
ABSTRACT
Circulating tumor cells (CTC) enter the blood from many carcinomas and represent a likely source of metastatic dissemination. In contrast to the peripheral circulation, KRAS mutation- positive CTC thrive in the portal venous blood of patients with pancreatic ductal adenocarcinoma (PDAC). To analyze the essential interactions that contribute to carcinoma CTC growth and immune resistance, portal venous blood was collected during pancreatico-duodenectomy in 41 patients with peri-ampullary pathologies (PDAC = 11; ampullary adenocarcinoma (AA) = 15; distal cholangiocarcinoma (CC) = 6; IPMN = 7; non-malignant pancreatitis = 2). FACS-isolated cell populations from the portal circulation were reconstituted ex vivo using mixed cell reaction cultures (MCR). During the first 48hr, PDAC, AA, and CC patient CTC were all highly proliferative (mean 1.7 hr/cell cycle, 61.5% ± 20% growing cells) and resistant to apoptosis (mean 39% ± 25% apoptotic cells). PDAC CTC proliferation and resistance to T cell cytotoxicity were decreased among patients who received pre-operative chemotherapy (p = 0.0019, p = 0.0191, respectively). After 7 days in culture, CTC from PDAC, CC, and AA patients recruited multiple immune cell types, including CD105 + CD14 + myeloid fibroblasts, to organize into spheroid-like clusters. It was only in PDAC and CC-derived MCR that cluster formation promoted CTC survival, growth, and fibroblast differentiation. FACS depletion of CTC or myeloid fibroblast cells eliminated cluster network formation, and re-introduction of these cell populations reconstituted such ability. Our findings suggest that PDAC and CC CTC survival within the portal venous circulation is supported by their interactions with immune cells within multi-cell type clusters that could represent vectors of local recurrence and metastatic progression.
Introduction
Peri-ampullary carcinomas, including pancreatic ductal adenocarcinoma (PDAC), distal bile duct cancer (or cholangiocarcinoma, CC), and ampullary adenocarcinoma (AA) arise in the vicinity of the duodenal papilla (ampulla of Vater) and are frequently characterized as aggressive tumors with elevated metastatic potentialCitation1. These tumors have insidious development and often very poor prognosisCitation2,Citation3. Endoscopic evaluation of pre-malignant conditions such as intraductal papillary mucinous neoplasm (IPMN) or pancreatic intraepithelial neoplasia (PanIN) is invasive and not conducive to routine screening of the general populationCitation4.
Surgical resection of the primary cancer is currently the only potentially curative treatment for peri-ampullary cancers. In the case of PDAC, less than 20% of patients are typically eligible for surgical resection with curative intent. Combined with complete surgical resection, chemotherapy and radiation therapy have shown modest improvements on progression free survival, but little effect on overall survivalCitation3. Though immune responses to these tumors are specific and effective in vitro, immunosuppression is a major factor in its failure to deter cancer cell regrowth in vivoCitation5,Citation6. To date, none of the current T cell-directed checkpoint blockade therapies to release immunosuppression have proven effective in peri-ampullary cancer treatmentCitation5. Pre-operative administration of chemotherapy has shown improved resectability in some PDAC patientsCitation7, but circulating tumor cells (CTC) seem to remain viable and metabolically active even when there is objective tumor response to such therapiesCitation6.
Because of the inherent aggressive biologic behavior of peri-ampullary carcinomas, there is a great need to accurately assess and treat the residual tumor cell burden during or immediately following surgical resection.
Recent work with patient-tumor derived organoid and spheroid tissue cultures have shown the power of creating a patient-specific ex vivo tumor analysis system with the potential for clinical application in deriving individualized treatment regimensCitation8. These systems offer individual testbeds for tumor characterization and effectiveness of treatment. The main drawback of organoid systems is that they are derived from the resected primary tumor which may not necessarily reflect the viable remnant tumor cell population which is the true metastatic vector through proliferation, invasion and distant-organ seeding. The second major impediment to the use of organoids in clinical decision-making is that they are time-consuming and usually take weeks to months to establish; a time that may be too long to obtain a clinically significant benefit from their analysisCitation8.
Our previous characterization of CTC in the portal circulatory compartmentCitation6 suggested that the portal venous blood provides an essential environment for harboring tumor cells and possibly enabling distant metastasis in PDAC patients. Though the origin of these cells is not known, they may spring from primary tumor or local lymphoid reservoirs feeding into the portal circulation. Using aseptic, high speed fluorescence activated cell sorting (FACS), we have developed patient-derived culture platforms. Our model includes a rapid ex vivo CTC-based mixed cell reaction (MCR) culture to characterize the remnant tumor cell population found in the portal venous circulation following carcinoma surgical resection. In the current study, we propose these patient-derived platforms may be especially useful clinically as treatment testbed culture systems akin to primary tumor organoid or stem cell ‘spheroid’ cultures established in other tumor types, useful in mutation profiling and the design of individualized post-operative treatmentCitation8. These ex-vivo platforms may be used to analyze the role portal blood CTC aggregation and interaction play in CTC survival and development of distant metastasis.
Results
Proliferation and apoptosis in MCR
We generated a series of ex vivo patient-derived cultures using FACS-isolated portal blood mononuclear cells (PoBMC) to reconstitute CTC and immune cell interactions with defined circulating cell populations, including T cells, dendritic cells (DC), myeloid-derived suppressor cells (MDSC), fibroblasts (FB) and myeloid-derived fibroblasts (MFB). CTC cultured alone were capable of high doubling rates, averaging division every 1.7 hours within the first 16–60 hr ex vivo (p < 0.0001, r2 = 0.774). In the first 24 hours in MCR culture, portal blood CD44 + CD147 + EPCAM + CD45- cells sorted from patients with PDAC, CC, and AA had 1.5-fold increase in cell cycling rate compared to CD44 + CD147 + EPCAM + CD45- cells collected from patients with non-malignant pancreatitis and IPMN (Mann Whitney U test, p = 0.0042). CD44 + CD147 + EPCAM + CD45- cells detected in non-carcinoma patient samples may represent false positive collection of cells. Carcinoma CTC cell division time continued to increase gradually over time in culture. This high CTC replication rate led to rapid proliferation, outstripping T cell responses and cytotoxic killing (). The presence of MDSC suppressed T cell proliferation compared to that seen in cultures containing CTC-primed DC (p = 0.0200, ), suggesting ongoing immune-suppression that favors CTC proliferation. Moreover, cultured CTC exhibited a robust resistance to apoptosis which remained unaltered in the presence of tumor recognizing T cells with or without primed DC present ().
Figure 1. Proliferation and Apoptosis of CTC and T cells in MCR. PoBMC CTC and T cells isolated from the portal blood of 27 patients by FACS and labeled with CSFE prior to addition to cultures with and without other cell isolates (MDSC, DC, Fibroblasts) from the same patient’s PoBMC sample. DC were primed with sonicated CTC antigens prior to addition to the MCR. MCR cultures were held for 16–60 hr at 37C 5%CO2 and then harvested for flow cytometric analysis of cell cycle, T cell (white bars) and CTC (grey shaded bars) (A) proliferation by CFSE fluorescence dilution and (B) apoptosis induction by Annexin V-PE binding and 7AAD intercalation. (A) Addition of immune cells to the CTC MCR cultures did not significantly alter the rate of CTC cell division. (B) CTC (gray shaded bars) were found to be resistant to apoptosis with ≤ 10% increase in cell death seen with addition of T cells (CTCT) with or without CTC primed DC (CTCDCT) or other immune cells (MDSC, DC, MFB) present. In contrast, T cells (white bars) from the portal blood were highly apoptotic and had only a moderate activation increase in proliferation seen when CTC primed DC and CTC were present in the culture (A). However, T cell proliferation was significantly decreased by the presence of MDSC in place of DC as antigen presenting cells (p = 0.0200, A). Columns represent mean of data set and error bars represent standard error from the mean. Multiple variable analyses were done using a one-way ANOVA with Bonferroni Pairwise comparisons and pairwise comparisons were due using Mann-Whitney U test.
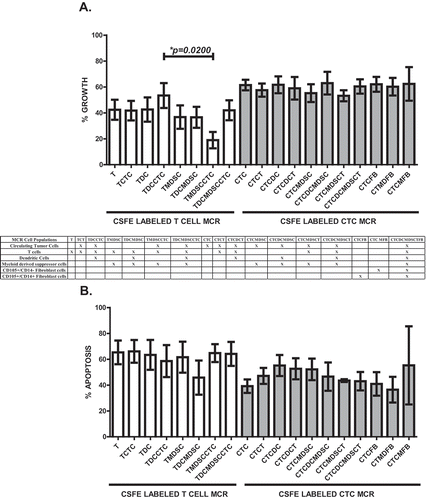
Immune responses to CTC in the portal blood
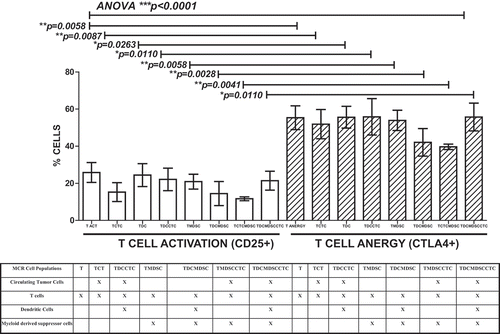
T cells added to CTC-containing cultures could recognize and kill their tumor targets; however, they themselves appear to be strongly anergized (). T cell cytolytic response in the ex vivo MCR () when cultured with CTC primed DC (TDCCTC), suggest a robust memory T response is present in the portal blood containing CTC. However, T cell activation (as measured by CD25 upregulation) was accompanied by a concurrent increase in CTLA-4 expression (), suggesting that activation-induced anergy and apoptosis reflect T cell interactions with MDSC and DC within the CTC-rich portal circulation. In our ex vivo MCR platform, MDSC appear to augment CTC proliferation and apoptosis resistance both through T cell-mediated and T cell-independent mechanisms (). Similarly, CTC-primed DC may promote CTC evasion of T cell effector killing through CTLA4 engagement and T cell suppression via regulatory T cell activation ().
Figure 2. T cell activation and anergy in MCR. PoBMC T cell populations isolated from 13 patients (5 PDAC, 2AA, 3 CC, 2 IPMN, 1 non-malignant pancreatitis) by FACS and added to cultures with and without other cell isolates (CTC, MDSC, DC) from the same patient’s PoBMC sample. DC were primed with sonicated CTC antigens prior to addition to the MCR. MCR cultures were held for 16–60 hr at 37C 5%CO2 and then harvested for flow cytometric analysis of CD3 + T cell CD25 surface expression as a biomarker for T cell activation and CTLA4 surface expression as a biomarker for T cell anergy. CTLA-4 expression was analyzed with or without concomitant CD25 expression (p < 0.001). T cell activation was not significantly improved by the presence of CTC or CTC-primed DC. Columns represent mean of data set and error bars represent standard error from the mean. Multiple variable analyses were done using a one-way ANOVA with Bonferroni Pairwise comparisons and pairwise comparisons were due using Mann-Whitney U test.
Tumor type affects CTC survival and immune responsiveness
Our study included portal blood samples from patients with a variety of peri-ampullary pathologies, including 15 AA (7 intestinal type/6 pancreatico-biliary type/2 mixed morphology), 6 CC (bile duct carcinoma), 11 PDAC, 7 IPMN and 2 non-malignant pancreatitis. In comparing the proliferative and apoptotic characteristics of CD47 + CD147 + EPCAM + CD45- cells (CTC) in PoBMC from patients with different pathologies, PDAC, typically the most aggressive of the peri-ampullary carcinomas, had the highest proliferation (p = 0.0301, ). All tumor types had relatively high resistance to apoptosis compared to IPMN and non-malignant tissue; however, these differences did not reach statistical significance (data not shown).
Figure 3. PDAC CTC are the most proliferative and most resistant to T cell cytotoxicity in MCR compared to other Peri-ampullary tumor types. Comparison of 23 patients’ CTC proliferation in culture alone (white bars) or in MCR with T cells (+T, hatched bars) indicated that T cells had little to no effect on CTC proliferation in any tumor type tested (PDAC (n = 7; 4 with pre-operative chemotherapy treatment (PDAC NEO), Ampullary (AA, n = 11), Cholangiocarcinoma (CC, n = 2), and intrapapillary mucinous neoplasm (IPMN, n = 3)). PDAC CTC proliferation was significantly greater than all other tumor types tested (p = 0.0301). Pre-operative chemotherapeutic treatment (PDAC NEO) of patients prior to surgery significantly decreased PDAC CTC proliferation with or without T cells present (p = 0.0019, p = 0.0191, respectively). Columns represent mean of data set (duplicate wells/sample whenever possible) and error bars represent standard error from the mean. Multiple variable analyses were done using a one-way ANOVA with Bonferroni Pairwise comparisons and pairwise comparisons were due using Mann-Whitney U test, as appropriate.
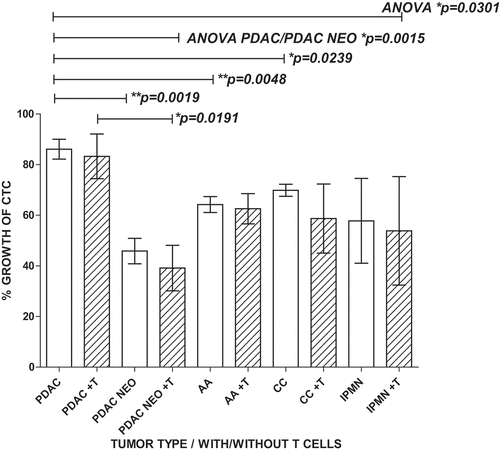
Portal Blood CTC from all analyzed tumor types were not strongly affected by T cell-mediated cytotoxicity, especially CTC from patients with PDAC (). PDAC patients treated with preoperative chemotherapy displayed a significant decrease in portal blood CTC proliferation rate both with (p = 0.0191) and without (p = 0.0019) T cells as compared to PDAC that did not receive chemotherapy prior to surgery ().
CTC cluster formation in ex vivo pobmc cultures
PoBMC containing CTC cultures from patients with carcinomas clustered spontaneously into ‘spheroid-like’ foci with fibroblast-like cells integrating into the clusters and surrounding the proliferating CTC (). PoBMC cultures were grown for 7 or more days in rich medium with or without the presence of supplements reported to support cluster formation and fibroblast differentiation from myeloid cells: 50% MatrigelCitation8,Citation9, 10 µg/ml human fibronectinCitation10, 10 µg/ml human adiponectinCitation11, or 0.5µg/ml macrophage colony stimulating factor (MCSF)Citation12, These cultures developed multi-cell type aggregates enveloping proliferating CTC (). Analysis of cluster composition revealed multiple cell types including T cells, MDSC, DC, and CD105 + fibroblasts. Myeloid marker expressing cells including MDSC (p < 0.0001, r2 = 0.5928), DC (p < 0.0001; r2 0.6453), and CD105 + CD14 + fibroblasts (p < 0.0001, r2 = 0.5366), increased in number over time correlating directly with the increases of CTC seen in clusters; whereas, T cell populations and CD105 + CD14- fibroblasts did not. There was no quantitative difference among the tested culture treatments (i.e., media only, fibronectin, adiponectin, Matrigel, or M-CSF) in the overall ability of the cells to form of these spheroid-like clusters or promote CTC proliferation within them. (). Clusters in non-malignant and most IPMN patient PoBMC cultures were found on flow analysis have few if any true CTC and to mainly be comprised of immune cells congregating around antigen presenting cells such as DC or monocytes since fewer if any CD47 + CD147 + EPCAM + CD45- cells were present in the original PoBMC or grow in culture ex vivo (). These immune dominated clusters disappeared as cells died off gradually in the cultures between 7 and 21 days.
Figure 4. CTC cluster formation in PoBMC culture. A) CTC containing PoBMC samples were noted to spontaneously formed clusters containing CTC when held in rich media culture at about 7–11 days in vitro. PoBMC isolated from ficoll histopaque gradients were cultured in rich media alone (A,13 days), or in media supplemented with 50% Matrigel (B, 7 days), 10µg/ml human fibronectin (C, 7 days), 10µg/ml human adiponectin (D, 12 days), or 0.5 µg/ml MCSF (E, 7 days). Fibroblast-like cells (B, arrow) became apparent in the cultures in 3–5 days and multi-cell type clusters were visible by 7 days in culture in monolayer cultures (A-E). At 11days, cultures began to exhibit fibroblast ‘networking’ reminiscent of stroma development (D). No quantitative differences were found among the treatments in their ability to support CTC cluster formation. Images representative of cultures from 31 patients as micrographs taken on as DIC images on a Zeiss Axiovert fluorescence DIC microscope. Size bars represent 200 pixels (34,000 microns).
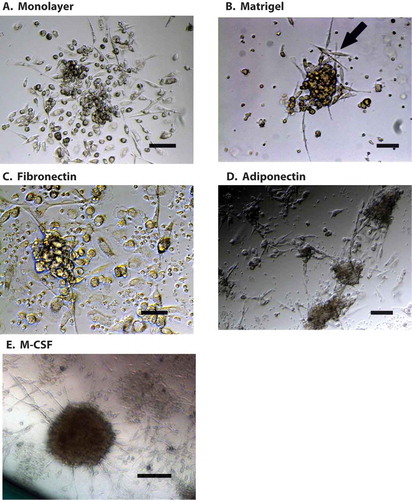
Figure 5. Portal blood CTC cluster formation promotes CTC proliferation in PDAC and cholangiocarcinoma. Flow cytometric analysis of 30 patients’ PoBMC (5 PDAC (2 with pre- operative (neo adjuvant) chemotherapy treatment (PDAC NEO), 15 AA, 4 CC, 6 IPMN) in CTC cluster cultures indicated that CTC proliferation was enhanced in PoBMC cluster cultures compare to the ex vivo input to the cultures (median counts listed in ). CTC proliferation in cluster cultures derived from patients with PDAC or CC was greater than PoBMC from patients with AA or IPMN (p values as indicated on the graph). Preoperative chemotherapy significantly decreased CTC proliferation in cluster cultures compared with PDAC patient without pre-operative treatment (p = 0.0485). No significant difference was seen between pancreatico-biliary cancer types and intestinal type ampullary adenocarcinomas in supporting CTC proliferation in CTC clusters in the portal blood (data not shown). Columns represent mean of data set and error bars represent standard error from the mean. Multiple variable analyses were done using a one-way ANOVA with Bonferroni pairwise comparisons and pairwise comparisons with Mann-Whitney U test, as appropriate.
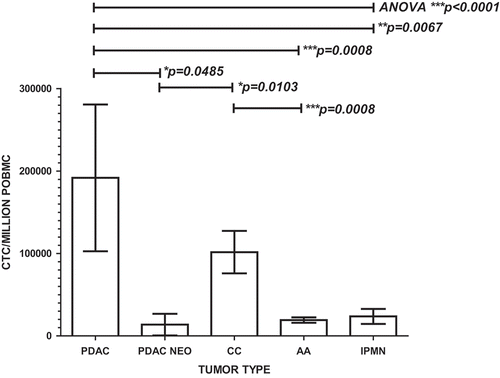
CTC numbers in clusters were significantly higher than that detected in the original ex vivo PoBMC used to seed the cultures (Wilcoxon rank test, p > 0.01). CTC numbers were significantly higher in PDAC and CC 7 day cluster cultures than the CTC numbers in cluster cultures from AA or IPMN (; ANOVA, p < 0.0001). However, CTC clusters from patients with PDAC that had received pre-operative chemotherapy (PDAC NEO) contained significantly less CTC than cultures from PDAC or CC patients who did not receive preoperative treatment (Mann Whitney U test; p = 0.0485, ).
Essential cellular components in PoBMC CTC cluster formation
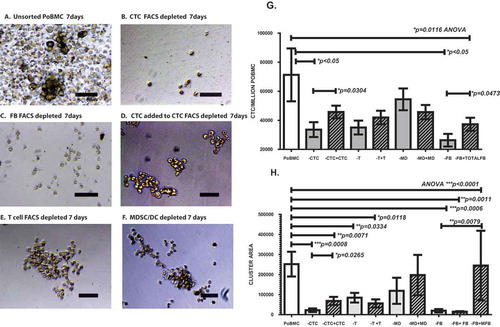
We identified populations of CTC, MDSC, DC, T cells, and fibroblasts within the cluster cultures by flow cytometric analysis. To test if any of the cell types identified in PoBMC CTC clusters were essential for the cluster formation and stability, we selectively depleted PoBMC of each cell population by FACS prior to their growth in ex vivo MCR cultures (). Intact (un-sorted) PoBMC formed CTC clusters that were significantly more supportive of CTC survival and proliferation than any of the depleted MCR cultures except for those depleted of MDSC and DC (-MD, and ). Removal of T cells, or DC and MDSC did not inhibit CTC cluster formation at 7 days in cultures ( and ). In contrast, cultures with CTC or CD105 + fibroblasts removed did not form clusters ( and ). Addition of FACS isolated CTC to CTC-depleted cultures () or CD105 + fibroblasts () to CD105 + fibroblast-depleted cultures reconstituted cluster formation, and significantly increased CTC proliferation (p = 0.304, p = 0.0473, respectively). Re-introduction of T, MDSC, DC, or CD105+/CD14- fibroblasts did not influence cluster-formation capacity or CTC proliferation ( and ). Analysis of subpopulations within the CD105 + fibroblasts indicated that addition of CD105+/CD14 + myeloid fibroblasts (but not CD105 + CD14-) fibroblasts could reconstitute cluster formation to the levels comparable to unsorted PoBMC (p = 0.0078, ). This suggests that the CD105 + CD14 + myeloid fibroblasts are vital components of the CTC cluster formation.
Figure 6. FACS depleted PoBMC cultures indicate that CTC and CD105 + /CD14 + myeloid fibroblasts are essential to CTC cluster formation. One to five million PoBMC were cultured for 7 days in rich medium (A) without FACS sorting (G,H; POBMC, white bars) and after FACS depletion of (B) CD44 + CD147 + EPCAM + CD45- CTC population, (C) CD105 + total fibroblast populations, (E) CD3 + CD45 + T cell populations, and F) CD33 + CD11b + CD14 + CD11c-MHCDR- MDSC and CD11c + MHCDR + DC populations (G,H; gray bars). Only the depletion of CTC (-CTC, B) and CD105 + fibroblast populations eliminated cluster formation (-FB, C). Images in panels A-F represent 20x magnification micrographs taken on as DIC images on a Zeiss Axiovert fluorescence DIC microscope. Size bar represents 200 pixels (34,000 microns). Addition of the FACS selected CTC population restored cluster formation (D, H, hatched bars, +CTC) and allowed for survival and growth of CTC (G, hatched bars) within the cluster culture. Addition of CD105 + fibroblasts restored CTC growth in cluster cultures (G, hatched bars, -FB, +total FB). On further analysis of CD105 + subpopulations (H), addition of CD105+/CD14- fibroblasts (+FB) to the depleted PoBMC did not restore clustering; however, addition of CD105 + /CD14 + myeloid fibroblasts (+MFB) did. Bars on graphs (G,H) represent mean of data set and error bars represent standard error from the mean 7 days after ex vivo PoBMC culture. Mean input ex vivo CTC numbers for 7 runs of these studies was 4,425 ± 11,002 CTC per million PoBMC as determined by FACS isolated CTC analysis prior to culture. Statistical analyses yielding the significance values depicted on graphs of CTC growth (G) and cluster area (H) included one-way ANOVA multiple variant analysis with Bonferroni pairwise comparisons and pairwise comparisons with Mann-Whitney U test, as appropriate.
Discussion
Successful treatment of peri-ampullary carcinomas is largely contingent on the combination of complete surgical removal with effective systemic chemotherapy and radiation therapy strategies. Currently, only a minority of PDAC and CC patients are candidates for multi-modality therapy including surgical resection as tumor burden is often advanced at the time of clinical detection. Furthermore, viable tumor cells often remain in the circulation and target organs following complete primary tumor resection leading to loco-regional recurrence and distant metastasis that negatively impact patient survivalCitation3.
Our earlier findings pointed to the permanence of viable CTC in the portal venous blood after primary tumor resection suggesting a potential role for portal CTC in metastatic progression among patients with peri-ampullary carcinomasCitation6.
In the current study, we sought to elucidate the interactions of portal circulation CTC with other cell types that may promote an immune-quiescent environment and enable tumor cell dissemination. Our results suggest that T cell anti-tumor immune responses though present in the portal circulation are suppressed under the influence of CTC (–). Portal blood T cells appear suppressed by MDSC and susceptible to apoptosis in the presence of CTC. CTC in turn, rapidly proliferate and recruit suppressive immune cells developing a multicellular cluster around them that promotes CTC growth and immune resistance ().
Rapid proliferation of portal blood CTC in patient-derived ex vivo platforms supports our previous observations of rapid re-growth or re-emergence of covert tumor reservoirs in the portal venous circulatory compartment following primary tumor surgical removalCitation6.
Analysis of various tumor types arising from the peri-ampullary region treated with pancreatico-duodenectomy, associate PDAC and CC to highly proliferative portal vein CTC that spontaneously constitute multi-cellular clusters when cultured ex vivo ( and )
Neoadjuvant chemotherapy and/or radiation treatments have been used in borderline resectable or unresectable tumors in the hopes of decreasing primary tumor size and enabling complete surgical resection. Our previous study showed that viable, transcriptionally active CTC are still present in the portal circulation even in patients with favorable response to pre-operative treatmentCitation6. Our current data suggest that cytotoxic and perhaps immunomodulator effects of pre-operative chemotherapy may be influencing the proliferative rate of portal blood CTC re-growth in clusters ( and ). A better understanding of the underlying mechanisms of preoperative chemotherapy effects on portal CTC and stem cell viability are needed to optimize the combination of systemic treatment modalities with surgical resection.
The role of stromal tissue in primary tumor support and immune resistance has been the subject of much debate and studyCitation13. If the stromal component of pancreatic tumors is vital to its immune evasion and/or its nutrient supply for rapid growth, it begs the question how then do CTC survive in the immune rich environs of the portal blood without a stromal support? A portion of CTC survival may be granted to the immunosuppressive microenvironment that the gastrointestinal tract, microbiome, and caustic enzymatic nature of the localeCitation14. However, this does not explain the ability of CTC to survive without stromal metabolic and structural support as they migrate through the blood and on to re-grow in a metastatic site.
Myeloid cells are known for their abilities to differentiate and re-differentiate into a variety of important cell morphologies when exposed to specific growth factorsCitation10,Citation11,Citation15,Citation16. The ability of myeloid precursors to form fibroblasts in blood offers a possible way for CTC to attract and subvert immune cells into forming a stromal-like support for their survival and development. Our observations of CD105 + CD14 + fibroblast cells being essential for CTC cluster formation suggest that myeloid cells may be influenced by the presence of CTC within the portal blood microenvironment to differentiate into stromal-like fibroblasts that predominate within CTC-induced clusters. These cells may support de novo conversion of proliferating CTC into multicell foci, able to not only survive in the portal blood, but also migrate out of the circulation into distant metastatic site such as the liver. Recent studies have given evidence that not only do naturally forming CTC clusters represent the most likely source of tumor remnant survivalCitation9,Citation17,Citation18, but that they have the capability of re-assembling themselves into linked linear associations that can extravasate and invade distant tissues to form metastatic lesionsCitation18. This ability point to CTC clusters as prime candidates for surrogate biomarkers of metastatic potential. If cluster development and integrity ex vivo can be associated to PDAC and CC metastatic potential, the CTC interaction with myeloid fibroblasts may provide a therapeutic target for effective attenuation of local re-growth and metastatic potential directed at the portal venous circulation.
Previous clinical trials in Japan targeted treatment of pancreatic cancer and hepatic metastases via direct portal vein has been attempted using either perfusion of 5-FluorouracilCitation19 or injection of Gemcitabine aloneCitation20 or in mouse tumor models, Gemcitabine in combination with IL-2 as an immunostimulatory agentCitation21. These small pilot studies resulted in modest extension of progression free survival but added caution due to potentially high toxicity in the case of 5-FUCitation19. The mouse model studies indicated that immune stimulation improved the effectiveness of gemcitabine not only in tumor growth suppression but also through its inhibition of MDSC suppressor function in the tumor microenvironmentCitation21. Our MCR culture could provide a test platform to testbed more effective immune modulation therapies directed at breaking tumor supportive immune cell-CTC interactions in the portal circulation. Our preliminary MCR and CTC cluster culture studies focusing on myeloid cell directed therapies, anti-inflammatory agents and lipid signaling and metabolism have shown promise; whereas, T-cell directed checkpoint inhibitors, mirroring clinical outcomes with these agents, have not.
Our findings support the use of that patient-derived platforms as tools for development of a combination therapies for post-operative care and in future finding those that can be safely applied directly to the portal venous circulation CTC reservoir while it is accessible during pancreatico-duodenectomy or in regional treatment strategies using interventional endoscopy. Potential uses of ex vivo portal blood CTC culture systems include analyses for predicting outcomes of surgical patients, as an aid in testbed trials for tailoring post-surgical treatment design, and as an indicator of the ‘window’ of opportunity for development of intra-operative and peri-operative treatment options to prevent both local recurrence and distant metastases.
Materials and methods
Patient study population
Patients being evaluated for surgical treatment of localized PDAC and other peri-ampullary pathologies were offered participation in this study during their pre-operative outpatient visit at Florida Hospital Cancer Institute in Orlando, Florida. All study procedures conformed to the relevant regulatory standards required for ethical research involving volunteer human patients as approved by the Florida Hospital equivalent of an Ethics Committee for Human Experimentation, i.e., the Institutional Review Board (IRB). Informed consent was obtained under Florida Hospital IRB approved linked protocols 507397 and 946704. Demographic information on the study patient population is listed in . Patient samples were processed for the study without knowledge of their final diagnosis and the results were segregated to tumor subtype groups after laboratory data collection. This caused a random and blinded distribution of samples to the analyses available. Due to volume limitations in portal blood collection and patient variation for quantitative FACS isolation of CTC from mononuclear cell isolates (PoBMC), not all analyses were run on every patient sample. Sample size for each set of analyses is listed in the descriptive text or figure legends.
Table 1. Demographic information for study participants & median CD47 + CD147 + EPCAM + CD45- potential CTC/million cells isolated from ex vivo portal venous blood samples.
Patient sample collections
Blood samples were collected from a total of 41 participants undergoing open pancreatico-duodenectomy for various pathologies. () Portal blood samples (10-20ml) were obtained by direct intra-operative venipuncture of the portal vein with a 21G needle and 20 cc syringe immediately after resection of the duodeno-pancreas. The venipuncture site was then oversewn with 5–0 polypropylene suture. These blood samples were stored in heparin coated vacutainers and kept on ice until further processing. Specimens were used for isolation of CTC and immune cells by high speed aseptic FACS using a Beckman Coulter MoFlo XDP sorting system and used for culture as Mixed Cell Reactions (MCR) reconstitutions. Potential ‘CTC’ cell counts were based on sorted CD147 + CD44 + EPCAM + CD45- cells collected in aseptic FACS. Sorted counts for all 41 samples analyzed gave a mean 4425 ± 11,002 CTC/million PoBMC sorted with a range from 0 to 58,738 CTC/million PoBMC, median 403. CTC and other cell type counts varied with primary cancer type and with individual patient variation (median, range listed in ).
High speed aseptic FACS isolation of CTC and immune cells
PoBMC were separated from red blood cells on ficoll histopaque gradients (Pharmacia/Life Technologies, cat# 17144003) by centrifugation. The PoBMC layer near the top of the gradient was collected, counted for viable cells, and washed with rich medium (RPMI 1640 (Mediatech Cellgro, cat#10–040), 10% medium 199 (Gibco, Life Technologies, cat# SH30253.FS), 10% fetal calf serum (Mediatech Cellgro, cat#35–016-CV), 2% antibiotic-antimycotic mix (Sigma-Aldrich, cat# A5955-100ML) Between 10 and 100 million PoBMC were immunologically stained with antibody fluorescent conjugates directed against CD45 (BD Biosciences, cat#347464), EpCAM (BD Biosciences, cat#347198), CD44 (Beckman Coulter, cat# IM1219U), and CD147 (Abcam, cat# ab108317). CTC collection used the immunological profile of CD44+, CD147+, EpCAM+, CD45- as the isolation FACS sort criteria on a 6-color channel high speed Beckman-Coulter MoFlo XDP FACS instrument housed within a Walker HEPA filtered laminar flood hood and using 0.2micron filtered sterile PurFlo sheath fluid (Beckman-Coulter, MoFlo XDP). CTC sorting by this profile typically ranged between 65% and 85% efficiency when profiled for purity (isolated cell) selection at flow rates of 20,000–100,000 cells per second. Immune cells also isolated by FACS included dendritic cells (DC, CD11c+, DR+, CD14-, CD11b-), myeloid-derived suppressor cells (MDSC, CD11b+, CD33+, CD14+, CD11c-, DR-, (antibody conjugates from BD Biosciences; cat# BD 555388, 551378, 340585, 561355, 559866), and T lymphocytes (T cells, CD3+, CD4+, CD8+, CD45+, (antibody conjugates from BD Biosciences; cat# BD 555342,561005,561949, 347464). In addition, epithelial–mesenchymal transition (EMT) fibroblasts (FB, CD105 + CD14-) and EMT myeloid fibroblasts (MFB, CD105 + CD14+) were similarly FACS isolated (antibody conjugates from Abcam cat# ab201236, BD Biosciences, BD cat# 562408,347493). Sorted cells and specific cell population FACS depleted PoBMC cell populations (-CTC, -T, -MDSC/DC, -FB/MFB) were collected and washed with the same rich medium, then cultured and analyzed immediately.
Mixed cell reaction (MCR) cultures
Ficoll-isolated PoBMC, FACS sorted PoBMC subpopulations, and specific cell population FACS-depleted PoBMC were washed in rich medium, centrifuged at 600xg, 5min, 25°C and resuspended in fresh rich medium for culture. In MCR cultures with FACS isolated cells, portions of the isolated CTC and T cells were labeled by CFSE (Molecular Probes, cat#C34554) uptake for 10min in 1XPBS (Corning, cat#20–031), 1%dimethyl sulfoxide (DMSO, Sigma Aldrich, cat#D2650) for 10min at 37C/5%CO2, then washed in rich medium, centrifuged at 600xg, 5min, 25°C and resuspended in rich medium for culture. CSFE labeled Cells were cultured alone (optimized at 5,000 CTC per well), or together in MCR containing unlabeled cell numbers proportional to that seen in the patient blood PoBMC, relative to the CTC population isolated in FACS. On average, PoBMC samples tested yielded 4,422 ± SD11,142 CTC PoBMC, 20,949 ± SD 49,213 MDSC, 6,491 ± SD 17,089 DC, 132,644 ± SD 222,414 T cells, and 49,612 ± SD111,899 CD105 + fibroblasts per million PoBMC sorted; yielding average ratios of 1 CTC to 5 MDSC, 1.5 DC, 30 T cells, and 11 CD105 + fibroblasts per MCR test 96 or 48well plate well. MCR culture mixes included T cells + CTC, DC + T cells, DC + T cells + CTC, MDSC + DC + T cells, MDSC + DC + T cells + CTC, CTC + MDSC, CTC + MDSC + DC, CTC + FB/MFB, and CTC + MDSC + DC + T + FB/MFB. For doubling time analyses, CFSE labeled CTC were cultured alone for 16–60hrs at 37C/5%CO2, with separate culture wells of the same patient used for each MCR. Whenever possible, MCR cultures were run in duplicate or triplicate to allow for analysis of T cell activation (CD25, CD28, CD3, CD4, CD8 (antibody conjugates from BD Biosciences and Beckman-Coulter) BD cat# 555431, 561793,555342, 561005, 557086) and anergy (CD95, CTLA-4, PD1/PD1L-1 (antibody conjugates from BD Biosciences and Abcam; BD# 555673, 560939, Abcam cat# ab220301, ab209960) cell surface marker analysis in addition to CFSE, Annexin V, and 7AAD staining.
Cell viability, proliferation & apoptosis analyses
PoBMC were analyzed for viability (Trypan Blue Exclusion, Gibco/Life Technologies, Grand Island, NY). PoBMC and sorted cell populations were analyzed for apoptosis/cell death using Annexin-V/7AAD fluorescent reagents (BD Biosciences, Beckman-Coulter; cat# 559763, BC cat# IM3546, IM3422) and detection by an Accuri C6 cytometer (BD BioSciences, Accuri C6). No significant differences were seen in viability or apoptotic cells among any of the 41 PoBMC samples tested prior to culture. Proliferation of sorted CTC and T cells was analyzed by CFSE vital dye dilution fluorescence (Molecular Probes, cat#C34554) detected by flow cytometric analysis on an Accuri C6 cytometer.
CTC cluster formation analyses
PoBMC cultures were seeded at 1–5 million total cells per well were maintained up to 21 days in rich medium (RPMI 1640, 10% medium 199, 10% fetal calf serum, 2% antibiotic-antimycotic mix), with some cultures supplemented with 10µg/ml fibronectin (Sigma-Aldrich, cat#F0895), 10µg/ml adiponectin (Sigma-Aldrich, cat#SRP4901), 0.5µg/ml MCSF (BioSource, cat# PHG6054) or 50% Matrigel (Sigma-Aldrich, cat# E1270-5X10ML). Cultures were re-supplied with media every 3 days. At 7 days in culture, cell clusters were harvested and dissociated by vigorous pipetting after incubation in Cell Stripping Buffer (Cellgro, Mediatech; cat#25–056-CI) for 30min at 4°C. Collected cells were analyzed by flow cytometry for viability by trypan blue exclusion, and for identification of cell types within the clusters and for the growth of CTC numbers within the clusters, using the same antibody conjugates as described above for FACS identification and isolation. Cluster cultures were monitored by photo-microscopy for development of fibroblast-like cells and stromal-like network formation over 21 days and then cultures were cryogenically persevered in 90% fetal calf serum, 10% dimethyl sulfoxide (DMSO) (Sigma Aldrich, cat#D2650) and stored at −80°C. Cluster size changes over time were quantified by area using NIH Image J software. Thaw of cryogenic samples resulted in loss of cluster formation capability and subsequent loss of tumor cell viability in most cultures. Thaw of cytogenetically preserved PDAC CTC cluster cultures resulted in viable monolayer cultures of CTC; however, these cells did not reform clusters.
FACS depleted and add-back analyses
Ten to one hundred million cells negatively selected from PoBMC isolates subjected to FACS depletion of specific cell types (i.e., residual cells after isolation and removal of CTC, T, MDSC, DC, and/or CD 105/CD14 positive fibroblasts as described above) were collected and cultured at 0.5–1 million negatively sorted cells/well for 7 days in rich medium (RPMI 1640, 10% medium 199, 10% fetal calf serum, 2% antibiotic-antimycotic mix) either as is or with FACS isolated cell populations re-added to the cultures at the ratios originally seen in the ex vivo PoBMC. At 7days in culture, the cultures were harvested after incubation in Cell Stripping Buffer (Cellgro, Mediatech; cat#25–056-CI) for 30min at 4°C. Collected cells were analyzed for viability by trypan blue exclusion, and by flow cytometry for identification of cells and the growth of CTC numbers within the clusters. Cultures were monitored by photo-microscopy for development of clusters containing fibroblast-like cells. Cluster size changes over time were quantified using NIH Image J software.
DNA and mRNA analysis
PoBMC and FACS sorted CTC samples were collected as described above for use in DNA and mRNA analysis. For mRNA analyses, samples were diluted 1–2 volumetrically in RNALater (Ambion cat# AM7020) and stored at 4°C for later RNA extraction. mRNA samples were analyzed by Quantitative RT-PCR using TaqMan primer sets (Ambion/Life Technologies, cat# 4402954 and Qiagen, cat# 13323) specific for K-RASwt (UniGene ID: Hs.505033), K-RAS mut12exon (5ʹ ACC TTA TGT GTG ACA TGT TCT AAT ATA GT3ʹ and 5R’ GCA CTC TTG CCT ACG CGA T3R’, with probe FAM 5ʹCCT GCT GAA AAT GAC TGA ATA TAA ACT TGT GG-MGB for exon 12 12Ala, 12Arg, 12asp, 12Cyc, 12Ser, 12Val, and 13Asp mutations, and mutation 12D blocker 5ʹCCT ACG CCA GCT3ʹ), and GAPDH (UniGene ID:Hs.544577). Results from quantitative RT-PCR analyses of samples available from 32 patients were compared using ΔΔCt values of the K-RASmut gene expression compared to that of the GAPDH control. Two of the nine (22%) PDAC PoBMC samples examined and one out of six (17%) cholangiocarcinoma samples examined were positive for the K-RAS gene mutation, and none of 12 AA, 7 IPMN or 2 non-malignant samples tested contained the K-RAS gene mutation or expression.
Statistical analysis
Multi-variant, ANOVA with Bonferroni Pairwise comparisons, where appropriate, and pair-wise comparison (Student T test, Welsh corrected T test, and Mann-Whitney U test) statistical analyses were performed using Prism 5 (GraphPad Software, Inc. 2015) and Stata ICE v.12 (Stata Corp. 2012). Correlation analyses were done using Pearson’s Correlative analysis and Wilcoxon rank test, where appropriate. Statistical method used in each analysis is listed in the appropriate figure legends. Statistical significance limit was set at p < 0.05 with a 95% confidence interval.
Acknowledgments
The authors gratefully acknowledge support by the Phi Beta Psi Philanthropic Sorority, the Fraternal Order of Eagles, and the generosity of donors to the Florida Hospital Foundation.
Disclosure Statement
The authors have no conflicts of interest to report for this work.
Additional information
Funding
References
- Hatzaras I, George N, Muscarella P, Melvin WS, Ellison EC, Bloomston M. Predictors of survival in periampullary cancers following pancreaticoduodenectomy. Ann Surg Oncol. 2010;17(4):991–997. doi:10.1245/s10434-009-0883-9.
- Kanarajah SK. Pancreaticoduodenectomy for periampullary tumours: a review article based on Surveillance, End Results and Epidemiology (SEER) database. Clin Transl Oncol. 2018. doi:10.1007/s12094-018-1832-5.
- Ansari D, Friess H, Bauden M, Samnegard J, Anderson R. Pancreatic cancer: disease dynamics, tumor biology and the role of the microenvironment. Oncotarget. 2018;9(5):6644–6651. doi:10.18632/oncotarget.24019.
- Eshlerman JR, Norris AL, Sadakai Y, Debeljak M, Borges M, Harrington C, Lin E, Brant A, Barkley T, Almario JA, et al. KRAS and guanine nucleotide-binding protein mutations in pancreatic juice collected from the duodenum of patients at high risk for neoplasia undergoing endoscopic ultrasound. Clin Gastroenterol Hepatol. 2015;13(5):963–969.e4. doi:10.1016/j.cgh.2014.11.028.
- Foley K, Kim V, Jaffee E, Zheng L. Current progress in immunotherapy for pancreatic cancer. Cancer Lett. 2016;381(1):244–251. doi:10.1016/j.canlet.2015.12.020.
- Arnoletti JP, Zhu X, Almodovar AJO, Veldhuis PP, Sause R, Griffith E, Corpus G, Chang JCC, Fanaian N, Litherland SA. Portal venous blood circulation supports immunosuppressive environment and pancreatic cancer circulating tumor cell activation. Pancreas. 2017;46(1):p 116–123. doi:10.1097/MPA.0000000000000667.
- Epelboym I, DiNorcia J, Winner M, Lee MK, Lee JA, Schrope BA, Chabot JA, Allendorf JD. Neoadjuvant therapy and vascular resection during pancreaticoduodenectomy: shifting the survival curve for patients with locally advanced pancreatic cancer. World J Surg. 2014;38:1184–1195. doi:10.1007/s00268-013-2384-z.
- Baker LA, Tiriac H, Clevers H, Tuveson DA. Modeling pancreatic cancer with organoids. Trends in Cancer/Cell. 2016;2(4):176–190. doi:10.1016/j.trecan.2016.03.004.
- Cheung KJ, Ewald AJ. A collective route to metastasis: seeding by tumor cell clusters. Science. 2016;352(6282):167–169. doi:10.1126/science.aaf6546.
- Pilling D, Gomer RH. Differentiation of circulating monocytes into fibroblast-like cells. Methods Mol Biol. 2012;904:191–206. doi:10.1007/978-1-61779-943-3_16.
- Yang J, Lin S-C, Chen G, He L, Hu Z, Chan L, Trial J, Entman ML, Wang Y. Adiponectin promotes monocyte-to-fibroblast transition in renal fibrosis. J Am Soc Nephrol. 2013;24(10):644–1659. doi:10.1681/ASN.2013030217.
- Metcalf D. The molecular biology and function of the granulocyte-macrophage colony-stimulating factors. Blood. 1986;67(2):257–267.
- Sherman MH, Yu RT, Tseng TW, Sousa CM, Liu S, Truitt ML, He N, Ding N, Liddle C, Atkins AR, et al. Stromal cues regulate the pancreatic cancer epigenome and metabolome. PNAS. 2016;114(50):1129–1134. doi:10.1073/pnas.1620164114.
- Dimeloe S, Mehling M, Frick C, Loeliger J, Bantug GR, Sauder U, Fischer M, Belle R, Develioghi L, Tay S, et al. The immune-metabolic basis of effector memory CD4 + T cell function under hypoxic conditions. J Immunol. 2016;196(1):106–114. doi:10.4049/jimmunol.1501766.
- Eom DS, Parichy DM. A macrophage relay for long-distance signaling during postembryonic tissue remodeling. Science. 2017;355(6331):1317–1319. doi:10.1126/science.aal2745.
- Alvarez-Errico D, Vento-Tormo R, Sieweke M, Ballestar E. Epigenetic control of myeloid cell differentiation, identity and function. Nat Rev Immunol. 2015;15:7–17. doi:10.1038/nri3777.
- Sarioglu AF, Aceto N, Kojic N, Donaldson MC, Zeinali M, Hamza B, Engstrom A, Zhu H, Sundaresan TK, Miyamoto DT, et al. A microfluidic device for label-free, physical capture of circulating tumor cell-clusters. Nat Methods. 2015;12(7):685–691. doi:10.1038/nmeth.3404.
- Au SH, Storey BD, Moore JC, Tang Q, Chen Y-L, Javaid S, Sarioglu AF, Sullivan R, Madden MW, O’Keefe R, et al. Clusters of circulating tumor cells traverse capillary-size vessels. PNAS. 2016;113(18):4947–4952. doi:10.1073/pnas.1524448113.
- Kurosaki I, Kawachi Y, Nihei K, Tsuchiya Y, Aono T, Yokoyama N, Shimizu T, Hatakeyama K. Liver perfusion chemotherapy with 5-Fluorouracil followed by systemic gemcitabine administration for resected pancreatic cancer: preliminary results of a prospective phase 2 study. Pancreas. 2009;38(2):161–167. doi:10.1097/MPA.0b013e31818815f7.
- Kitami C, Kurosaki I, Kawachi Y, Nihei K, Tsuchiya Y, Nomura T, Minagawa M, Takano K, Hatakeyama K. Niigata study group of pancreatic cancer. Portal vein infusion chemotherapy with gemcitabine after surgery for pancreatic cancer. Surg Today. 2013;43:33–39. doi:10.1007/s00595-012-0179-8.
- Ito Y, Aiura K, Ueda M, Kitajima M, Kitagawa Y. Establishment of combined immune-chemotherapy with systemically administered gemcitabine and intra-portal administration of interleukin-2 in murine models of liver metastases of pancreatic cancer. Int J Oncol. 2008;33:49–58.