
ABSTRACT
The PI3K/mTOR pathway is one of the most frequently aberrantly activated pathways in human malignancies, such as renal cell carcinoma (RCC), and contributes to resistance to antitumor therapies. Thus, PI3K/mTOR is an attractive target for the development of antitumor agents. In this study, we evaluated the preclinical effects of a novel inhibitor SN202. We examined Akt/mTOR activities in renal cancer cells after SN202 treatment. The preclinical effects of SN202 on tumor growth were evaluated in renal cancer cells in vitro and in murine xenografts in vivo. SN202 inhibits PI3Kα, PI3Kγ, and mTOR, the corresponding IC50 values were 3.2, 3.3, and 1.2 nM, respectively. In A498, 786-0, and ACHN renal cancer cell lines, SN202 inhibits cell proliferation in a dose-dependent manner and significantly inhibits 786-0 cell growth. Western blot analysis revealed that SN202 decreases the phosphorylation of PI3K downstream signaling molecules, Akt and S6K, in 786-0 renal cancer cells. Furthermore, oral administration of SN202 results in significant inhibition in human renal carcinoma xenografts in nude mice and favourable pharmacokinetic properties in rats. These results suggest that SN202 might be a promising therapeutic agent against RCC as a dual PI3K/mTOR inhibitor.
Introduction
The PI3K/AKT signaling pathway plays a key role in carcinogenesis as an activator of its downstream effectors, such as the mammalian target of rapamycin (mTOR). mTOR is associated with numerous cellular events, including cell cycle progression, cellular proliferation, differentiation, apoptosis, and angiogenesis.Citation1, Citation2 Class I PI3K catalyses the conversion of phosphatidylinositol 4,5-biphosphate (PIP2) to the second messenger phosphatidylinositol 3,4,5-triphosphate (PIP3).Citation3 Interaction between PIP3 and Akt provokes conformational changes in Akt, resulting in Akt activation through phosphorylation at Thr308 by phosphatidylinositol-dependent kinase 1 (PDK1).Citation4 Maximal activation can be reached by additional phosphorylation at Ser473 by PDK2.Citation5 The mTOR signaling pathway is a major compensatory pathway that confers resistance to numerous cancer drugs,Citation6 including signal transduction inhibitors, such as small-molecule tyrosine kinase inhibitors and antibodies.Citation7-11 The PI3K/mTOR signaling pathway is one of the most frequently aberrantly activated pathways in human cancer. In renal cell carcinoma (RCC), PI3K overexpression was confirmed as crucial to tumor progression, and the activation of PI3K protein was found to be significantly associated with reduced survival times.Citation12 Although the mTOR inhibitors everolimus and temsirolimus are approved for the treatment of RCC, significant benefits are seen in a minority of patients because these mTORC1 inhibitors can active PI3K/Akt and mitogen-activated protein kinase or can upregulate mTORC2.Citation13 Small-molecule drugs formulated for inhibiting both PI3K and mTOR in this pathway, such as PF-04691502 and NVP-BEZ235, which may establish superior antitumor effects are currently being tested in various preclinical and clinical trials.Citation14-19 Unfortunately, a dose-escalation phase Ib trial of BEZ235 showed significant toxicity without objective responses.Citation13
We are interested in the identification of novel small molecule inhibitors of the PI3K/mTOR pathway because such molecules may be suitable drug candidates in the treatment of RCC. In this work, we describe a novel compound, SN202 (A), which was originally discovered in targeting PI3K/mTOR kinase screening.
Figure 1. Chemical structure and biochemical activity of SN202. (A) The chemical structure of SN202. Inhibition rate of SN202 on kinase activity of mTOR (B), PI3Kα (C) and PI3Kγ (D).
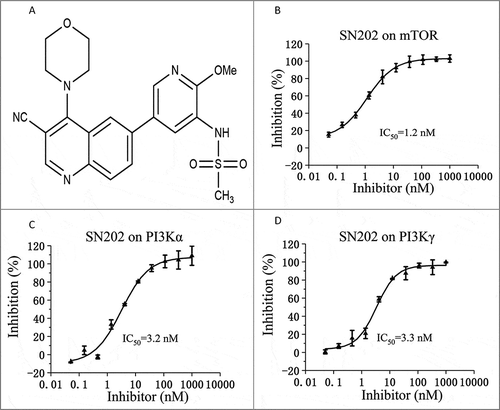
Results
SN202 is an ATP-competitive PI3K/mTOR inhibitor
To explore kinase selectivity, inhibitory activities of SN202 against PI3K/mTOR were first measured. Recombinant human p110α/85α, p110γ, and mTOR were used in an ATP-competitive assay in the presence of 6–25 μM ATP. In this assay, SN202 showed inhibition against PI3K and mTOR kinases. The IC50 values for PI3K isoform p110α/85α, PI3K isoform p110γ, and mTOR were 3.2, 3.3, and 1.2 nM, respectively (). The kinase inhibitory activities were dependent on concentration (B–D) and were stronger than that of the tool compound PI-103 (). The results indicate that SN202 is an ATP-competitive PI3K/mTOR dual inhibitor.
Table 1. In vitro IC50 values for SN202 against recombinant PI3-kinases and mTOR
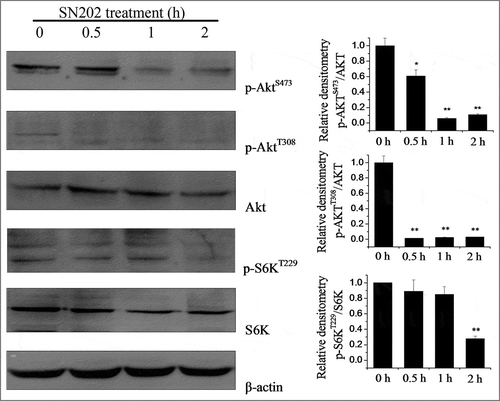
SN202 inhibits the phosphorylation of S6K and Akt in 786-0 cells
Akt is a direct target of the phosphatidylinositol 3-kinase.Citation20 Furthermore, the mTOR kinase contains two complexes, mTORC1 and mTORC2,Citation21-23 which phosphorylate S6K and Akt, two crucial downstream signaling molecules of the PI3K pathway. We therefore analyzed the phosphorylation of S6K and Akt in 786-0 cells by Western blot using antibodies specific for either serine- or threonine-phosphorylated S6K and Akt, as well as antibodies that recognize all S6K or Akt forms. As shown in , SN202-treated (10 μM) 786-0 cells result in a time-dependent inhibition of Akt phosphorylation at Ser473 and Thr308 and S6K phosphorylation at Thr229.
Figure 2. Inhibition of Akt and S6K phosphorylation by SN202. 786-0 cells were incubated for the indicated time periods with 10 µM SN202. Zero time samples were treated with DMSO. These cells were harvested and analyzed by Western blot using antibodies specific for either serine- or threonine-phosphorylated S6K and Akt. β-actin was used as a loading control. Results are expressed as the mean ± SEM of three independent experiments. One-way ANOVA for SN202 treatment versus the control group: *P < 0.05, **P< 0.01.
SN202 inhibits cell proliferation and impedes cell cycle progression in renal cancer cells
The PI3K/AKT/mTOR pathway is frequently aberrantly activated in human cancer and is involved in different biological effects, such as cell cycle progression and cell proliferation. To assess the inhibition potential of SN202 against cancer cell proliferation, three renal cancer cell lines were used in a Cell Counting Kit-8 cell viability assay. SN202 inhibited cell proliferation of 786-0, A498 and ACHN in a dose-dependent manner (), with IC50 values of 0.486, 0.176, and 0.298 μM (). These IC50 values were obviously lower than that of the compounds BEZ235 and BKM120, which may be promising candidates for development as new drugs targeting the PI3K pathway. However, the maximum inhibition of SN202 against cell proliferation in the 786-0 line (89.10%) was higher than that in the ACHN and A498 cell lines (58.76% and 55.56%, respectively).
Figure 3. Inhibition potential of SN202 against RCC. The inhibitory effect of SN202 on cell proliferation was determined by CCK-8 assay. 786-0 (A), ACHN (B) and A498 (C) cells were treated with varying concentrations of SN202, BKM120 and BEZ235. Data are presented as the means of three independent experiments.
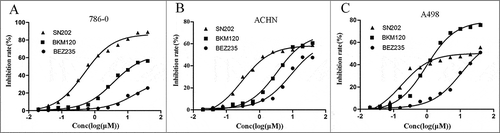
Table 2. In vitro IC50 values for SN202 against three renal cancer cell lines
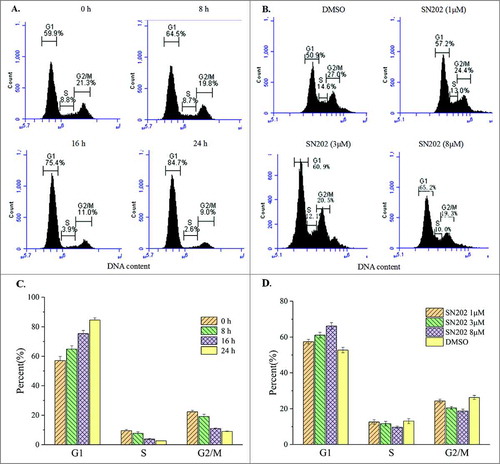
Subsequently, cell cycle analysis was performed in the 786-0 cells by flow cytometry. Results showed that SN202 inhibited cell cycle progression via G0/G1 arrest ().
Figure 4. Inhibition of 786-0 cell cycle progression by SN202. (A) and (C) 786-0 cells were treated with 8 μM SN202 for 8, 16, and 24 h. (B) and (D) 786-0 cells were incubated in the presence of 1, 3, and 8 μM SN202 for 16 h. Cell cycle was determined by a flow cytometry using propidium iodide staining. Results are representative of three independent experiments at minimum.
SN202 is effective as a single agent in a renal cancer xenograft model
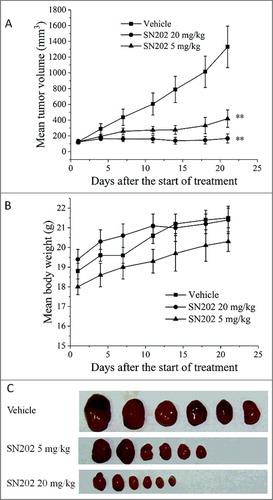
The effect of SN202 on the growth of renal cancer xenograft was evaluated using an in vivo nude mice model. As shown in , SN202 significantly reduced the growth of the 786-0 renal carcinoma xenografts by oral administration. Compared with the control group (vehicle) at 21 days after treatment, the decreases in tumor weight (IR) were 68.61% (p < 0.01) for SN202 5 mg/kg group, 88.99% (p < 0.01) for SN202 20 mg/kg group. The mice body weights of the SN202-treated groups were not significantly affected and no apparent toxicity was observed (data not shown), indicating the relative safety of these dose regimens.
Figure 5. Inhibition of tumor growth by SN202 in 786-0 xenograft model. (A) Mean tumor volume after the start of treatment. (B) Mean body weight of mice after the start of treatment. (C) Tumors resected from nude mice on 21 d. Renal carcinoma 786-0 cells (106 cells) were implanted into the left armpit of each nude mouse to develop tumors. Mice bearing tumors around 120 mm3 were orally treated with SN202 (5 mg/kg and 20 mg/kg) once per day for 21 days. Tumor volume and body weight were recorded twice per week. Data are expressed as mean ± SEM. **P< 0.01 versus the vehicle group.
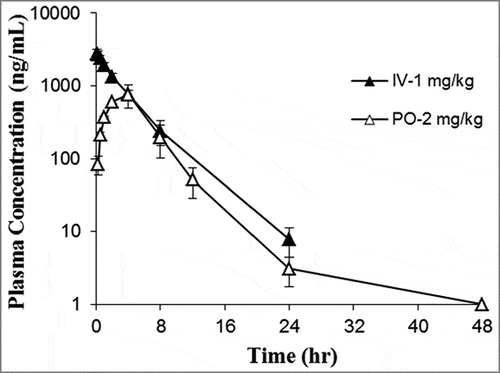
Pharmacokinetic properties of SN202
The mean plasma concentration versus time profile is presented in . In male Sprague Dawley rats, following a single intravenous (iv) bolus of 1 mg/kg, SN202 exhibited an elimination half-life (t1/2) of 3.06 ± 0.234 h, an apparent distribution volume (Vss) of 0.357 ± 0.0235 L/kg, and an AUC0-∞ of 10152 ± 1659 h ng/mL. Oral administration of a 2 mg/kg dose resulted in a tmax of 3.33 ± 1.15 h, a Cmax of 786 ± 227 ng/mL, an t1/2 of 3.55 ± 1.39 h, and an AUC0-∞ of 4805 ± 1394 h ng/mL. Consequently, oral bioavailability was 23.7 ± 6.87%.
Figure 6. Mean plasma concentration-time profile of SN202 after iv or oral administration. Rats were given a single iv bolus at 1 mg/kg or a single oral administration at 2 mg/kg. At the indicated times after dosing, blood samples were obtained via tail vein of individual rats. Plasma was separated and then determined by LC/MS/MS.
Discussion
The PI3K/mTOR signaling pathway is constitutively activated in numerous types of cancer and involved in cell growth, survival, and resistance to chemotherapy and radiotherapy.Citation24 Thus, PI3K/mTOR is an advantageous target for the development of antitumor agents. PI3K/mTOR dual inhibitors are considered more effective against human cancers than PI3K- or mTOR-specific inhibitors.Citation18 Our study focused on screening PI3K/mTOR dual inhibitors and eventually led to the identification of SN202, a novel small molecule with excellent growth inhibitory effects on several human cancer cell lines, including lung, gastric, colon, and breast cancer cell lines (data not shown).
In RCC, though huge progresses have made by targeted therapies, the twelve FDA approved drugs mainly focusing on VEGF pathway did not overcome drug resistance or achieve long-term patient survival. The activation of PI3K/mTOR signaling pathway contributes to RCC progression.Citation25-27 Several inhibitors targeting PI3K/mTOR have entered clinical trials and revealed their efficacy in the treatment of renal cancer patients,Citation28 as well as in combination therapy with other antitumor agents, such as sorafenib.Citation29
In this study, SN202 was determined to be an ATP-competitive PI3K/mTOR dual inhibitor, and was shown to significantly inhibit PI3Kα and PI3Kγ activity. Class I PI3Ks consists of a regulatory subunit and one of four catalytic subunits: p110α, p110β, and p110δ and p110γ. PI3Kα has been linked to the activation of the PI3K signaling pathway, and PI3Kγ is required for AKT activation in many cancers. Considering the two isoform's prominent role in cancers, this may provide a therapeutic target in solid tumor. However, the development of small molecules targeting PI3Kβ, and δ was considered in some tumors, especially in haematological malignancies. Thus, the isoform-selective inhibitory effect of SN202 on PI3Kβ and δ remain to be determined. In addition, the ATP-binding sites of most kinases are highly conservative, inhibitors designed based on ATP-binding domain may be of poor selectivity and strong cytotoxicity. Notably, a research report based on 243 clinical kinase inhibitors showed that the number of targets for a given drug differed substantiallyCitation30. Therefore, confirmation of a large panel of kinase selectivity profile of SN202 will be needed.
Our data showed that SN202 exhibits similar biological activities to other inhibitors and is more potent in biochemical and antiproliferative cellular assays than PI-103 and certain reference inhibitors,Citation31 such as NVP-BEZ235 and BKM120. In the RCC 786-0 cell line, SN202 suppresses cell growth by inducing cell cycle arrest. In mechanistic assays, SN202 can significantly inhibit AKT phosphorylation at Ser-473 and Thr-308 in 786-0 cells. The function of crucial downstream target Akt exerts through mTOR by the activation of p70 robosomal S6 kinase (S6K), thus, we detected S6K phosphorylation in 786-0 cells. Subsequently, a decrease in S6K phosphorylation at Thr-229 induced by the treatment with SN202 was observed. Based on these results, it is rational that SN202 produced a higher maximum inhibition against cell proliferation in 786-0 than that in ACHN and A498. Regardless of the VHL gene status, 786-0 cells are PTEN negative and have a higher phospho-Akt level at both Thr308 and Ser473 compared with ACHN and A498, suggesting that the constitutive activation of this pathway is more referential than the VHL status for RCC treatment by PI3K/mTOR inbitors.
Antitumor efficacy was demonstrated in a preclinical renal carcinoma xenograft model and the pharmacokinetic profiles were reported in this study. In previous studies, significant toxicity in preclinical models was the main issue of PI3K/mTOR dual inhibitors.Citation32 In contrast to this, SN202 showed a dose-dependent inhibition of tumor growth but did not cause body weight loss and obvious adverse effects over a relatively wide dose range in mice. Good safety as well as favourable exposure was also observed after orally administration in rats. Despite these advantageous pharmacologic profile and druggability, given the negative clinical trial results of BEZ-235, the more comprehensive safety evaluation for SN202 will be needed.
In summary, we showed a structurally novel PI3K/mTOR dual inhibitor SN202 with marked antitumor effects against RCC in both in vitro and in vivo experiments. This compound also demonstrated to be safe and displayed favourable pharmacokinetic properties in preclinical species. Thus, SN202 is promising for further preclinical and clinical development to investigate the therapy value in renal cell carcinoma.
Materials and methods
Compounds and reagents
SN202 with a molecule weight of 439.5 was synthesized and provided by Sichuan Sinovation Bio-technology Co., LTD (Chengdu, PR China). Structure and purity were analysed using mass spectrometry (Supplemental Figure S1), 1HNMR spectroscopy (Supplemental Figure S2) and High Performance Liquid Chromatography (HPLC, Supplemental Figure S3). SN202 displayed over 99% purity in HPLC analysis. PI103, BEZ235, and BKM120 of more than 99% purities were purchased from Abmole Bioscience. In the in vitro analysis, a 10 mmol/L stock solution in 100% DMSO was diluted to an appropriate concentration in a culture medium prior to addition to the cells. The final DMSO concentration was maintained at 0.1% in the control and the compound-treated cells. Primary antibodies directed against phospho-Akt (Ser473/Thr308), Akt, phospho-S6K (Ser371/Thr229), and S6K were purchased from Cell Signaling Technologies. The other reagents used in the study were obtained from Sigma and Cell Signaling Technologies.
PI3K/mTOR kinase assay
The inhibition of PI3K kinase activity by SN202 was tested using the Kinase-Glo Luminescent Kinase Assay (Promega). The enzyme reaction involved 1.65 nM recombinant human PI3Kα (Invitrogen, Cat. No. PV4788) or 7.6 nM p110γ (Invitrogen, Cat. No. PR8641C), 50 μM PIP2, SN202 (0.05-1000 nM), and 25 μM ATP (Sigma, Cat. No. A7699-1G). The reaction was initiated by the addition of ATP and terminated by the addition of a 10 μL Kinase-Glo reagent. The 96-well plate was shaken for 10 min and incubated for 60 min before reading in a plate reader (SynegyMx, Biotek) for luminescence.
The inhibition of mTOR kinase activity by SN202 was tested using the LANCE Ultra phosphatase assay (Perkin Elmer). The enzyme reaction contained 4 nM mTOR (Millipore, Cat. No. 14–770), 50 nM ULight-4E-BP1 (PE, Cat. No. TRF0128-M), SN202 (0.05-1000 nmol/L), and 6 μM ATP (Sigma, Cat. No. A7699-1G). The reaction mixture was shaken for 10 min and terminated by the addition of 2 nM Eu-anti-P-4E-BP1 (PE, Cat. No. TRF0216-M). The 96-well plate was incubated for 60 min before reading in a plate reader (Envision, Perkin Elmer) for luminescence.
Cell culture and cell proliferation assay
All cell lines were obtained from ATCC and maintained at 37 °C and 5% CO2 in a DMEM medium supplemented with 10% fetal bovine serum.
Cells (2.5 × 105 per well) were seeded in 96-well plates and treated with various concentrations of SN202 for 48 or 72 h. At the end of the incubation period, 10 μL of the cell proliferation reagent Cell Counting Kit-8 (Dojindo Labrotories) was added to each well and incubated for 1 h. Optical density was measured at 570 nm in a plate reader (Spectramax M5), and then the proliferation activity curve was plotted to determine the IC50 value. Each experiment was performed in triplicate. A final concentration of 0.1% DMSO was used as negative control. Two PI3K/mTOR inhibitors, NVP-BEZ235 and BKM120, were used as positive control.
Flow cytometric analysis
SN202-treated cells were harvested at a semiconfluent stage and stained with a solution containing 50 μg/mL propidium iodide (Key GEN Biotech, KGA214). Flow cytometry analysis was conducted in a Accuri™ C6 (BD Biosciences) flow cytometer, and data were analyzed using FlowJo software.
Western blot analysis
After SN202 treatments, the cells were washed with PBS and lysed with lysis buffer with a protease inhibitor. The protein concentration in the cell lysates was determined by BCA assay, and 15 μg of protein from a sample were loaded onto 10% SDS–polyacrylamide gels. Proteins were transferred to nitrocellulose membranes, and Western blot analysis was performed. The primary antibodies used in the study were those specific for either serine- or threonine-phosphorylated S6K and Akt and those for S6K and Akt. All antibodies were used as described by the manufacturers. Western blot membranes were imaged, and results were normalized to total S6K or Akt intensity using densitometry.
Tumor growth inhibition in xenograft model
Female nude mice (7–8 weeks old) were obtained from SIPPR-BK Labs (Shanghai). Animals were maintained under standard cleanroom conditions. The 786-0 cells (1 × 107 cells) were resuspended in a serum-free medium and then implanted subcutaneously into the left armpit of the nude mice (n = 6). Treatments were initiated when tumors reached a mean volume of around 120 mm3. SN202 was formulated in 0.5% carboxymethyl cellulose (w/v) and administered orally at 5 and 20 mg/kg once per day for 21 days. Mice in the control group were dosed with an equivalent volume of the vehicle. Tumor volume and animal weight were measured every 3–4 days. At the end of the experiment, the tumors were excised and then weighed. Tumor volume (TV) was calculated by the formula: TV = Length × Width2/2. The tumor growth inhibition rate (IR) on the basis of tumor weight was calculated from IRtw = (1 -TWt/TWc) × 100%, where TWt represents the mean tumor weight of a treated group and TWc indicates that of the control group. Data are presented as mean ± SEM. Student's t test was used to determine the P value for the comparisons between the treatment and vehicle groups.
Pharmacokinetic study in rats
The male Sprague Dawley rats (6–8 weeks old, 220–250 g) used in the PK experiment were obtained from Shanghai SLAC Laboratory Animal Co., Ltd. The intravenous injection solution was prepared by a vehicle consisting of 10% DMSO, 20% propylene glycol, 20% PEG400, and 10% Solutol HS15 in water (w/v). For oral administration, SN202 was suspended in 0.5% carboxymethyl cellulose in water (w/v). Blood samples (approximately 150 μL) were collected via tail vein at 0.083 (just iv group), 0.25, 0.5, 1, 2, 4, 8, 12, 24, and 48 (just oral group) h after a single iv bolus or oral gavage. Blood samples stored in tubes containing sodium heparin as anticoagulant were separated by centrifugation and then frozen at -70 °C until analysis. After treatment with acetonitrile containing diclofenac as internal standard and centrifugation, 3 μL of the supernatant was injected onto a HPLC/MS/MS triple quadrupole mass spectrometer (AB-sciex5500, Applied Biosystems). A Waters BEH C18 column (2.1 × 50 mm, 1.7 µm) was used to separate the samples. A non-compartmental module of WinNonlin® Professional 6.2 (Pharsight, USA) was used to calculate the parameters. All the animal studies were approved and supervised by Sichuan University Animal Ethics Experimentation Committee following the regulation of People's Republic of China on Use of Experimental Animals.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
supp_mat_1470733_KCBT.zip
Download Zip (246.2 KB)Acknowledgments
We express our gratitude for excellent technical assistance provided by Dr. Sheng Zhang, Dr. Wei Fu and Dr. Xiaoping Gao, Biprotein Biotechnology, Chengdu, China. This work was supported by Sichuan Science and Technology Program (Grant No. 2018JY0188), the Fundamental Research Funds for the Central Universities (Grant No. 2018SCU12043) the Postdoctoral Foundation of Sichuan University.
References
- Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genetics. 2006;7(8):606–619. PMID: 16847462; doi: https://doi.org/10.1038/nrg1879.
- Dey N, Sun Y, Carlson JH, Wu H, Lin X, Leyland-Jones B, De P. Anti-tumor efficacy of BEZ235 is complemented by its anti-angiogenic effects via downregulation of PI3K-mTOR-HIF1alpha signaling in HER2-defined breast cancers. Am J Cancer Res. 2016;6(4):714–746. PMID: 27186427.
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase–AKT pathway in human cancer. Nat Rev Cancer. 2002;2(7):489–501. PMID: 12094235; doi: https://doi.org/10.1038/nrc839.
- Vara JÁF, Casado E, de Castro J, Cejas P, Belda-Iniesta C, González-Barón M. PI3K/Akt signalling pathway and cancer. Cancer Treatment Rev. 2004;30(2):193–204. PMID: 15023437; doi: https://doi.org/10.1016/j.ctrv.2003.07.007
- Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PRJ, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr Biol. 1997;7(4):261–269. PMID: 9094314; doi: https://doi.org/10.1016/S0960-9822(06)00122-9.
- Guri Y, Hall MN. mTOR signaling confers resistance to targeted cancer drugs. Trends Cancer. 2016;2(11):688–697. PMID: 28741507; doi: https://doi.org/10.1016/j.trecan.2016.10.006.
- Ding J, Romani J, Zaborski M, MacLeod RAF, Nagel S, Drexler HG, Quentmeier H. Inhibition of PI3K/mTOR overcomes Nilotinib resistance in BCR-ABL1 positive leukemia cells through translational down-regulation of MDM2. Plos One. 2013;8(12):e83510. PMID: 24349524; doi: https://doi.org/10.1371/journal.pone.0083510.
- Sun Z, Li Q, Zhang S, Chen J, Huang L, Ren J, Chang Y, Liang Y, Wu G. NVP-BEZ235 overcomes gefitinib-acquired resistance by down-regulating PI3K/AKT/mTOR phosphorylation. OncoTargets Therapy. 2015;8:269–277. PMID: 25674002; doi: https://doi.org/10.2147/OTT.S62128.
- Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, Linn SC, Gonzalez-Angulo AM, Stemke-Hale K, Hauptmann M, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of Trastuzumab resistance in breast cancer. Cancer Cell. 2007;12(4):395–402. PMID: 17936563; doi: https://doi.org/10.1016/j.ccr.2007.08.030.
- Park BH, Davidson NE. PI3 kinase activation and response to Trastuzumab therapy: What's neu with Herceptin resistance? Cancer Cell. 2007;12(4):297–299. PMID: 17936554; doi: https://doi.org/10.1016/j.ccr.2007.10.004.
- Gajria D, Chandarlapaty S. HER2-amplified breast cancer: mechanisms of trastuzumab resistance and novel targeted therapies. Expert Rev Anticancer Therapy. 2011;11(2):263–275. PMID: 21342044; doi: https://doi.org/10.1586/era.10.226.
- Merseburger AS, Hennenlotter J, Kuehs U, Simon P, Kruck S, Koch E, Stenzl A, Kuczyk MA. Activation of PI3K is associated with reduced survival in renal cell carcinoma. Urologia Internationalis. 2008;80(4):372–377. PMID: 18587247; doi: https://doi.org/10.1159/000132694.
- Carlo MI, Molina AM, Lakhman Y, Patil S, Woo K, Deluca J, Lee CH, Hsieh JJ, Feldman DR, Motzer RJ. A Phase Ib Study of BEZ235, a dual inhibitor of phosphatidylinositol 3-Kinase (PI3K) and Mammalian Target of Rapamycin (mTOR), in patients with advanced renal cell Carcinoma. Oncologist. 2016;21(7):787–788. PMID: 27286790; doi: https://doi.org/10.1634/theoncologist.2016-0145.
- Maira S-M, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, Brachmann S, Chène P, De Pover A, Schoemaker K, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Therapeutics. 2008;7(7):1851–1863. PMID: 18606717; doi: https://doi.org/10.1158/1535-7163.MCT-08-0017.
- Yuan J, Mehta PP, Yin M-J, Sun S, Zou A, Chen J, Rafidi K, Feng Z, Nickel J, Engebretsen J, et al. PF-04691502, a potent and selective oral inhibitor of PI3K and mTOR kinases with antitumor activity. Mol Cancer Therapeutics. 2011;10(11):2189–2199. PMID: 21750219; doi: https://doi.org/10.1158/1535-7163.MCT-11-0185.
- Pal I, Mandal M. PI3K and Akt as molecular targets for cancer therapy: current clinical outcomes. Acta Pharmacologica Sinica. 2012;33(12):1441–1458. PMID: 22983389; doi: https://doi.org/10.1038/aps.2012.72.
- Britten CD, Adjei AA, Millham R, Houk BE, Borzillo G, Pierce K, Wainberg ZA, LoRusso PM. Phase I study of PF-04691502, a small-molecule, oral, dual inhibitor of PI3K and mTOR, in patients with advanced cancer. Investigational New Drugs. 2014;32(3):510–517. PMID: 24395457; doi: https://doi.org/10.1007/s10637-013-0062-5.
- Gravina GL, Mancini A, Scarsella L, Colapietro A, Jitariuc A, Vitale F, Marampon F, Ricevuto E, Festuccia C. Dual PI3K/mTOR inhibitor, XL765 (SAR245409), shows superior effects to sole PI3K [XL147 (SAR245408)] or mTOR [rapamycin] inhibition in prostate cancer cell models. Tumour Biol. 2016;37(1):341–351. PMID: 26219891; doi: https://doi.org/10.1007/s13277-015-3725-3.
- Serra V, Markman B, Scaltriti M, Eichhorn PJ, Valero V, Guzman M, Botero ML, Llonch E, Atzori F, Di Cosimo S, et al. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Research. 2008;68(19):8022–8030. PMID: 18829560; doi: https://doi.org/10.1158/0008-5472.CAN-08-1385.
- Datta K, Bellacosa A, Chan TO, Tsichlis PN. Akt is a direct target of the phosphatidylinositol 3-kinase activation by growth factors, v-src and v-ha-ras, in sf9 and mammalian cells. J Biol Chem. 1996;271(48):30835–30839. doi: https://doi.org/10.1074/jbc.271.48.30835.
- Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6(9):729–734. PMID: 16915295; doi: https://doi.org/10.1038/nrc1974.
- Guertin DA, Sabatini DM. An expanding role for mTOR in cancer. Trends Mol Med. 2005;11(8):353–361. PMID: 16002336; doi: https://doi.org/10.1016/j.molmed.2005.06.007.
- Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12(1):9–22. PMID: 17613433; doi: https://doi.org/10.1016/j.ccr.2007.05.008.
- Wang Z, Huang Y, Zhang J. Molecularly targeting the PI3K-Akt-mTOR pathway can sensitize cancer cells to radiotherapy and chemotherapy. Cell Mol Biol Letters. 2014;19(2):233–242. PMID: 24728800; doi: https://doi.org/10.2478/s11658-014-0191-7.
- Cho DC, Cohen MB, Panka DJ, Collins M, Ghebremichael M, Atkins MB, Signoretti S, Mier JW. The efficacy of the novel dual PI3-Kinase/mTOR inhibitor NVP-BEZ235 compared with rapamycin in renal cell Carcinoma. Clin Cancer Res. 2010;16(14):3628–3638. PMID: 20606035; doi: https://doi.org/10.1158/1078-0432.ccr-09-3022.
- Cho D. Novel targeting of Phoshatidylinositol-3 Kinase and Mammalian Target of Rapamycin (mTOR) in renal cell carcinoma. Cancer J. 2013;19(4):311–315. PMID: 23867512; doi: https://doi.org/10.1097/PPO.0b013e31829d5cea.
- Santoni M, Pantano F, Amantini C, Nabissi M, Conti A, Burattini L, Zoccoli A, Berardi R, Santoni G, Tonini G, et al. Emerging strategies to overcome the resistance to current mTOR inhibitors in renal cell carcinoma. Biochimica Et Biophysica Acta (BBA) – Rev Cancer. 2014;1845(2):221–231. PMID: 24480319; doi: https://doi.org/10.1016/j.bbcan.2014.01.007.
- Conti A, Santoni M, Amantini C, Burattini L, Berardi R, Santoni G, Cascinu S, Muzzonigro G. Progress of molecular targeted therapies for advanced renal cell carcinoma. Biomed Res Int. 2013;2013(12):1–9. PMID: 24093097; doi: https://doi.org/10.1155/2013/419176.
- Roulin D, Waselle L, Dormond-Meuwly A, Dufour M, Demartines N, Dormond O. Targeting renal cell carcinoma with NVP-BEZ235, a dual PI3K/mTOR inhibitor, in combination with sorafenib. Mol Cancer. 2011;10(1):90. PMID: 21791089; doi: https://doi.org/10.1186/1476-4598-10-90.
- Klaeger S, Heinzlmeir S, Wilhelm M, Polzer H, Vick B, Koenig P-A, Reinecke M, Ruprecht B, Petzoldt S, Meng C, et al. The target landscape of clinical kinase drugs. Science. 2017;358(6367):eaan4368. PMID: 29191878; doi: https://doi.org/10.1126/science.aan4368.
- Cheng H, Li C, Bailey S, Baxi SM, Goulet L, Guo L, Hoffman J, Jiang Y, Johnson TO, Johnson TW. Discovery of the highly potent PI3K/mTOR dual inhibitor PF-04979064 through structure-based drug design. Acs Medicinal Chem Letters. 2012;4(1):91–97. PMID: 24900568; doi: https://doi.org/10.1021/ml300309h.
- Elfiky AA, Aziz SA, Conrad PJ, Siddiqui S, Hackl W, Maira M, Robert CL, Kluger HM. Characterization and targeting of phosphatidylinositol-3 kinase (PI3K) and mammalian target of rapamycin (mTOR) in renal cell cancer. J Translational Med. 2011;9:133–133. PMID: 21834980; doi: https://doi.org/10.1186/1479-5876-9-133.