
ABSTRACT
The targeted treatment of advanced non-small cell lung cancer (NSCLC) harboring genomic rearrangement of ALK is a paradigm for personalized oncology. More than 15 different ALK fusion partners have been discovered in NSCLC patients. Most of these ALK fusions responded well to the ALK inhibitor crizotinib. Crizotinib is an oral MET/ALK inhibitor used as first-line therapy in the treatment of advanced NSCLC harboring ALK rearrangement. An understanding of the mechanisms by which tumors harbor primary drug resistance or acquired resistance to targeted therapies is critical for predicting which patients will respond to a specific therapy and for the identification of additional targetable pathways to maximize clinical benefits. Cap methyltransferase 1(CMTR1) also known as hMTr1, which is translate a human cap1 2ʹ-o-ribose methyltransferase. Here, we report the newly found ALK fusion, CMTR1-ALK, in a patient who has no response to the ALK inhibitor crizotinib. The results remind us that detecting ALK status is important, but that determining the ALK fusion type and function may be more important for patient.
Introduction
Non-small-cell lung cancer (NSCLC) accounts for approximately 80–85% of lung cancer deaths and is a leading cause of cancer-related mortality for both men and women worldwid.Citation1–Citation5 Anaplastic lymphoma kinase (ALK) rearrangements have been identified in 5–6% of NACLC.Citation6 The targeted treatment of advanced NSCLC harboring genomic rearrangement of ALK is a paradigm in personalized oncology.Citation7
The Echinoderm microtubule-associated protein-like 4 (EML4-ALK) fusion gene is found in 3–5% of NSCLC. EML4-ALK fusion in NSCLC occurs in at least 23 variants. All known variants have a breakpoint in intron 19 of ALK, but a variety of introns in EML4 experience rearrangement, which results in inclusion of differing fragments of the N-terminal region of EML.Citation8 Hybrid-capture-based genomic sequencing that targets intron 19 of ALK can capture all possible breakpoint events involving this intron and unequivocally identify the partner gene. In vitro modeling of EML4-ALK demonstrates that it is an oncogenic driver that is sensitive to multiple ALK kinase inhibitors such as the approved drug crizotinib, consistent with in vivo studie.Citation9 Clinical trials have established ALK inhibitors as standard treatment for patients with advanced ALK-rearranged NSCLC, with crizotinib response rates of approximately 74% and median progression-free survival (PFS) of 10.9 months, compared with approximately 45% and 7.0 months, with pemetrexed plus either cisplatin or carboplatin, respectivel.Citation6
Detection of ALK rearrangements in patients with NSCLC has recently become a routine pathological diagnosis. There are three major conventional diagnostics for ALK fusion: fluorescence in situ hybridization (FISH), real time reverse-transcriptase polymerase chain reaction (RT-PCR), and immunohistochemistry (IHC). Next generation sequencing has displayed impressive capability to detect FISH-negative and novel fusion type ALK-rearranged NSCLCitation10 and has developed into a clinically practical alternative for diagnosis of ALK rearrangements in addition to FISH, IHC, and RT-PCR based techniques.
Besides EML4, there are other partner genes that can be fused with ALK. Currently, more than 15 different ALK fusion partners have been discovered in NSCLC including EML4, KIF5B, KLC1, and TFG. Most of these ALK fusions in NSCLC patients responded well to the ALK inhibitor crizotinib. Recently, we reported a novel ALK fusion partner gene CMTR1 in an NSCLC patient who has no response to the ALK inhibitor crizotinib. CMTR1 also known as hMTr1, which is translate a human cap1 2ʹ-o-ribose methyltransferas.Citation11 We analyzed the CMTR1-ALK fusion gene sequence and determined that it is a null fusion type. This result suggests that while the detection of ALK status is important, confirming the fusion gene production may be more important.
Case presentation
A 75-year-old Chinese man with a 1-pack-year smoking history of 50 years and a history of early-stage NSCLC (adenocarcinoma of lung T1N0M0) diagnosed 7 years before the patient presented with mediastinal adenopathy with probable metastatic disease within multiple lymph nodes in the left side of his neck. The patient had a lung resection on Apr. 2010. After surgery, the disease was under control until July 2014 when a lung lesion was found by computed tomography scan. The patient had signs and symptoms of dysphagia, choking, and hoarseness in September 2014. He took 5 cycles of radiation treatment (Dt 32.5 gry) starting in October 2014(Figure S1). Needle aspiration biopsy of the cervical lymph node in July 2016 revealed that the tumor was a metastatic, poorly differentiated carcinoma (). This sample was identified as TTF1 () and was napsin A expression-positive by IHC staining. Ventana IHC (D5F3) analyses of ALK protein expression were negative ().
Figure 1. Hematoxylin and eosin(HE) staining, TTF1 staining, Ventana IHC(D5F3), and FISH staining slides from patient.
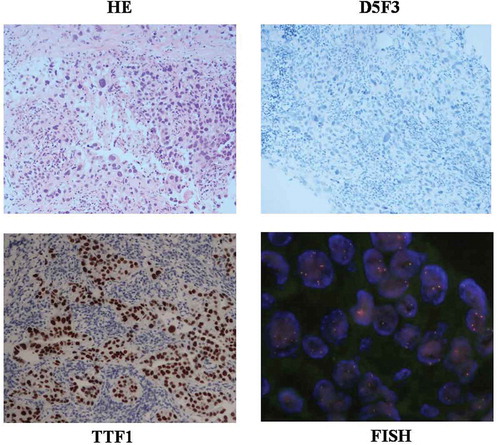
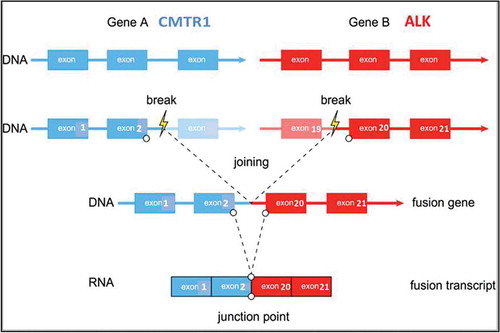
In order to take molecular targeted therapeutic drugs, the lymph node FFPE specimen was subjected to qRT-PCR, FISH, and genomic profiling using a clinical grade NGS assay (Illumina Hiseq 4000 NGS platforms). Results showed that this specimen was PCR-positive, FISH-negative, and NGS-positive (). NGS result revealed that this tumor specimen harbored a previously unknown CMTR1-ALK fusion variant () involving rearrangement and fusion of exon 1 to 2 of CMTR1 to exon 20 to 29 of ALK (C2; A20). This fusion comprises only 3% of the sequencing runs where tumor purity in the biopsy sample was estimated to be 40% ().
Table 1. Pathologic characteristics and molecular test results of the patient.
Figure 2. Schematic structure of the genomic DNA and RNA sequence.
Based on these results, treatment was initiated with 250 mg crizotinib per day starting in September 2016. The disease still developed 1 month after drug treatment showing that this patient has no response to crizotinib (Figure S1). Then the patient dropped out of the crizotinib treatment and was treated with pemetrexed in November 2016, which delayed disease progression. The disease has remained under control for approximately 13 months at this time.
Discussion
In recent years, genetic alterations that are responsible for initiation and maintenance of the malignant phenotype have been identified in several cancers including NSCLC. Drugs targeting these alterations, often named “driver” genetic alterations, can provide significant clinical benefit. One of the genetically defined subsets of NSCLC is the ALK positive subset. In recent years, major therapeutic advances have occurred in this subset of NSCLC.
In 2007, Soda and colleagues first identified the EML4-ALK oncogenic fusion gen.Citation12 It is now recognized that there are several different variants of EML4-ALK translocation that are dependent on variations in the length of the EML4 gene involved in the fusion. In addition, in less than 5% of ALK+ NSCLCs, the fusion partner is not EML4 and instead is one of more than 15 genes such as KIF5B, TFG, KLC1, TPR, HIP1, BIRC6, and BCL11.Citation11,Citation13–Citation18
Advances in the management of ALK+ NSCLC started with the recognition that crizotinib can provide clinical benefit in these patients. Recently, Yoshida et al. reported the variability of crizotinib activity in tumors with different ALK variant.Citation19 The variants are defined by the size of the EML4 gene involved in the ALK fusion. In a relatively small series, Yoshida and colleagues demonstrated that the median PFS was 11 months in variant 1, the most common variant, but was only 4.2 months in patients with tumors that were not variant 1. These data suggest that better understanding of the biological factors that influence the activity of crizotinib may allow a more personalized approach to treating ALK+ NSCLC patient.Citation20
For this patient, we first used RT-PCR to detect the ALK gene and found that he was ALK-positive. After one month of taking crizotinib, the patient was found to have primary drug resistance. To analyze the cause of primary drug-resistance, we used three other methods to detect the ALK-status. The results revealed that the patient was ALK-positive on the DNA level (PCR and NGS) but negative on the protein level (IHC-D5F3). Additionally, the “gold standard” FISH assay result was ALK-negative. The positive result from PCR was confusing, because the commercial PCR kit we used does not include the fusion type of this patient (Table S1). In future research, we will explore the underlying mechanism for this result using different methods.
The NGS result revealed a novel CMTR1-ALK fusion gene in NSCLC, which we reported in another paper (in preparation). In that study we analyzed the CMTR1-ALK fusion gene using online software PRABI (http://doua.prabi.fr/software/cap3), which indicated that the coiled-coil structure of CMTR1-ALK protein has a weakened dipolymerization capacity which partly causes reduced ALK protein kinase activity.
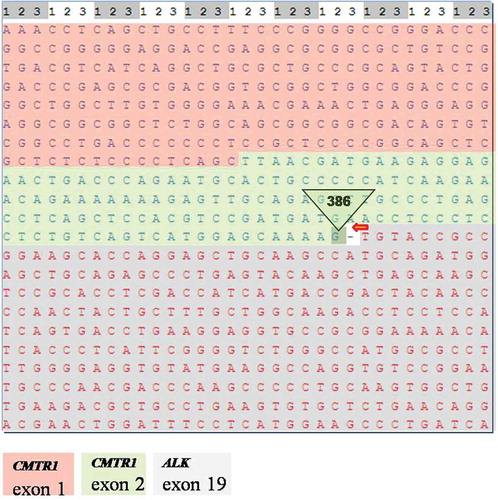
To confirm our speculation, we simulated the structure of this novel fusion protein. Based on NGS, this tumor specimen harbored a new CMTR1-ALK fusion variant involving rearrangement and fusion of exon 1 to 2 of CMTR1 to exon 20 to 29 of ALK (C2; A20). The breakpoint of the fusion gene is in intron 2 of the CMTR1 genomic DNA. During the process of building the fusion protein structure, we analyzed the CMTR1 mRNA sequence and found that exons 1 and 2 of CMTR1 contain 386 nucleotides, which is not a multiple of 3 required for a codon. This means that in exons 1 and 2 of the CMTR1 gene replication and expression are linked together with follow-up exons. Once they fuse with ALK, the last 2 nucleotides of CMTR1 will occupy 1 nucleotide of ALK ().
Figure 3. mRNA sequence of CMTR1-ALK fusion gene.
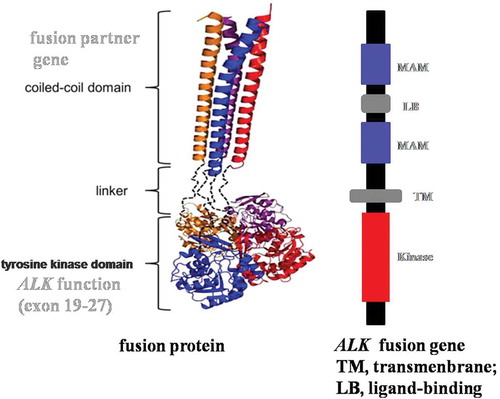
The normal structure of ALK (,Citation21 together with its fusion pattern, may function as a cell surface receptor for specific ligands that regulate the proliferation or differentiation of neural cell.Citation22 In this case, the CMTR1-ALK fusion gene caused an ALK gene frameshift mutation, which means that the fusion gene cannot translate kinase-active ALK fusion protein, explaining the IHC ALK-negative result.
Figure 4. Schematic illustration of ALK and the general fusion strategy.
According to China Food and Drug Administration (CFDA), when ALK fusion status is positive by any of the three platforms – FISH, IHC (Ventana), or RT-PCR – there is evidence for using targeted drug therapy in that patient. Despite the excellent efficacy of crizotinib in most ALK-positive lung cancers, heterogeneous responses have been reported and the reason for this variability is unknow.Citation9,Citation23,Citation24 The mechanism of primary resistance is complicated and deserves more detailed exploratio.Citation25 According to previous studies, there are three major mechanisms of resistance to targeted drug therapy: genetic alteration in the target; activation of bypass tracks or phenotypic change in the tumor such as development of epithelial mesenchymal transition; or limited penetration of “sanctuary” sites such as the central nervous syste.Citation20,Citation26 All three mechanisms of resistance have been identified in patients treated with crizotini.Citation20,Citation27,Citation28 We are the first to report intrinsic resistance to crizotinib caused by ALK gene frameshift mutation. In this case, the ALK fusion status was positive, but the patient had primary resistance to ALK inhibitor. By NGS and analysis of the fusion gene sequence, we found that the ALK fusion type is a null fusion which cannot translate “driven” protein that cause cancer incidence. This result shows that when the detection of ALK status is positive in a patient with drug resistance, we should pay more attention to the underlying mechanism.
Conflicts of interest
The authors have no conflicts of interest to declare.
Supplemental Material
Download MS Word (469.7 KB)Additional information
Funding
Related Research Data
References
- Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. Cancer J Clinicians. 2015;65(2):87–108. doi:10.3322/caac.21262.
- Jemal A, Center MM, DeSantis C, Ward EM. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol Biomarkers Prev. 2010;19(8):1893–1907. doi:10.1158/1055-9965.epi-10-0437.
- Wen M, Wang X, Sun Y, Xia J, Fan L, Xing H, Zhang Z, Li X. Detection of EML4-ALK fusion gene and features associated with EGFR mutations in Chinese patients with non-small-cell lung cancer. OncoTargets Ther. 2016:1989. doi:10.2147/ott.s100303.
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. Cancer J Clinicians. 2013;63(1):11–30. doi:10.3322/caac.21166.
- Shaw AT, Engelman JA. ALK in lung cancer: past, present, and future. J Clin Oncol. 2013;31(8):1105–1111. doi:10.1200/jco.2012.44.5353.
- Devarakonda S, Morgensztern D, Govindan R. Genomic alterations in lung adenocarcinoma. Lancet Oncol. 2015;16(7):E342–E351. doi:10.1016/S1470-2045(15)00077-7.
- Shaw AT, Hsu PP, Awad MM, Engelman JA. Tyrosine kinase gene rearrangements in epithelial malignancies. Nat Rev Cancer. 2013;13(11):772–787. doi:10.1038/nrc3612.
- Heuckmann JM, Balke-Want H, Malchers F, Peifer M, Sos ML, Koker M, Meder L, Lovly CM, Heukamp LC, Pao W, et al. Differential protein stability and ALK inhibitor sensitivity of EML4-ALK fusion variants. Clin Cancer Res. 2012;18(17):4682–4690. doi:10.1158/1078-0432.ccr-11-3260.
- Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, De Pas T, Besse B, Solomon BJ, Blackhall F, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385–2394. doi:10.1056/NEJMoa1214886.
- Ali SM, Hensing T, Schrock AB, Allen J, Sanford E, Gowen K, Kulkarni A, He J, Suh JH, Lipson D, et al. Comprehensive genomic profiling identifies a subset of crizotinib-responsive ALK-rearranged non-small cell lung cancer not detected by fluorescence in situ hybridization. Oncologist. 2016;21(6):762–770. doi:10.1634/theoncologist.2015-0497.
- Takeuchi K, Choi YL, Togashi Y, Soda M, Hatano S, Inamura K, Takada S, Ueno T, Yamashita Y, Satoh Y, et al. KIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancer. Clin Cancer Res. 2009;15(9):3143–3149. doi:10.1158/1078-0432.ccr-08-3248.
- Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara SI, Watanabe H, Kurashina K, Hatanaka H, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–U3. doi:10.1038/nature05945.
- Wong DWS, Leung ELH, Wong SKM, Tin VPC, Sihoe ADL, Cheng LC, Au JSK, Chung LP, Wong MP. A novel KIF5B-ALK variant in nonsmall cell lung cancer. Cancer. 2011;117(12):2709–2718. doi:10.1002/cncr.25843.
- Togashi Y, Soda M, Sakata S, Sugawara E, Hatano S, Asaka R, Nakajima T, Mano H, Takeuchi K, Lo AWI. KLC1-ALK: A novel fusion in lung cancer identified using a formalin-fixed paraffin-embedded tissue only. Plos One. 2012;7(2):e31323. doi:10.1371/journal.pone.0031323.
- Choi YL, Lira ME, Hong M, Kim RN, Choi SJ, Song JY, Pandy K, Mann DL, Stahl JA, Peckham HE, et al. A novel fusion of TPR and ALK in lung adenocarcinoma. J Thoracic Oncol. 2014;9(4):563–566. doi:10.1097/jto.0000000000000093.
- Fang DD, Zhang B, Gu QY, Lira M, Xu Q, Sun HY, Qian MX, Sheng WQ, Ozeck M, Wang ZX, et al. HIP1-ALK, a novel ALK fusion variant that responds to crizotinib. J Thoracic Oncol. 2014;9(3):285–294. doi:10.1097/jto.0000000000000087.
- Shan L, Jiang PD, Xu F, Zhang WL, Guo L, Wu J, Zeng YX, Jiao YC, Ying JM. BIRC6-ALK, a novel fusion gene in ALK break-apart FISH-negative lung adenocarcinoma, responds to crizotinib. J Thoracic Oncol. 2015;10(6):E37–E39. doi:10.1097/jto.0000000000000467.
- Tian Q, Deng WJ, Li ZW. Identification of a novel crizotinib-sensitive BCL11A-ALK gene fusion in a nonsmall cell lung cancer patient. Eur Respir J. 2017;49(4) doi:10.1183/13993003.02149-2016.
- Ruano-Ravina A, Provencio-Pulla M. Differential crizotinib response duration among ALK fusion variants in ALK-positive non-small-cell lung cancer. Transl Cancer Res. 2017; 6:S54–S56. doi:10.21037/tcr.2017.02.26.
- Gadgeel SM. Sequencing of ALK inhibitors in ALK plus non-small cell lung cancer. Curr Treat Options Oncol. 2017;18(6):12. doi:10.1007/s11864-017-0479-8.
- Diener M, Kopka B, Pohl M, Jaeger K-E, Krauss U. Fusion of a coiled-coil domain facilitates the high-level production of catalytically active enzyme inclusion bodies. ChemCatChem. 2016;8(1):142–152. doi:10.1002/cctc.201501001.
- Mano H. ALKoma: a cancer subtype with a shared target. Cancer Discovery. 2012;2(6):495–502. doi:10.1158/2159-8290.cd-12-0009.
- Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa K, Mekhail T, Felip E, Cappuzzo F, Paolini J, Usari T, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer (vol 371, pg 2167, 2014). N Engl J Med. 2015;373(16):1582. doi:10.1056/NEJMx150034.
- Ou SHI, Ahn JS, De Petris L, Govindan R, Yang JCH, Hughes B, Lena H, Moro-Sibilot D, Bearz A, Ramirez SV, et al. Alectinib in crizotinib-refractory ALK-rearranged non-small-cell lung cancer: a phase II global study. J Clin Oncol. 2016;34(7):661-+. doi:10.1200/jco.2015.63.9443.
- Toyokawa G, Seto T. ALK inhibitors: what is the best way to treat patients with ALK(+) non-small-cell lung cancer? Clin Lung Cancer. 2014;15(5):313–319. doi:10.1016/j.cllc.2014.05.001.
- Camidge DR, Pao W, Sequist LV. Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat Rev Clin Oncol. 2014;11(8):473–481. doi:10.1038/nrclinonc.2014.104.
- Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, Jessop NA, Wain JC, Yeo AT, Benes C, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci Transl Med. 2012;4(120):120ra17–120ra17. doi:10.1126/scitranslmed.3003316.
- Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, Kondo KL, Linderman DJ, Heasley LE, Franklin WA, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012;18(5):1472–1482. doi:10.1158/1078-0432.ccr-11-2906.