total of 5 × 106 H1299 cells were subcutaneously injected into the right flank of nude mice. When tumors became palpable at 10 days after transplantation, the mice were randomized into 3 groups (7 mice/group): vehicle control (0.1% Pluronic F68), 2.5 mg/kg cisplatin, and 12 mg/kg A-macB, which was diluted in equivalent amounts of Pluronic F68 and DMSO. Vehicle or drug was injected intraperitoneally every other day, and mouse weight and tumor volume were measured before injection. Tumor volume was calculated using the formula V =. When the tumor volume in the vehicle control group reached the ethical limit (2000 mm3), mice were euthanized, and the tumors were harvested, weighed, and photographed. Tumor inhibition was calculated using the following equation.Citation49,Citation50
Statistical analysis
All the in vitro experiments were repeated in triplicate, and the data are presented as the mean ± SEM. Statistical analysis was performed using GraphPad Prism 5 (GraphPad Software Inc., La Jolla, CA, USA). One-way ANOVA followed by Dunnett's post hoc test was used for multiple comparisons. Independent t-tests were performed to compare the treated and control groups. p < 0.05 indicated statistical significance.Citation51
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Ethical approval
The in vivo animal experiments were approved by the Cancer Institute and Hospital of the Chinese Academy of Medical Sciences Institutional Animal Care and Use Committee (IACUC; permission number: NCC2015A095). All applicable institutional guidelines for the care and use of animals were followed. This article does not contain any studies with human participants performed by any of the authors.
2017CBT10926R-s02.docx
Download MS Word (1.2 MB)Acknowledgments
The authors thank X. Zeng (WuXi AppTec., Shanghai) for his help and support in the animal experiments.
Additional information
Funding
References
- Chen Z, Fillmore CM, Hammerman PS, Kim CF, Wong KK. Non-small-cell lung cancers: A heterogeneous set of diseases. Nat Rev Cancer. 2014;14:535–46. doi:10.1038/nrc3775. PMID:25056707.
- Ettinger DS, Wood DE, Akerley W, Bazhenova LA, Borghaei H, Camidge DR, Cheney RT, Chirieac LR, D'Amico TA, Dilling TJ, et al. NCCN guidelines insights: Non-small cell lung cancer, version 4.2016. J Natl Compr Canc Netw. 2016;14:255–64. doi:10.6004/jnccn.2016.0031. PMID:26957612.
- Xiong Y, Huang BY, Yin JY. Pharmacogenomics of platinum-based chemotherapy in non-small cell lung cancer: Focusing on DNA repair systems. Med Oncol. 2017;34:48. doi:10.1007/s12032-017-0905-6. PMID:28215024.
- Tan WL, Jain A, Takano A, Newell EW, Iyer NG, Lim WT, Tan EH, Zhai W, Hillmer AM, Tam WL, et al. Novel therapeutic targets on the horizon for lung cancer. Lancet Oncol. 2016;17:e347–62. doi:10.1016/S1470-2045(16)30123-1. PMID:27511159.
- D'Addario G, Fruh M, Reck M, Baumann P, Klepetko W, Felip E, Group EGW. Metastatic non-small-cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2010;21 Suppl 5:v116–9. doi:10.1093/annonc/mdq189.
- Tam K, Daly M, Kelly K. Treatment of Locally Advanced Non-Small Cell Lung Cancer. Hematol Oncol Clin North Am. 7;31:45–57. doi:10.1016/j.hoc.2016.08.009.
- Mondal S, Bandyopadhyay S, Ghosh MK, Mukhopadhyay S, Roy S, Mandal C. Natural products: Promising resources for cancer drug discovery. Anticancer Agents Med Chem. 2012; 12: 49–75. doi:10.2174/187152012798764697. PMID:21707502.
- Mishra BB, Tiwari VK. Natural products: An evolving role in future drug discovery. Eur J Med Chem. 2011;46:4769–807. doi:10.1016/j.ejmech.2011.07.057. PMID:21889825.
- Wan J, Liu M, Jiang HY, Yang J, Du X, Li XN, Wang WG, Li Y, Pu JX, Sun HD. Bioactive ent-kaurane diterpenoids from Isodon serra. Phytochemistry. 2016;130:244–51. doi:10.1016/j.phytochem.2016.05.014. PMID:27298277.
- Sun HD, Huang SX, Han QB. Diterpenoids from Isodon species and their biological activities. Nat Prod Rep. 2006;23:673–98. doi:10.1039/b604174d. PMID:17003905.
- Cheng PY, Lin YL, Xu GY. New diterpenoids of rabdosia macrocalyx: the structure of macrocalin A and macrocalin B. Yao Xue Xue Bao. 1984;19:593–8. PMID:6549528.
- Hou AJ, Li ML, Jiang B, Lin ZW, Ji SY, Zhou YP, Sun HD. New 7,20:14,20-diepoxy ent-kauranoids from Isodon xerophilus. J Nat Prod. 2000;63:599–601. doi:10.1021/np9903705. PMID:10843567.
- Yang Y, Sun H, Zhou Y, Ji S, Li M. Effects of three diterpenoids on tumour cell proliferation and telomerase activity. Nat Prod Res. 2009;23:1007–12. doi:10.1080/14786410802295149. PMID:19521915.
- Yan S, Sorrell M, Berman Z. Functional interplay between ATM/ATR-mediated DNA damage response and DNA repair pathways in oxidative stress. Cell Mol Life Sci. 2014;71:3951–67. doi:10.1007/s00018-014-1666-4. PMID:24947324.
- Park GB, Kim YS, Lee HK, Song H, Kim S, Cho DH, Hur DY. Reactive oxygen species and p38 MAPK regulate Bax translocation and calcium redistribution in salubrinal-induced apoptosis of EBV-transformed B cells. Cancer Lett. 2011;313:235–48. doi:10.1016/j.canlet.2011.09.011. PMID:22056078.
- Ma YC, Ke Y, Zi X, Zhao W, Shi XJ, Liu HM. Jaridonin, a novel ent-kaurene diterpenoid from Isodon rubescens, inducing apoptosis via production of reactive oxygen species in esophageal cancer cells. Curr Cancer Drug Targets. 2013;13:611–24. doi:10.2174/15680096113139990030. PMID:23597192.
- Liao YJ, Bai HY, Li ZH, Zou J, Chen JW, Zheng F, Zhang JX, Mai SJ, Zeng MS, Sun HD, et al. Longikaurin A, a natural ent-kaurane, induces G2/M phase arrest via downregulation of Skp2 and apoptosis induction through ROS/JNK/c-Jun pathway in hepatocellular carcinoma cells. Cell Death Dis. 2014;5:e1137. doi:10.1038/cddis.2014.66. PMID:24651440.
- Sun X, Wang W, Chen J, Cai X, Yang J, Yang Y, Yan H, Cheng X, Ye J, Lu W, et al. The natural diterpenoid isoforretin a inhibits thioredoxin-1 and triggers potent ros-mediated antitumor effects. Cancer Res. 2017;77:926–36. doi:10.1158/0008-5472.CAN-16-0987. PMID:28011619.
- Redza-Dutordoir M, Averill-Bates DA. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta. 2016;1863:2977–92. doi:10.1016/j.bbamcr.2016.09.012. PMID:27646922.
- Simon HU, Haj-yehia a and levi-schaffer F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis. 2000;5:415–8. doi:10.1023/A:1009616228304. PMID:11256882.
- Son Y, Cheong YK, Kim NH, Chung HT, Kang DG, Pae HO. Mitogen-activated protein kinases and reactive oxygen species: How can ROS Activate MAPK 7athways? J Signal Transduct. 2011;2011:792639. doi:10.1155/2011/792639. PMID:21637379.
- Wu J, Zhang H, Xu Y, Zhang J, Zhu W, Zhang Y, Chen L, Hua W, Mao Y. Juglone induces apoptosis of tumor stem-like cells through ROS-p38 pathway in glioblastoma. BMC Neurol. 2017;17:70. doi:10.1186/s12883-017-0843-0. PMID:28388894.
- Wang H, Ye Y, Chui JH, Zhu GY, Li YW, Fong DW, Yu ZL. Oridonin induces G2/M cell cycle arrest and apoptosis through MAPK and p53 signaling pathways in HepG2 cells. Oncol Rep. 2010;24:647–51. PMID:20664969.
- Ma YC, Su N, Shi XJ, Zhao W, Ke Y, Zi X, Zhao NM, Qin YH, Zhao HW, Liu HM. Jaridonin-induced G2/M phase arrest in human esophageal cancer cells is caused by reactive oxygen species-dependent Cdc2-tyr15 phosphorylation via ATM-Chk1/2-Cdc25C pathway. Toxicol Appl Pharmacol. 2015;282:227–36. doi:10.1016/j.taap.2014.11.003. PMID:25450480.
- Sun WJ, Huang H, He B, Hu DH, Li PH, Yu YJ, Zhou XH, Lv Z, Zhou L, Hu TY, et al. Romidepsin induces G2/M phase arrest via Erk/cdc25C/cdc2/cyclinB pathway and apoptosis induction through JNK/c-Jun/caspase3 pathway in hepatocellular carcinoma cells. Biochem Pharmacol. 2017;127:90–100. doi:10.1016/j.bcp.2016.12.008. PMID:28012958.
- Landau HJ, McNeely SC, Nair JS, Comenzo RL, Asai T, Friedman H, Jhanwar SC, Nimer SD, Schwartz GK. The checkpoint kinase inhibitor AZD7762 potentiates chemotherapy-induced apoptosis of p53-mutated multiple myeloma cells. Mol Cancer Ther. 2012;11:1781–8. doi:10.1158/1535-7163.MCT-11-0949. PMID:22653969.
- Dolado I, Swat A, Ajenjo N, De Vita G, Cuadrado A, Nebreda AR. p38alpha MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell. 2007;11:191–205. doi:10.1016/j.ccr.2006.12.013. PMID:17292829.
- Xu HG, Zhai YX, Chen J, Lu Y, Wang JW, Quan CS, Zhao RX, Xiao X, He Q, Werle KD, et al. LKB1 reduces ROS-mediated cell damage via activation of p38. Oncogene. 2015;34:3848–59. doi:10.1038/onc.2014.315. PMID:25263448.
- Park SG, Kim SH, Kim KY, Yu SN, Choi HD, Kim YW, Nam HW, Seo YK, Ahn SC. Toyocamycin induces apoptosis via the crosstalk between reactive oxygen species and p38/ERK MAPKs signaling pathway in human prostate cancer PC-3 cells. Pharmacol Rep. 2017;69:90–6. doi:10.1016/j.pharep.2016.10.014. PMID:27912102.
- Yarden RI, Pardo-Reoyo S, Sgagias M, Cowan KH, Brody LC. BRCA1 regulates the G2/M checkpoint by activating Chk1 kinase upon DNA damage. Nat Genet. 2002;30:285–9. doi:10.1038/ng837. PMID:11836499.
- Busino L, Chiesa M, Draetta GF, Donzelli M. Cdc25A phosphatase: Combinatorial phosphorylation, ubiquitylation and proteolysis. Oncogene. 2004;23:2050–6. doi:10.1038/sj.onc.1207394. PMID:15021892.
- Rossi A, Sacco PC, Santabarbara G, Sgambato A, Casaluce F, Palazzolo G, Maione P, Gridelli C. Developments in pharmacotherapy for treating metastatic non-small cell lung cancer. Expert Opin Pharmacother. 2017;18:151–63. doi:10.1080/14656566.2017.1280460. PMID:28067062.
- Deng R, Tang J, Xia LP, Li DD, Zhou WJ, Wang LL, Feng GK, Zeng YX, Gao YH, Zhu XF. ExcisaninA, a diterpenoid compound purified from Isodon MacrocalyxinD, induces tumor cells apoptosis and suppresses tumor growth through inhibition of PKB/AKT kinase activity and blockade of its signal pathway. Mol Cancer Ther. 2009;8:873–82. doi:10.1158/1535-7163.MCT-08-1080. PMID:19372560.
- Liu M, Wang WG, Sun HD, Pu JX. Diterpenoids from isodon species: an update. Nat Prod Rep. 2017;34:1090–140. doi:10.1039/C7NP00027H. PMID:28758169.
- Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361:1570–83. doi:10.1056/NEJMra0901217. PMID:19828534.
- Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9:537–49. doi:10.1038/nrc2694. PMID:19629069.
- Valdiglesias V, Giunta S, Fenech M, Neri M, Bonassi S. Gammah2ax as a marker of DNA double strand breaks and genomic instability in human population studies. Mutat Res. 2013;753:24–40. doi:10.1016/j.mrrev.2013.02.001. PMID:23416207.
- Reinhardt HC, Yaffe MB. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr Opin Cell Biol. 2009;21:245–55. doi:10.1016/j.ceb.2009.01.018. PMID:19230643.
- Sur S, Agrawal DK. Phosphatases and kinases regulating CDC25 activity in the cell cycle: Clinical implications of CDC25 overexpression and potential treatment strategies. Mol Cell Biochem. 2016;416:33–46. doi:10.1007/s11010-016-2693-2. PMID:27038604.
- Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 2010;108:73–112. doi:10.1016/B978-0-12-380888-2.00003-0. PMID:21034966.
- Gavet O, Pines J. Progressive activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev Cell. 2010;18:533–43. doi:10.1016/j.devcel.2010.02.013. PMID:20412769.
- Chou YW, Zhang L, Muniyan S, Ahmad H, Kumar S, Alam SM, Lin MF. Androgens upregulate Cdc25C protein by inhibiting its proteasomal and lysosomal degradation pathways. PLoS One. 2013;8:e61934. doi:10.1371/journal.pone.0061934. PMID:23637932.
- Thompson R, Meuth M, Woll P, Zhu Y, Danson S. Treatment with the Chk1 inhibitor Go6976 enhances cisplatin cytotoxicity in SCLC cells. Int J Oncol. 2012;40:194–202. PMID:21894433.
- Reinhardt HC, Aslanian AS, Lees JA, Yaffe MB. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell. 2007;11:175–89. doi:10.1016/j.ccr.2006.11.024. PMID:17292828.
- Zhang J, Nannapaneni S, Wang D, Liu F, Wang X, Jin R, Liu X, Rahman MA, Peng X, Qian G, et al. Phenformin enhances the therapeutic effect of selumetinib in KRAS-mutant non-small cell lung cancer irrespective of LKB1 status. Oncotarget. 2017;8:59008–22. PMID:28938614.
- Yang L, Zhou Y, Li Y, Zhou J, Wu Y, Cui Y, Yang G, Hong Y. Mutations of p53 and KRAS activate NF-kappaB to promote chemoresistance and tumorigenesis via dysregulation of cell cycle and suppression of apoptosis in lung cancer cells. Cancer Lett. 2015;357:520–6. doi:10.1016/j.canlet.2014.12.003. PMID:25499080.
- Flatmark K, Maelandsmo GM, Martinsen M, Rasmussen H, Fodstad O. Twelve colorectal cancer cell lines exhibit highly variable growth and metastatic capacities in an orthotopic model in nude mice. Eur J Cancer. 2004;40:1593–8. doi:10.1016/j.ejca.2004.02.023. PMID:15196545.
- Kumar D, Das B, Sen R, Kundu P, Manna A, Sarkar A, Chowdhury C, Chatterjee M, Das P. Andrographolide analogue induces apoptosis and autophagy mediated cell death in U937 Cells by inhibition of PI3K/Akt/mTOR pathway. PLoS One. 2015;10:e0139657. doi:10.1371/journal.pone.0139657. PMID:26436418.
- Sanceau J, Poupon M-F, Delattre O, Sastre-Garau X, Wietzerbin J. Strong inhibition of ewing tumor xenograft growth by combination of human interferon-alpha or interferon-beta with ifosfamide. Oncogene. 2002;21:7700. doi:10.1038/sj.onc.1205881. PMID:12400012.
- Leuraud P, Taillandier L, Medioni J, Aguirre-Cruz L, Criniere E, Marie Y, Kujas M, Golmard JL, Duprez A, Delattre JY, et al. Distinct responses of xenografted gliomas to different alkylating agents are related to histology and genetic alterations. Cancer Res. 2004;64:4648–53. doi:10.1158/0008-5472.CAN-03-3429. PMID:15231677.
- Guo Q, He J, Shen F, Zhang W, Yang X, Zhang C, Zhang Q, Huang JX, Wu ZD, Sun XC, et al. TCN, an AKT inhibitor, exhibits potent antitumor activity and enhances radiosensitivity in hypoxic esophageal squamous cell carcinoma in vitro and in vivo. Oncol Lett. 2017;13:949–54. doi:10.3892/ol.2016.5515. PMID:28356983.