
ABSTRACT
The receptor tyrosine kinase MET is frequently involved in malignant transformation and inhibiting its activity in MET-dependent cancers is associated with improved clinical outcomes. Emerging evidence also suggests that mitochondria play an essential role in tumorigenesis and Dynamin Related Protein (DRP1), a key component of the mitochondrial fission machinery, has emerged as an attractive therapeutic target. Here, we report that inhibiting MET activity with the tyrosine kinase inhibitor MGCD516 attenuates viability, migration, and invasion of non-small cell lung cancer (NSCLC) and malignant pleural mesothelioma (MPM) cell lines in vitro, and significantly retards tumor growth in vivo. Interestingly, MGCD516 treatment also results in altered mitochondrial morphology in these cell lines. Furthermore, inhibiting MET pharmacologically or knocking down its expression using siRNA, decreases DRP1 activity alluding to possible crosstalk between them in these two cancers. Consistently, a combination of MGCD516 and mdivi-1, a quinazolinone reported to inhibit mitochondrial fission, is more effective in attenuating proliferation of NSCLC and MPM cell lines than either drug alone. Considered together, the present study has uncovered a novel mechanism underlying mitochondrial regulation by MET that involves crosstalk with DRP1, and suggests that a combination therapy targeting both MET and DRP1 could be a novel strategy for NSCLC and MPM.
Introduction
The receptor tyrosine kinase (RTK) MET plays a crucial role in cancer biology including cell survival, cell growth, angiogenesis, and metastasis.Citation1–Citation5 Thus, MET is overexpressed or activated in many types of cancer including non-small cell lung cancer (NSCLC).Citation6 NSCLC which constitutes ~ 85% of all lung cancer, is the most common cause of death worldwide.Citation7 Receptor tyrosine kinase (RTK) inhibitors that target MET are quite effective in patients with NSCLC.Citation8–Citation10 However, MET (and other RTKs) often activates signaling pathways in cancer cells that converge on common downstream effectors resulting in resistance to RTK-based treatment and hence, limiting their continued use.Citation6,Citation8
The redundancy of RTK-transduced signaling in cancer cells, and the wide-ranging effects of RTK ligands that lead to drug resistance, can be overcome through combinations of targeted agents.Citation11,Citation12 MET itself can interact with other RTKs such as EGFR and RON. In particular, a combination of HGF and EGFR TKI showed a synergistic apoptotic effect in preclinical studies in NSCLCCitation13 and malignant pleural mesothelioma (MPM).Citation14 Further, MET amplification in NSCLC is implicated in acquired resistance to EGFR inhibitors and has been reported in approximately 20% of cases with EGFR inhibitor resistance.Citation1,Citation15,Citation16 This provides further therapeutic rationale for combination RTK inhibitor therapies to treat selected patients with NSCLC although, how these different mechanisms may affect treatment outcomes in response to mono- or combination therapies remains poorly understood.
MET is also upregulated in MPM,Citation17,Citation18 an aggressive form of cancer with median overall survival ranging from 9 to 17 months, regardless of stage. Indeed, MPM remains a serious public health problem. The number of worldwide cases has steadily increased over the past decade and unfortunately, is predicted to rise further.Citation19,Citation20 Moreover, MPM is extremely difficult to treat due to limited treatment options and therefore, MET is an attractive therapeutic target both in MPM as well as in NSCLC. However, in light of the resistance that patients tend to develop when treated with MET inhibitors, we have explored a novel approach for these two cancers by evaluating the effect of inhibiting MET as well as mitochondrial dynamics and morphology in NSCLC and MPM.
Mitochondria are highly dynamic organelles that play a critical role in cellular physiology. Alterations in mitochondrial structures and functions have been linked to a wide variety of cancers.Citation21–Citation24 Cancer cells can alter the dynamics, bioenergetics, and biosynthetic properties of mitochondria to support their proliferation, migration and resistance to therapeutics.Citation25,Citation26 The mechanochemical GTPase, dynamin related protein 1 (DRP1), is a key regulator of mitochondrial fission that, in turn, regulates the structure and functions of mitochondria. Changes in DRP1 expression or activation are linked to various cancers suggesting that DRP1 can be a promising target for cancer therapy.Citation27–Citation31
Mdivi-1 (mitochondrial division inhibitor) is a quinazolinone originally described as a selective inhibitor of Drp1 over other dynamin family members that is reported to inhibit mitochondrial fission.Citation32 Although, it is reported to inhibit mitochondrial division in mammalian cells by attenuating DRP1 self-assembly the exact mechanism remains poorly understood. Indeed, a recent study noted that mdivi-1 poorly inhibits recombinant DRP1 GTPase activity (Ki > 1.2 mM) and therefore, it is likely that mdivi-1 may not be a specific DRP1 inhibitor.Citation33 On the other hand, there is also a large body of literature using mdivi-1 in vitro with effects that are predictive of inhibiting DRP1 and mitochondrial fission.Citation34
We recently showed that mdivi-1 is efficacious in MPM cell lines.Citation35 Furthermore, we showed that activation of the MET pathway induced changes in mitochondrial morphology and affected mitochondrial staining intensity in HEK-293 cellsCitation36 alluding to possible crosstalk between MET signaling and mitochondrial morphology and functions. Therefore, in this manuscript we explored the effect of combining MET and DRP1 inhibitors on NSCLC and MPM. First, we demonstrate that pharmacologically inhibiting MET with the orally-active tyrosine kinase inhibitor MGCD516 attenuates viability migration and invasion of NSCLC and MPM cells in vitro, and significantly retards tumor growth in vivo. Next, we show that inhibiting MET decreases DRP1 activity via the MAP kinase pathway and significantly alters mitochondrial morphology. Finally, we show that a combination of MGCD516 and mdivi-1 is more effective in attenuating proliferation of NSCLC and MPM cells than either drug alone. Thus, the present study has uncovered a novel mechanism that involves crosstalk between an RTK (MET) and a mitochondrial fission-related protein (DRP1), and suggests that a combination therapy targeting both MET and DRP1 could be a novel strategy for NSCLC and MPM.
Results
MET is overexpressed in NSCLC and MPM cell lines and inhibiting its activity attenuates their growth in vitro
We first determined MET protein expression in NSCLC and MPM cell lines by immunoblotting. Total cell lysates prepared from the NSCLC cell lines A549 and H1993, and the MPM cell lines H2373 and H2461 were probed with an anti-MET antibody as described in the Materials and Methods. The non-tumorigenic lung epithelial cell line BEAS-2B, and the mesothelioma cell line MeT-5A, were used as respective controls. As can been seen from , expression of MET protein was significantly higher both in NSCLC (panel A) and the MPM cell lines (panel C) compared to the respective control cell lines.
Figure 1. Validation of MET expression in cell lines and effect of MGCD516 in NSCLC and mesothelioma cells. A. Expression level of MET in NSCLC cell lines (A549, H1993) and lung epithelial cell line BEAS-2B were determined by immunoblotting. B. A549, H1993 and BEAS-2B were treated with increasing concentrations of MGCD516 for 72 hours. C. Expression level of MET in MPM cell lines (H2373, H2461) and mesothelioma control cell line MeT-5A were determined by immunoblotting. D. H2373, H2461 and MeT5A cell lines were treated with increasing concentrations of MGCD516 for 72 hours.
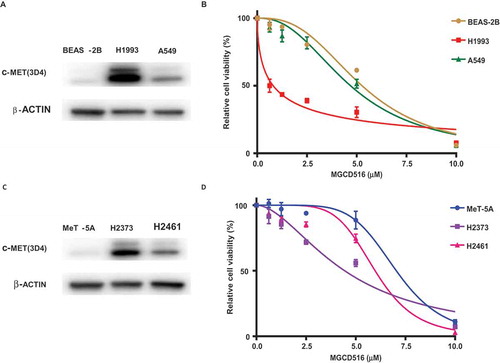
Having conformed MET upregulation, next, we determined the effect of inhibiting MET kinase activity with MGCD516. The same cell lines that were used in the immunoblotting experiment were treated with increasing concentrations of MGCD516 for 72 h. As indicated in , the various cell lines exhibited differential sensitivities to this particular RTK inhibitor. Thus, in the case of NSCLC, while the H19193 cell line with higher MET expression was quite sensitive (IC50 0.8μM), the A549 cell line with a significantly lower MET protein level, was fairly resistant to MGCD516 (IC50 4.7μM). Similarly, in the case of MPM cell lines, the H2373 cell line with higher MET expression was more sensitive to MGCD516 inhibition (IC50 4.4μM) than was the H2461 cell line (IC50 6.0μM) that had significantly lower MET expression.
MGCD516 attenuates migration and invasion of NSCLC and MPM cell lines
Since we observed a differential effect of MET inhibition on cell proliferation that correlated with the extent of MET expression, we asked if there was a similar correlation with regard to the migration and invasion capabilities of these cell lines. Cell migration was determined using a wound healing assay. DMSO or MGCD516 pretreated cells were seeded on 8W1E arrays as confluent monolayer. Cell monolayers were electrically wounded as described in Materials and Methods cellular resistance were recorded. The wound was then allowed to heal from cells surrounding the small active electrode that did not undergo the elevated voltage pulse. As shown in , MGCD516 treatment delayed wound healing in the case of the NSCLC cell line H1993 as well as the MPM cell line H2373 compared to vehicle (DMSO) control.
Figure 2. MGCD516 attenuates wound healing and invasion in NSCLC and MPM cells. A. DMSO or drug pretreated cells were seeded on 8W1E arrays as confluent monolayer. Cell monolayer was electrically wounded at 3.5 V for 30 sec for each well. Impedance and resistance of the cell layer were immediately recorded for a period of 25-40h. The wound was then allowed to heal from cells surrounding the small active electrode that did not undergo the elevated voltage pulse. MGCD516 treatment delayed wound healing compare to DMSO control for H1993 cells. b. MGCD516 treatment showed delay of wound healing compare to DMSO control for H2373 cells. c&e. Cells were treated with 2μM MGCD516 for 24 h and assayed as described in material and methods. Representative images show cells that penetrated Matrigel with or without MGCD516 treatment. d&f. Invasive cells were counted and analyzed by Prism software. Error bars indicate standard error of the mean. *p < 0.05, **p < 0.01, ***p < 0.001.
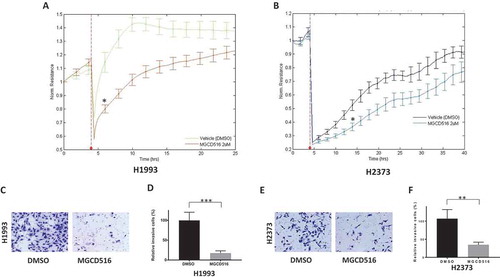
To assess the effect of MGCD516 on cell invasion, cells were treated with 2μM MGCD516 for 24 h and assayed using BioCoat™ Matrigel® Invasion Chambers and quantitated by counting the invasive cells as described in Material and Methods. Again, as can be seen from , MGCD516 dramatically attenuated the invasiveness of the H1993 (17.6%, p = 0.0002) and the H2373 (35.0%, p = 0.0088) cells compared to pretreatment with the DMSO vehicle control.
MGCD516 inhibits DRP1 activation and regulates mitochondrial dynamics and morphology
To investigate whether MGCD516 targets the MET-Erk1/2 signaling pathway and affects mitochondrial dynamics, we treated H1993 and H2373 cells with MGCD516 for 24 h and assayed changes in levels and/or activation of MET, Erk1/2, DRP1, and Fis1 by immunoblotting. In both cell lines, MGCD516 significantly lowered the steady state levels of phosphorylated MET, Erk1/2 and DRP1, and also reduced the level of total Fis1 () confirming crosstalk between MET and DRP1 that we suspected in these two cancers.
Figure 3. MGCD516 inhibits DRP1 activation and regulates mitochondrial morphology. a&d. MGCD516 inhibited HGF induced activation of DRP1 (phosphorylation at Ser616) in H1993 and H2373 cells. The cells were treated with 2µM of MGCD516 or DMSO for 24 hours, followed by stimulation with 100 ng/ml HGF for 15 min. Treated cells were then analyzed by Western blot. b&e. Changes of mitochondrial morphology in cells treated with MGCD516 or DMSO. Representative images of one H1993 and one H2373 cell either treated with DMSO or MGCD516 are shown. Inlets represent an enlarged part of the cells to show details of mitochondrial morphology. c&f. Quantification of mitochondrial lengths. Length of a single mitochondrion was defined from either one end to the next branching point or from one branching point to another. Measurements were carried out using ImageJ software. The numbers showed the averages of at least 20 measurements per cell from 50 cells. Data analysis were carried out by Prism software. Error bars indicate standard error of the mean. *p < 0.05, **p < 0.01, ***p < 0.001.
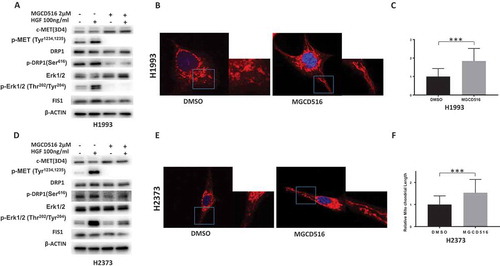
Next, we asked if MGCD516 targeted ligand-dependent or ligand-independent activation of DRP1 by MET (phosphorylation at Ser616) in H1993 and H2373 cells. Toward this end, cells were treated with MGCD516 or DMSO for 24 h, followed by stimulation with 100 ng/ml HGF for 15 minutes to activate MET. Treated cells were then stained using antibodies and visualized by immunofluorescence microscopy. The results revealed decreased fluorescence in the distribution of phospho-MET and phospho-DRP1 (Figure S1(a,c) but did not affect total MET or DRP1 (Figure S1(b,d)) indicating that MGCD561 targeted ligand-dependent MET activation.
To assess the effect of MGCD516 on mitochondrial morphology, we examined cells stained with MitoTracker using confocal microscopy. In control cells NSCLC and MPM cells treated with just the vehicle (DMSO), the mitochondria appeared rounded and clustered around perinuclear regions. However, in MGCD516-treated cells the mitochondria appeared more elongated and showed a scattered cytoplasmic distribution (). The relative average length of the mitochondria in MGCD516-treated cells was significantly longer in both cancer cells than in DMSO-treated controls ().
Since MGCD516 can also inhibit other RTKs that interact with MET, it is quite possible that the effect of this drug that we observed on mitochondrial dynamics and morphology may have been indirect. To rule out this possibility, we selectively silenced MET expression both in NSCLC and MPM cell lines by transfecting them with MET-specific siRNA. As shown in , knocking down MET downregulated DRP1 protein in H1993 and H2373 cells compared to the control siRNA. Furthermore, the steady state levels of phospho-DRP1 and phospho-ERK1/2 proteins in the case of the MET-specific siRNA were also significantly decreased compared to the control siRNA.
Figure 4. MET regulates DRP1 activation and mitochondrial morphology. a&d. Knockdown of MET downregulated activated DRP1 in H1993 and H2373 cells. b&e. Knockdown of MET induces elongated mitochondria in H1993 and H2373 cells. Inlets represent an enlarged part of the cells to show details of mitochondrial morphology. c&f. Quantification of mitochondrial lengths. The numbers showed the averages of at least 20 measurements per cell from 50 cells. Data analysis were carried out by Prism software. Error bars indicate standard error of the mean. *p < 0.05, **p < 0.01, ***p < 0.001.
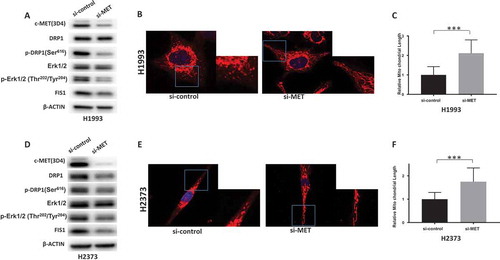
We also assessed the effect of knocking down MET expression on mitochondrial morphology by observing MitoTracker-stained cells by confocal microscopy. In both NSCLC and MPM cell lines transfected with a scrambled (control) siRNA, mitochondria appeared rounded and clustered around perinuclear regions while in the cells transfected with the MET-specific siRNA, the mitochondria appeared elongated and exhibited a more scattered distribution () as was seen when we inhibited MET with MGCD516. The relative average length of the mitochondria in MET-specific siRNA cells was significantly longer than in cells transfected with the control siRNA (). Considered together, these observations not only confirmed that there is crosstalk between MET signaling and mitochondrial morphology but also underscore the functional link between MET and DRP1 in NSCLC and MPM.
Kinomics Analysis:
To discern kinase activity dynamics of control and drug-treated cells, raw PamGene data were preprocessed as follows. “N/A”, negative and zero values were set to missing; the values greater than 0 were log transformed. The Log values of the peptides representing the same gene were added up. We shall refer to these values as gene-level measurements in this manuscript. Gene-level measurements were considered significant if FDR < 0.05 (Supplemental Table 1) where LogFC = Log(treatment) – Log(control). The statistically-significant gene-level measurements are also shown on heat maps for the two cell lines (Supplemental Figure 2).
When significant measurements were analyzed using Metacore to discover “pathway map folders”, i.e., groups of pathway maps linked by some common attribute, 35 folders with various properties were obtained. Of these, the “Lung Cancer” folder was chosen for further analysis. The maps within the “Lung Cancer” folder were ordered by the fraction of affected proteins for H1993 and H2373. Interestingly, the top map observed were “chemotaxis”, “HGF signaling”, “VEGF signaling” and “FGFR signaling”. An additional analysis was performed to investigate involvement of MET (the target of the drug). Specifically, from the list of statistically significant pathway maps derived from the data, the maps were also filtered by the presence of MET. Supplemental Figure 3 showed that MET inhibitory treatment of NSCLC and MPM cells related to “cell adhesion” and “neoplasms/carcinogenesis” indicating that the inhibition of PDK phosphorylation by MET upstream of inhibition of apoptosis is very likely the cause for reduced survival of the neoplastic cells.Citation37
MGCD516 treatment results in significant tumor growth inhibition in vivo models
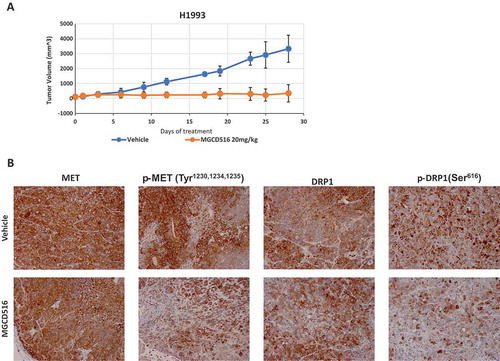
To evaluate the effect of inhibiting MET on DRP1 in vivo, we created a mouse xenograft model of NSCLC. Mice were injected with H1993 cells and dosed with an orally-active MGCD516 preparation or vehicle for 25 days after tumor volume reached an average of 200 to 300 mm3. As can be seen from , drug-treatment dramatically inhibited tumor growth compared vehicle-treated controls. Furthermore, immunohistochemistry of the xenograft tumor tissue revealed that the levels of both phospho-Met and phospho-DRP1 were significantly reduced in the drug-treated samples compared to the vehicle-treated controls ().
Figure 5. MGCD516 treatment results in significant tumor growth inhibition in vivo models.a. Tumor growth curve of H1993 xenograft. H1993 xenograft was treated with MGCD516 20mg/kg or vehicle for 25 days after tumor volume reached an average of 200 to 300 mm3. b. Activation of MET and DRP1 were evaluated by IHC staining.
A combination of MGCD516 and mdivi-1 is more effective than either drug alone in NSCLC and MPM
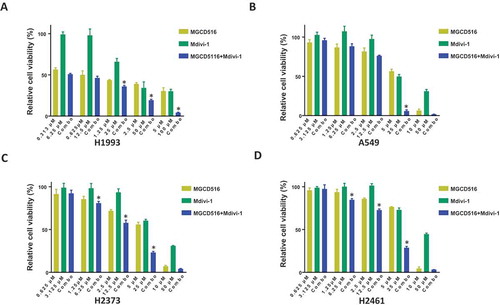
Having established a functional link between MET and DRP1 in NSCLC and MPM both in vitro and in vivo, we next determined the effect of disrupting the cross talk between them by inhibiting either or both protein activities in these two cancers using MGCD516 and mdivi-1, respectively. H1993 and A549, and H2373 and H2461 cell lines were treated with indicated in concentrations for 72 h and cell viability was determined as described in Materials and Methods. As shown in , while both NSCLC and MPM cell lines were sensitive to the highest dose of MGCD516, mdivi-1 by itself was relatively ineffective. However, when combined, the two drugs were far more efficacious than either drug alone, underscoring further the functional link between MET and DRP1.
Figure 6. Combination of MGCD516 and Mdivi-1 showed higher efficacy than either drug alone in NSCLC and MPM. a to d. NSCLC cells (H1993 and A549) and MPM cells (H2373 and H2461) were treated with indicated concentrations for 72h. Data analysis were carried out by Prism software. Error bars indicate standard error of the mean. *p < 0.05.
Of note, other than the RTK pathway, such as MET pathway, DRP1 is known to be regulated by other pathways such as CDK, PKA, PKC, and GSK3.Citation38 Moreover, DRP1 inhibition may be compensated by upregulation of other mitochondria proteins, such as Fis1. Interestingly, we found that Fis1 is also regulated by MET () demonstrating the benefit of simultaneously inhibiting both MET and DRP1 in NSCLC and MPM.
Discussion
Despite intense research efforts, NSCLC and MPM have remained notoriously resilient to therapy. Furthermore, the emergence of drug resistance against the popular RTK inhibitors that are effective in patients with elevated or genetically altered RTKs that drive proliferation, has added to the burgeoning problem. MET is a well-known drug target in NSCLC and also in MPM where it plays a crucial role in disease pathology including cell survival, cell growth, angiogenesis, and metastasis.Citation1–Citation5
We had previously showed that activation of the MET pathway induced mitochondrial morphology change and affected mitochondrial staining intensity in HEK-293 cells,Citation36 suggesting that MET may be regulating mitochondrial morphology and functions. In this study, we have provided evidence indicating that indeed, in NSCLC and MPM, MET does regulate mitochondrial structure and function. We have also shown that this regulation acts primarily through the mitochondrial fission proteins DRP1 and to a lesser extent Fis1. As far as we are aware, the present study is the first to demonstrate that a receptor tyrosine kinase regulates DRP1 in a ligand-dependent manner which is critical for DRP1 function in cancer cells. The fact that, hepatocyte growth factor (HGF), is also upregulated in these patientsCitation8,Citation39,Citation40 lends further credence to our observation.
In a recent report, the non-receptor tyrosine kinase c-Abl was shown to directly phosphorylate DRP1 to regulate its GTPase activity and promote oxidative stress-induced mitochondrial fragmentation and neuronal cell death.Citation41 Although the exact role of DRP1 phosphorylated by MET in cancer remains unknown, inhibiting DRP1 in conjunction with MET appears promising. Unfortunately, as pointed out earlier, the specificity of mdivi-1 as well as its mode of action remain equivocal, and attempts to develop small molecule inhibitors that specifically inhibit DRP1 have not been successful. However, a small peptide inhibitor that selectively inhibits DRP1 enzyme activity and disrupts the DRP1-Fis1 interaction has been identified.Citation42 The availability of such a compound coupled with new technologies that facilitate therapeutic peptide deliveryCitation43 would open new possibilities of combination therapies targeting MET and DRP1 as a novel therapeutic approach for NSCLC and MPM.
Materials and methods
Cell lines and Reagents: Lung cancer cell lines A549, H1993 and lung epithelial cell line BEAS-2B were from American Type Culture Collection (ATCC) (Manassas, VA, USA). Mesothelioma cell lines H2373, H2461 and one nonmalignant transformed mesothelioma control cell line (MeT-5A) were also from ATCC. Met-5A cells were cultured in M199 media supplemented with various growth factors according to manufacturer’s instructions (ATCC). All other cells were cultured in RPMI 1640 medium (Gibco/BRL) supplemented with 10% (v/v) fetal bovine serum (FBS), L-glutamine and 1% penicillin-streptomycin at 37°C with 5% CO2. Recombinant human HGF was purchased from R&D Systems (Minneapolis, MN, USA). MGCD516, provided by Mirati therapeutics, was dissolved in dimethyl sulfoxide (DMSO) for in vitro experiments. Mdivi-1 was purchased from Sigma (St. Louis, MO, USA).
Antibodies: Phospho-MET (Tyr1234/1235), phospho-DRP1 (Ser616), phospho-Erk1/2 (Thr202/Tyr204), and Erk1/2 antibodies were purchased from Cell Signaling (Danvers, MA, USA). Antibodies against Fis1 (c-10) were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Anti-DRP1 antibodies were purchased from Abcam (Cambridge, MA, USA) and the β-actin antibody was purchased from Sigma (St. Louis, MO, USA). Antibodies against MET (3D4) and phospho-MET (Tyr1230, 1234, 1235) were purchased from Invitrogen (Carlsbad, CA, USA).
Cell Viability Assay: To determine specific cytotoxicity, we used Cell Counting Kit-8 (CCK-8) purchased from Dojindo Molecular Technologies. Cells were seeded in 96-well plates and allowed to adhere in normal growth media for 24 h. The test compounds were added to 100 µl of media at the indicated concentrations for 72 h. 10 μl of the CCK-8 solution was added to each well of the plate and absorbance at 450 nm was measured using a Tecan Spark 10M multimode microplate reader.
Immunoblotting: Whole cell lysates were prepared using RIPA lysis buffer. Protein samples were run on 4–15% Mini-protean TGX gels (Bio-Rad) and transferred onto ImmobilonTM membranes (Millipore). Blots were blocked using 5% BSA in TBST for 1 h and probed with primary antibody overnight at 4°C. After washing 3 times in TBST, blots were incubated with HRP-conjugated secondary antibodies for 1 h at room temperature. The blots were then washed again and visualization of immuno-reactive bands was achieved using LumiGLO® enhanced chemiluminescence (Cell Signaling Technology) and imaged using the ChemiDoc MP imager (Bio-Rad).
Electrical cell-based impedance sensing (ECIS) wound-healing assays: Wound-healing assays were done with ECIS array (Applied Biophysics, Troy, NY) technology. Confluent cell monolayers cultured on ECIS plates were submitted to an elevated voltage pulse of 40-kHz frequency, 3.5-V amplitude, and 30-s duration, which led to death and detachment of cells present on the small active electrode, resulting in a wound normally healed by cells surrounding the small active electrode that have not been submitted to the elevated voltage pulse. Wound healing was then assessed by continuous resistance measurements for 24 or more hours.
Invasion assay: Cell invasion assays were evaluated using Corning® BioCoat™ Matrigel® Invasion Chambers with 8.0μm PET Membrane (Corning, NY, USA). After overnight serum starvation, cells were treated with 2Μm MGCD516 in serum-free medium for 24 h. Cells were prepared as mentioned above in 24-well plates following the manufacturer’s instructions. 10% serum was added as a chemoattractant. After 24 h incubation, invasive cells were stained by Diff-Quik kit and counted under a microscope.
Kinomics analysis: PamGene technology using the PamChip Tyrosine Kinase Array was used to detect phosphorylation of peptides. This technology was used to measure the activity of purified kinases and the effects of kinase inhibitors as described elsewhere.Citation10,Citation44 The lysates of cell lines H1993 and H2373 were directly treated with 1 uM of MGCD516. Image quantification and data processing were conducted with the Evolve and BioNavigator software package (PamGene). The peptides that were significantly differentially affected and signaling pathways were analyzed using Ingenuity Pathway Analysis (IPA) (Redwood City, CA).
Immunofluorescence: The cells were grown on glass coverslips coated with 1% gelatin solution in 6-well plates. Cells were fixed with 3.7% formaldehyde in phosphate-buffered saline (PBS), pH 7.4, permeabilized in PBS containing 0.1% Tween and 0.25% Triton x-100. After washing with TBST, cells were blocked with 5% serum for 1 h. Primary antibody diluted in antibody diluent (Dako) was incubated at 4°C overnight. Secondary antibody conjugated with AlexaFluor 488 or AlexaFluor 568 was added after washing. Following washing, the coverslips were mounted immediately in Prolong Gold Antifade reagent with DAPI (Life Technologies). Images of labeled cells were acquired by a confocal microscope (Zeiss LSM880).
Transfection: MET and control siRNAs were purchased from OriGene (Rockville, MD, USA). JetPrime transfection reagent (Polyplus Transfection) was used to transfect siRNAs according to manufacturer protocol.
Confocal microscopy: Cells were cultured on 35mm glass dishes to desired density. After MGCD516 treatment for 24 h, 30 nM MitoTracker dye (Thermofisher Scientific) and 0.1µg/mL Hoechst 33342 DNA dye were added to stain cells for 15 min. After washing, cells were observed with a confocal microscope immediately. The images were then imported into ImageJ software (http://imagej.nih.gov/ij/index.html) for mitochondrial length measurement.
Xenograft mouse tumor models: Female homozygous athymic nude mice (5–6 weeks of age) were obtained from Harlan Laboratories (Indianapolis, IN) and cared for according to institutional guidelines under a protocol approved by the University of Chicago Institutional Animal Care and Use Committee. Matrigel (500 µl, BD Biosciences) containing H1993 cells (2.0 × 106) was inoculated subcutaneously in the right flank of each mouse. Tumor growth was measured with calipers and volume (mm3) calculated as (A2 × B x π/6). When the volume reached a mean of 200 mm3, mice were randomized into two groups (n = 5 mice/group) to receive vehicle alone or MGCD516 (20 mg/kg). Drugs were administered once a day for 25 days by oral gavage. Body weight and tumor volume were recorded every 3 days until the study was terminated. Mice were sacrificed and tumor tissues were excised, weighed, subsequently fixed in 10% buffered formalin and embedded in paraffin. For in vivo studies, 8 mg MGCD516 was dissolved in 100 µl DMSO and 900 µl of PEG400 was added to bring the volume to 1 ml. After mixing, 1 ml of saline was added to bring the final volume to 2 ml. 2ml is enough for 20, 20g mice.
Immunohistochemistry (IHC) staining: Histological sections at a thickness of 5 μm were deparaffinized with xylene and rehydrated through an alcohol gradient series to water. Antigens were retrieved and endogenous peroxidase activity was quenched using 3% hydrogen peroxide. After blocking, the sections were incubated with the following primary antibodies, respectively, incubated for 30 min or 1 hour at room temperature or at 4°C overnight dependent on the optimization result of each antibody. Specimens were then incubated with the EnVision+ System-HRP labeled polymer anti-mouse or anti-rabbit, correspondingly for 30 min at room temperature, followed by incubation with the Liquid DAB+ Substrate Chromogen System (DAKO) for 8 minutes at room temperature on a DAKO Autostainer. After washing, the specimens were then counterstained with hematoxylin and covered with coverslips.
Statistical analyses: For data analysis, experimental samples were compared to controls by unpaired Student’s t-test. For multiple group comparisons, a one-way variance analysis (ANOVA) was used. Differences between groups were considered statistically significant when P value was less than 0.05. All statistical analyses were performed using the GraphPad Prism program (GraphPad Software Inc., USA).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Download Zip (782.6 KB)Acknowledgments
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under award number P30CA033572. Additional financial support for this research was provided by Mirati Therapeutics Inc. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Supplementary material
Supplemental data for this article can be accessed www.tandfonline.com/kcbt.
Additional information
Funding
Related Research Data
References
- Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. 2012;12:89–103. doi:10.1038/nrc3205.
- Granito A, Guidetti E, Gramantieri L. c-MET receptor tyrosine kinase as a molecular target in advanced hepatocellular carcinoma. J Hepatocell Carcinoma. 2015;2:29–38. doi:10.2147/JHC.S77038.
- Szturz P, Raymond E, Abitbol C, Albert S, De Gramont A, Faivre S. Understanding c-MET signalling in squamous cell carcinoma of the head & neck. Crit Rev Oncol Hematol. 2017;111:39–51. doi:10.1016/j.critrevonc.2017.01.004.
- Bradley CA, Salto-Tellez M, Laurent-Puig P, Bardelli A, Rolfo C, Tabernero J, Khawaja HA, Lawler M, Johnston PG, Van Schaeybroeck S. Targeting c-MET in gastrointestinal tumours: rationale, opportunities and challenges. Nat Rev Clin Oncol. 2017;14:562–576. doi:10.1038/nrclinonc.2017.40.
- Xiang C, Chen J, Fu P. HGF/Met signaling in cancer invasion: the impact on cytoskeleton remodeling. Cancers (Basel). 2017;9:44. doi:10.3390/cancers9050044.
- Sadiq AA, Salgia R. MET as a possible target for non-small-cell lung cancer. J Clin Oncol. 2013;31:1089–1096. doi:10.1200/JCO.2012.43.9422.
- Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553:446–454. doi:10.1038/nature25183.
- Salgia R. MET in lung cancer: biomarker selection based on scientific rationale. Mol Cancer Ther. 2017;16:555–565. doi:10.1158/1535-7163.MCT-16-0472.
- Sattler M, Salgia R. MET in the Driver’s Seat: exon 14 skipping mutations as actionable targets in lung cancer. J Thorac Oncol. 2016;11:1381–1383. doi:10.1016/j.jtho.2016.07.003.
- Tan Y-HC, Mirzapoiazova T, Won BM, Zhu L, Srivastava MK, Vokes EE, Husain AN, Batra SK, Sharma S, Salgia R. Differential responsiveness of MET inhibition in non-small-cell lung cancer with altered CBL. Sci Rep. 2017;7:9192. doi:10.1038/s41598-017-09078-4.
- Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, Peng J, Lin E, Wang Y, Sosman J, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487:505–509. doi:10.1038/nature11249.
- Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J, Frederick DT, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500–504. doi:10.1038/nature11183.
- Puri N, Salgia R. Synergism of EGFR and c-Met pathways, cross-talk and inhibition, in non-small cell lung cancer. J Carcinog. 2008;7:9. doi:10.4103/1477-3163.44372.
- Kawaguchi K, Murakami H, Taniguchi T, Fujii M, Kawata S, Fukui T, Kondo Y, Osada H, Usami N, Yokoi K, et al. Combined inhibition of MET and EGFR suppresses proliferation of malignant mesothelioma cells. Carcinogenesis. 2009;30:1097–1105. doi:10.1093/carcin/bgp097.
- Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, Chitale D, Motoi N, Szoke J, Broderick S, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A. 2007;104:20932–20937. doi:10.1073/pnas.0710370104.
- Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale C-M, Zhao X, Christensen J, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi:10.1126/science.1141478.
- Mukohara T, Civiello G, Davis IJ, Taffaro ML, Christensen J, Fisher DE, Johnson BE, Jänne PA. Inhibition of the met receptor in mesothelioma. Clin Cancer Res. 2005;11:8122–8130. doi:10.1158/1078-0432.CCR-05-1191.
- Ou W-B, Hubert C, Corson JM, Bueno R, Flynn DL, Sugarbaker DJ, Fletcher JA. Targeted inhibition of multiple receptor tyrosine kinases in mesothelioma. Neoplasia. 2011;13; 12–22.
- Tsao AS, Wistuba I, Roth JA, Kindler HL. Malignant pleural mesothelioma. J Clin Oncol. 2009;27:2081–2090. doi:10.1200/JCO.2008.19.8523.
- Bibby AC, Tsim S, Kanellakis N, Ball H, Talbot DC, Blyth KG, Maskell NA, Psallidas I. Malignant pleural mesothelioma: an update on investigation, diagnosis and treatment. Eur Respir Rev. 2016;25:472–486. doi:10.1183/16000617.0063-2016.
- Srinivasan S, Guha M, Kashina A, Avadhani NG. Mitochondrial dysfunction and mitochondrial dynamics-The cancer connection. Biochim Biophys Acta. 2017;1858:602–614. doi:10.1016/j.bbabio.2017.01.004.
- Burke PJ. Mitochondria, bioenergetics and apoptosis in cancer. Trends Cancer. 2017;3:857–870. doi:10.1016/j.trecan.2017.10.006.
- Maycotte P, Marín-Hernández A, Goyri-Aguirre M, Anaya-Ruiz M, Reyes-Leyva J, Cortés-Hernández P. Mitochondrial dynamics and cancer. Tumour Biol. 2017;39:1010428317698391. doi:10.1177/1010428317698391.
- Trotta AP, Chipuk JE. Mitochondrial dynamics as regulators of cancer biology. Cell Mol Life Sci. 2017;74:1999–2017. doi:10.1007/s00018-016-2451-3.
- Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12:685–698. doi:10.1038/nrc3365.
- Senft D, Ronai ZA. Regulators of mitochondrial dynamics in cancer. Curr Opin Cell Biol. 2016;39:43–52. doi:10.1016/j.ceb.2016.02.001.
- Smirnova E, Shurland DL, Ryazantsev SN, van der Bliek AM. A human dynamin-related protein controls the distribution of mitochondria. J Cell Biol. 1998;143:351–358.
- Rehman J, Zhang HJ, Toth PT, Zhang Y, Marsboom G, Hong Z, Salgia R, Husain AN, Wietholt C, Archer SL. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. Faseb J. 2012;26:2175–2186. doi:10.1096/fj.11-196543.
- Zhao J, Zhang J, Yu M, Xie Y, Huang Y, Wolff DW, Abel PW, Tu Y. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene. 2013;32:4814–4824. doi:10.1038/onc.2012.494.
- Cai J, Wang J, Huang Y, Wu H, Xia T, Xiao J, Chen X, Li H, Qiu Y, Wang Y, et al. ERK/Drp1-dependent mitochondrial fission is involved in the MSC-induced drug resistance of T-cell acute lymphoblastic leukemia cells. Cell Death Dis. 2016;7:e2459. doi:10.1038/cddis.2016.370.
- Serasinghe MN, Chipuk JE. Mitochondrial fission in human diseases. Handb Exp Pharmacol. 2017;240:159–188. doi:10.1007/164_2016_38.
- Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14:193–204. doi:10.1016/j.devcel.2007.11.019.
- Bordt EA, Clerc P, Roelofs BA, Saladino AJ, Tretter L, Adam-Vizi V, Cherok E, Khalil A, Yadava N, Ge SX, et al. The Putative Drp1 Inhibitor mdivi-1 is a reversible mitochondrial Complex I inhibitor that modulates reactive oxygen species. Dev Cell. 2017;40:583.e6–594.e6. doi:10.1016/j.devcel.2017.02.020.
- Smith G, Gallo G. To mdivi-1 or not to mdivi-1: is that the question? Dev Neurobiol. 2017;77:1260–1268. doi:10.1002/dneu.22519.
- Lennon FE, Cianci GC, Kanteti R, Riehm JJ, Arif Q, Poroyko VA, Lupovitch E, Vigneswaran W, Husain A, Chen P, et al. Unique fractal evaluation and therapeutic implications of mitochondrial morphology in malignant mesothelioma. Sci Rep. 2016;6:24578. doi:10.1038/srep24578.
- Kawada I, Hasina R, Lennon FE, Bindokas VP, Usatyuk P, Tan Y-HC, Krishnaswamy S, Arif Q, Carey G, Hseu RD, et al. Paxillin mutations affect focal adhesions and lead to altered mitochondrial dynamics: relevance to lung cancer. Cancer Biol Ther. 2013;14:679–691. doi:10.4161/cbt.25091.
- Viticchiè G, Muller PAJ. c-Met and other cell surface molecules: interaction, activation and functional consequences. Biomedicines. 2015;3:46–70. doi:10.3390/biomedicines3010046.
- Hu C, Huang Y, Li L. Drp1-dependent mitochondrial fission plays critical roles in physiological and pathological progresses in mammals. Int J Mol Sci. 2017;18:E144. doi:10.3390/ijms18010144.
- Feng Y, Ma PCMET targeted therapy for lung cancer: clinical development and future directions. Lung Cancer (Auckl). 2012;3:53–67. doi:10.2147/LCTT.S23423.
- Gaudino G, Yang H, Carbone M. HGF/Met signaling is a key player in malignant mesothelioma carcinogenesis. Biomedicines. 2014;2:327–344. doi:10.3390/biomedicines2040327.
- Zhou L, Zhang Q, Zhang P, Sun L, Peng C, Yuan Z, Cheng J. c-Abl-mediated Drp1 phosphorylation promotes oxidative stress-induced mitochondrial fragmentation and neuronal cell death. Cell Death Dis. 2017;8:e3117. doi:10.1038/cddis.2017.518.
- Qi X, Qvit N, Su Y-C, Mochly-Rosen D. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J Cell Sci. 2013;126:789–802. doi:10.1242/jcs.114439.
- Acar H, Ting JM, Srivastava S, LaBelle JL, Tirrell MV. Molecular engineering solutions for therapeutic peptide delivery. Chem Soc Rev. 2017;46:6553–6569. doi:10.1039/c7cs00536a.
- Versele M, Talloen W, Rockx C, Geerts T, Janssen B, Lavrijssen T, King P, Göhlmann HWH, Page M, Perera T. Response prediction to a multitargeted kinase inhibitor in cancer cell lines and xenograft tumors using high-content tyrosine peptide arrays with a kinetic readout. Mol Cancer Ther. 2009;8:1846–1855. doi:10.1158/1535-7163.MCT-08-1029.