
ABSTRACT
Chemotherapy resistance represents a major issue associated with gastric cancer (GC) treatment, and arises through multiple mechanisms, including modulation of the cell-cycle check point. Several ubiquitin kinases, including RING finger protein 138 (RNF138), have been reported to mediate the G2/M phase arrest. In this study, we investigated the role of RNF138 in the development of cisplatin resistance of two GC cell lines. We show that RNF138 levels are higher in cisplatin-resistant cell lines, compared with cisplatin-sensitive cells, and RNF138 expression was elevated during drug withdrawal following the cisplatin treatment. Using gene overexpression and silencing, we analyzed the impact of altering RNF138 level on GC cell viability, apoptosis, and cell cycle phenotypes in two isogenic cisplatin-sensitive and resistant cell lines. We show that RNF138 overexpression increased GC cell viability, decreased apoptosis and delayed cell cycle progression in the cisplatin-sensitive GC cells. Conversely, RNF138 silencing produced opposite phenotypes in the cisplatin-resistant cells. Moreover, RNF138-dependent phosphorylation of Chk1 was seen in GC cells, indicating a novel connection between cisplatin-induced DNA damage and apoptosis. Collectively, these data suggest that RNF138 modulates the cisplatin resistance in the GC cells, thus serving as a potential drug target to challenge chemotherapy failure. In addition, RNF138 can also be used as a marker to monitor the development of cisplatin resistance in GC treatment.
Introduction
Gastric cancer (GC) is the third leading cause of cancer-related fatalities globally. The current standard treatments for GC patients include surgery combined with radiation and/or chemotherapyCitation1-Citation4. Cisplatin (cis-diamminedichloroplatinum) along with its analogs, oxaliplatin and carboplatin, are the most commonly used chemotherapeutic drugs for the treatment of a wide variety of solid tumors including GC. Despite an effective initial response, cisplatin resistance typically develops over time, resulting in treatment failure and cancer recurrenceCitation2,Citation3. Cisplatin generates DNA adducts, which results in strand cross-linking, and activates the DNA damage associated response, eventually leading to apoptosis. On the other hand, the DNA repair pathways are also activated in cancer cells exposed to cisplatin, resulting in inhibition of the apoptotic machinery and thereby increased GC cell survivalCitation5-Citation10.
Checkpoint kinase 1 (Chk1) plays a crucial role in cell cycle control, allowing cells to repair DNA lesions and preventing cells from moving forward with aberrant mitosisCitation11. If cells progress through G2 phase with impaired DNA that is beyond repair, cells typically undergo apoptosis due to genomic instabilityCitation5,Citation12–Citation15. Chk1 phosphorylation has been reported to be upregulated in cisplatin-resistant cells in multiple cancersCitation12,Citation15. Because of its significance in chemotherapy, the mechanism underlying Chk1’s action in platinum-based chemoresistance have attracted a lot of attentionCitation15-Citation17. Surprisingly, little is known of the role of Chk1 in GC. Therefore, our study examined whether Chk1 is differentially phosphorylated in the isogenic cisplatin-sensitive and resistant cell lines.
RING finger protein 138 (RNF138) is an E3 ligase that was first identified in the Wnt signaling pathwayCitation18. RNF138 consists of a RING domain, three ZNF domains, and an UIM-type domainCitation19-Citation21. Recent studies showed that RNF138 functions as a crucial homologous recombination-promoting factor important for the maintenance of chromosome integrity, and regulation of cell cycle delay via Chk1 signaling under DNA damage agentsCitation20,Citation22–Citation26. Depletion of RNF138 expression increased sensitivity towards different DNA-damaging agents, including cisplatin in mouse embryonic fibroblasts (MEFs)Citation23. Whether RNF138 contributes to GC development and chemotherapy resistance is largely unknown.
In this study, we hypothesized that RNF138 may regulate cisplatin resistance through regulation of the Chk1 signaling pathway. To the end, we examined RNF138 expression patterns, cell viability, apoptosis, and cell cycle profile, using two isogenic cisplatin-sensitive and resistant GC cell lines, namely AGS and AGS/DDP and SGC7901 and SGC7901/DDP cells. We further manipulated RNF138 expression by overexpression and gene silencing, and examined the impact of altering RNF138 level on the above phenotypes and the phosphorylation of Chk1.
Results
RNF138 is upregulated during acquiring cisplatin resistance in GC cells
To understand the role of RNF138 in cisplatin-induced stress in GC cells, we first treated AGS and SGC7901 cell lines with cisplatin for 24 hours, followed by drug withdrawal for another 24 hours, and examined RNF138 expression at different time points in the drug treatment and withdrawal. Real-time qPCR analysis show that RNF138 mRNA levels were not much altered during the cisplatin treatment but was significantly increased after drug withdrawal (, Supplement 1A, B). We also treated AGS and SGC7901 cell lines with increasing concentration of cisplatin for 24 hours, followed by drug withdrawal for another 24 hours, and examined RNF138 expression at 24 and 48 hour time point. We found that the increase of RNF138 level was significantly increased after drug withdrawal and was not much affected by the cisplatin concentration. (, Supplement 1C, D). Immunoblotting analysis demonstrated similar changes at protein level in these two GC cell lines treated with cisplatin and undergoing withdrawal ().
Figure 1. RNF138 is upregulated during acquiring cisplatin resistance in GC cells. (A and B) Cluster heatmap of RNF138 mRNA expression profiles were detected with real-time qPCR using 0.5 μg/ml in AGS and 0.25 μg/ml in SGC7901 cells for the indicated cisplatin treatment time (A) and detected with the indicated cisplatin doses for continuous 24 h treatment or 24h after replacement in AGS and SGC7901 GC cells (B). Cisplatin treatment represented as cisplatin continuous stress for indicated time. Withdrawal represented as cisplatin continuous stress for 24 hours, and then replaced to normal medium for indicated time. β-actin served as loading control. (C) The expression of RNF138 was determined in AGS and SGC7901 cells with indicated cisplatin treatment time by immunoblotting analysis. α-Tubulin was used as the loading control. (D and E) The expression of RNF138 was determined in AGS and AGS/DDP cell lines by immunoblotting analysis (D) and real-time qPCR analysis (E). α-Tubulin and β-actin were used as loading controls, respectively. (F and G) The expression of RNF138 was determined in SGC7901 and SGC7901/DDP cell lines by immunoblotting analysis (F) and real-time qPCR analysis (G). α-Tubulin and β-actin were used as loading controls, respectively. Graphs show the mean of three experiments, and error bars represent SD. Statistically significant differences are shown by *, P < 0.05; **, P < 0.01; ***, P < 0.001.
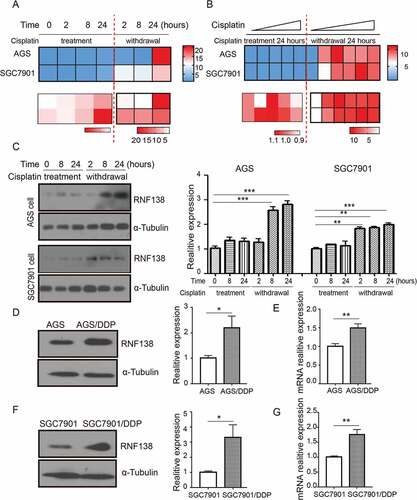
Together, these results strongly suggest that RNF138 mRNA levels were acutely and markedly up-regulated after cisplatin withdrawal, even in the cells that has been exposed to the lowest concentration of cisplatin. We then examined RNF138 expression in two isogenic GC cell lines, and found that RNF138 expression is significantly higher in the cisplatin-resistant AGS/DDP and SGC7901/DDP cells, compared with the two parental AGS and SGC7901 GC cells, which are cisplatin-sensitive, through both immunoblotting and real-time qPCR analysis (). These data together demonstrate a strong correlation between RNF138 expression level and cisplatin resistance in the GC cells and suggest that RNF138 may be involved in the development of cisplatin resistance.
RNF138 level determines the sensitivity of GC cells to cisplatin
To understand whether RNF138 plays a role in cisplatin resistance, we overexpressed RNF138 in AGS and SGC7901 cell lines using stable transfection. Cell viability was measured using the CCK-8 assay. Our results demonstrated that these overexpression cell lines displayed a significantly increased cell survival relative to the vector control cell lines treated with cisplatin (). The resistance index (RI) of the AGS and SGC7901 cell lines was 1.378 and 1.846, respectivelyCitation27. We next studied the long-term clonogenic survival and showed that the colony numbers were significantly increased in the RNF138 overexpression group, a finding consistent with the short-term cell survival CCK-8 assay (). These data indicated that RNF138 expression plays a critical role in the inhibition of cisplatin-induced cell death, and that upregulation of this gene can increase GC cell survival and result in the acquisition of certain resistance.
Figure 2. RNF138 level determines the sensitivity of GC cells to cisplatin. (A, B) IC50 values were calculated in AGS (A) and SGC7901 (B) cells transfected with the empty vector or RNF138 using cytotoxicity assay. (C) Colony formation was detected in AGS and SGC7901 cells transfected with RNF138 compared with empty control vector. The normalized ratio of clonogenic assay was shown in the histogram. (D, E) IC50 values were calculated in AGS/DDP (D) and SGC7901/DDP (E) cells transfected with the control or siRNF138 using cytotoxicity assay. (F) Colony formation was detected in AGS/DDP and SGC7901/DDP cells transfected with the control or siRNF138. The normalized ratio of clonogenic assay was shown in the histogram and error bars represent SD. Statistically significant differences are shown by *, P < 0.05.
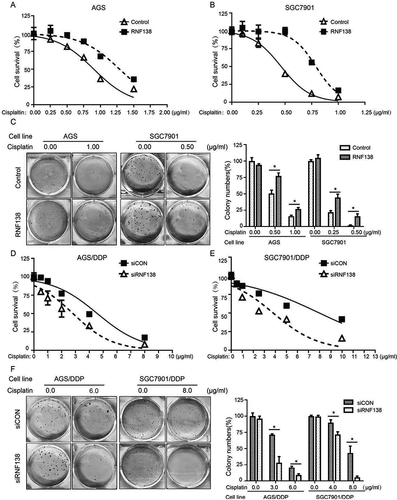
Conversely, knockdown of RNF138 expression using siRNA resulted in increased cisplatin sensitivity in AGS/DDP and SGC7901/DDP cell lines (). The RI of AGS/DDP and SGC7901DDP cells was measured to be 1.622 and 1.888, respectively. Likewise, colony formation experiments demonstrated that RNF138 suppression led to a reduction of cell colony numbers, as expected (). Altogether, these results support our hypothesis that RNF138 level positively affect cell proliferation, clonogenic survival and sensitivity to cisplatin.
RNF138 level determines cisplatin-induced cell apoptosis
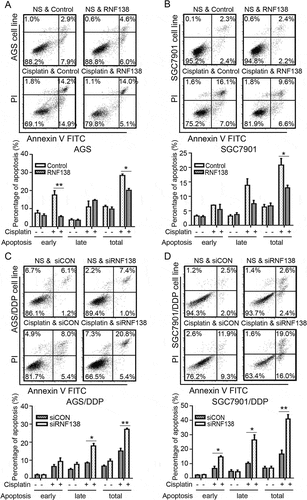
Because the cisplatin treatment is known to trigger apoptosis, we next examined whether altering RNF138 level might affect cisplatin-induced apoptosis. First, little apoptosis was seen in the two isogenic cisplatin-sensitive and resistant cell lines, which were not exposed to cisplatin. However, cisplatin treatment induced significant apoptosis in the cisplatin-sensitive, but not the cisplatin-resistant cells when treated with low concentration of cisplatin (data was not shown). Because RNF138 level is higher in the cisplatin-resistant cells, we next used gene silencing to deplete RNF138 in AGS/DDP and SGC7901DDP cells and overexpression to increase RNF138 in AGS and SGC7901 cells, respectively. Our data show that decreasing RNF138 level renders the cisplatin-resistant cells more susceptible to the drug induced apoptosis, while increasing RNF138 level in the cisplatin-sensitive cells reduced the percentage of apoptotic cells ().
Figure 3. RNF138 level determines cisplatin-induced cell apoptosis. (A, B) Percentage of apoptosis was detected in AGS (A) and SGC7901 (B) cells transfected with control vector or RNF138 with or without cisplatin treatment. Early apoptotic cells can be seen in the bottom right quadrant, late apoptotic cells can be seen in the top right quadrant, and the sum is the total apoptosis. The normalized ratio of apoptosis assay is shown in the histogram. (C, D) Cell apoptosis percentages of AGS/DDP and SGC7901/DDP cells transfected with control or siRNF138 combined with cisplatin treatment were detected. The normalized ratio of apoptosis assay was shown in the histogram and error bars represent SD. Statistically significant differences are shown by *, P < 0.05; **, P < 0.01.
RNF138 participates in the cisplatin-induced G2/M cell cycle arrest
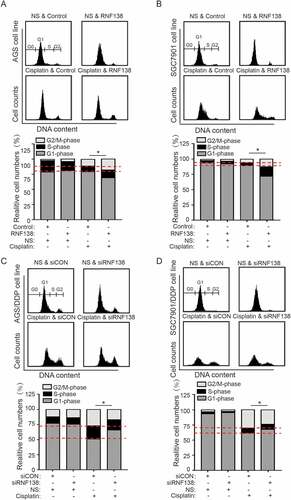
We next examined whether RNF138 level affects cell cycle progression in the AGS and SGC7901 cells treated with or without cisplatin. The cell cycle analysis demonstrated that the cell cycle distribution was not clearly altered by RNF138 levels in the absence of cisplatin. However, when combined with cisplatin stress, we found that the cells stably overexpressing RNF138 exhibited increased G2/M phase delays relative to control cells. Conversely, RNF138-depleted cells exhibited fewer G2/M cells, indicating a partially impaired cell cycle arrest (). These data demonstrate that RNF138 overexpression can promote cisplatin-induced G2/M phase cell cycle arrest, and the transition check point was partially abolished by the depletion of RNF138.
Figure 4. RNF138 participates in the cisplatin-induced G2/M cell cycle arrest. (A, B) The cell cycle distribution in AGS (A) and SGC7901 (B) cells transfected with control vector or RNF138 with cisplatin treatment was analyzed by flow cytometry. Normalized ratio of the cell cycle assay is shown in the histogram. (C, D) The cell cycle distribution of AGS/DDP (C) and SGC7901/DDP (D) cells transfected with control or siRNF138 was detected. Normalized ratio of the cell cycle assay was shown in the histogram and error bars represent SD. Statistically significant differences are shown by *, P < 0.05.
Cisplatin induced the phosphorylation of chk1 and chk1 phosphorylation is RNF138 dependent
The above data clearly demonstrate that RNF138 is involved in cisplatin resistance. The Chk1 signaling pathway is a critical regulator for G2/M transition check point. One previous study reported that RNF138 depletion inactivated Chk1 phosphorylation induced by DNA damage agent camptothecinCitation23, suggesting that RNF138 might regulate Chk1 phosphorylation. We first examined Chk1 phosphorylation during cisplatin treatment and drug withdrawal, and found that the phosphorylation of Chk1 and Cdc25C was increased as accompanied by increased RNF138 expression in a time-dependent manner in AGS and SGC7901 cells (). RNF138 expression and phosphorylation of Chk1 and Cdc25C are also higher in cisplatin-resistant AGS/DDP and SGC7901/DDP cells, compared with sensitive cells following the drug treatment. We did not observe any significant change in total Chk1 and Cdc25C protein levels, indicating that the increase in Chk1 phosphorylation was likely not a result of increase in total Chk1 protein. ().
Figure 5. Cisplatin induced the phosphorylation of Chk1 and Chk1 phosphorylation is RNF138 dependent. (A, B) Immunoblotting analysis of the RNF138 and phosphorylation of Chk1 signaling pathway level in AGS and SGC7901 cells treated with cisplatin. α-Tubulin was used as the loading control. (C, D) Immunoblotting analysis of the RNF138 and Chk1 signaling pathway level in isogenic AGS and AGS/DDP (C) and SGC7901 and SGC7901/DDP (D) cell lines treated with cisplatin. GAPDH was used as the loading control. (E) Chk1 signaling pathway was detected in AGS and SGC7901 stable cell lines treated with cisplatin by immunoblotting detection. α-Tubulin was used as the loading control. (F) Chk1 signaling pathway was detected in AGS/DDP and SGC7901/DDP cells transfected with siRNA treated with cisplatin by immunoblotting detection. α-Tubulin was used as the loading control. Data were normalized to control-treated cells. The numbers below the panels represent the normalized protein expression levels.
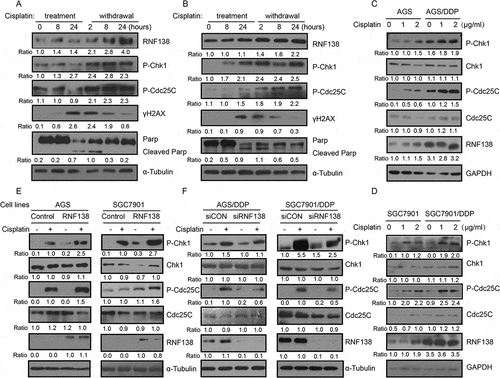
We further investigated whether manipulation of RNF138 expression levels affected p-Chk1 and p-Cdc25C levels. The cells stably overexpressing RNF138 demonstrated higher levels of phosphorylation of Chk1 and Cdc25C, relative to that observed in control cell lines upon cisplatin treatment (). Conversely, depletion of RNF138 reduced p-Chk1 and p-Cdc25C levels (). Total protein levels were not significantly altered with changes in gene expression (). These findings demonstrated that RNF138 expression was required to maintain the cisplatin-induced proper Chk1 phosphorylation in human GC cells.
The stability of RNF138 is increased in cisplatin-resistant GC cells
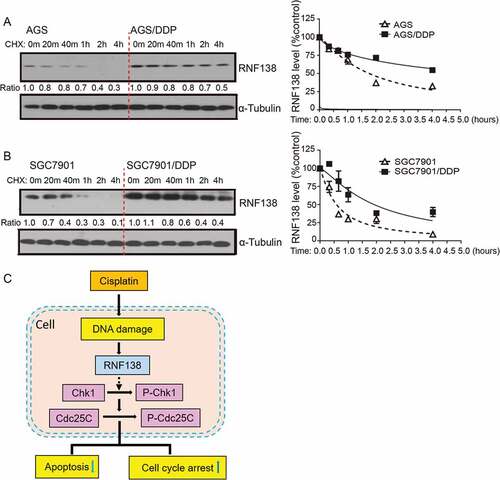
As described above, upregulated RNF138 expression conferred cisplatin resistance in GC cells. In particular, more significant increase in RNF138 expression level in protein than mRNA, was seen in AGS and SGC7901-based cell lines. (). These data suggested that post-transcriptional regulation mechanisms might account for the upregulation of RNF138. We next examined whether the stability of RNF138 protein was altered in cisplatin-resistant cells, compared with their parental cisplatin-sensitive cell lines. Cycloheximide chase experiments show that RNF138 in sensitive cell lines was degraded approximately 1.5- to 3-fold faster than in resistant cell lines (). Therefore, protein stabilization represents an important regulatory mechanism for the upregulation of RNF138 in cisplatin-resistant GC cells.
Figure 6. The stability of RNF138 is increased in cisplatin-resistant GC cells. (A, B) Half-life of RNF138 was detected in isogenic AGS, AGS/DDP and SGC7901, SGC7901/DDP GC cell lines by CHX assay. RNF138 and α-Tubulin signals in the immunoblotting detection were quantified by densitometry. The ratios of RNF138/α-Tubulin were normalized to time zero and plotted against time. (C) Schematic model for the mechanism by which RNF138 contributes to cisplatin resistance by facilitating activation of the Chk1 pathway in GC cells.
Discussion
Cisplatin and other platinum-based chemotherapy drugs are used in the clinic as first-line therapy treatments for GC patients Citation3,Citation11,Citation28,Citation29. Cisplatin and its analogues induce DNA damage, which subsequently leads to tumor cell apoptosis. However, over time, cancer cells are able to gradually adapt and develop resistance, resulting in chemotherapy failure.
In the current study, we investigated the role of RNF138 in the development of GC cisplatin resistance (). Cisplatin was found to induce RNF138 expression accompanied by the increase in phosphorylation of Chk1. Similar results were also found in other cisplatin-resistant cancer cells that were exposed to long-term, chronic cisplatin treatment. These data suggest that RNF138 may be used as a biomarker to monitor the development of cisplatin resistance. We further demonstrated that overexpression of RNF138 increased the phosphorylation of components of the Chk1 signaling pathway, which is accompanied by decreased apoptosis and a delayed G2/M transition in the presence of cisplatin, eventually leading to drug resistance in GC cells. Conversely, depletion of RNF138 reduced the Chk1 phosphorylation, increased apoptosis and thus reversed the resistant characteristics. Through a series of phenotypic and mechanistic analyses of the two isogenic cisplatin-sensitive and resistant cell lines, we suggest that a RNF138-Chk1 axis regulates the cisplatin-induced DNA damage and apoptosis. These findings further imply that RNF138 may be developed as a drug target for future treatments of GC.
We found that RNF138 promotes Chk1 phosphorylation. The underlying mechanism may rely on a higher total Chk1 expression or the increased phosphorylation. The immunoblotting analysis from our work demonstrated that total Chk1 protein expression levels did not significantly change (). Therefore, the alteration of Chk1 protein phosphorylation play a more important role. RNF138 is an ubiquitin E3 ligase that is highly conserved in higher eukaryotesCitation19,Citation23,Citation25. Recently, numerous E2s coordinate with E3 to promote Chk1 activation following treatment with DNA damage agents Citation25,Citation30. RNF138 has been shown to contain a RING domain, three zinc fingers, and an ubiquitin-interacting motif. It has been found to co-immunoprecipitate with an E2-UBE2D, and this is not dependent on whether RNF138 has a UIM or ZNF deletionCitation25. Defect in Chk1 phosphorylation observed in RNF138-depleted cells could not be rescued by ZNF domains with a mutation that causes it to be unable to localize to DNA damage sites Citation23. Thus, the likely mechanism could be that RNF138 is recruited to the site of damage by ZNF domains and then recruits UBE2D through the Ring domain, which synergistically promotes Chk1 phosphorylation associated with the UIM domain. Further studies are required to further dissect these mechanisms.
One of the key findings of our study is that RNF138 is upregulated upon withdrawal of cisplatin. RNF138 has been reported to be a critical DNA repair factor, particularly in the case of impaired double strands. Cisplatin is a strong DNA damage agent that induces the recruitment of key transcriptional factors following the DNA repair responseCitation20. The results from immunofluorescence demonstrated that part of RNF138 entered into the cell nucleus and then was recruited to the γH2AX damage locus when exposed to drug. Upregulated RNF138 expression was observed when drug withdrawal, which was referred as weakened γH2AX expression. Collectively, these data suggest that RNF138 plays an important role in the DNA repair response following the stress induced by cisplatin (Supplement 2A, B). The change of localization may be an important mechanism. Cisplatin can form intrastrand and interstrand crosslinks between purines. It is likely that these cisplatin–DNA adducts are unavailable for gene transcription, as they are needed to unwind and open the DNA double strandsCitation31. In addition, when cisplatin is withdrawn, cisplatin may be removed from the damaged DNA and some associated protein might induce RNF138 transcription. Thus, the transcriptional up-regulation of RNF138 could only take place after the drug withdrawal. Future studies will be needed to further dissect the mechanisms underlying the precise role of RNF138 in DNA repair response.
In addition, we showed that the different protein stability of RNF138 between resistant and parental sensitive cell lines. RNF138 is an E3 ligase, which can be ubiquitinated by itself or other proteins and plays a role in protein degradation via the proteasome. Regulation of RNF138 by protein stabilization that is associated with cisplatin resistance is hardly accidental. Similar related studies have reported altered stability of p53 in cisplatin-resistant A2780/CP70 cells compared with parental A2780 ovarian tumor cellsCitation32. Details regarding the molecular mechanism that modulates the half-life of RNF138 are unclear, as well as the mechanism by which RNF138 gene silencing impairs the Chk1 signaling pathway. Future studies are needed to provide mechanistic insight in this direction.
In conclusion, our findings defined a novel mechanism whereby RNF138 confers cisplatin resistance in GC cells by attenuating cisplatin-induced, Chk1-mediated cell cycle arrest and apoptosis. The elevated RNF138 expression may be potentially useful as a novel predictive and prognostic biomarker in the chemotherapy treatment of GC. Moreover, depletion or inhibition of RNF138 could be clinically beneficial to reverse chemotherapy resistance for the treatment of GC and other cancers.
Materials and methods
Cell culture and stable transfection
Human GC SGC7901 cells and its cisplatin-resistant SGC7901/DDP cells were kindly provided by Professor Jianwei Zhou (Nanjing Medical University)Citation33. AGS cells and its cisplatin-resistant AGS/DDP cells were obtained from Professor Chengchao Shou (Peking University Cancer Hospital & Institute)Citation27. The cisplatin-resistant SGC7901/DDP and AGS/DDP cells were developed from the parental SGC7901 and AGS cells that were subjected to persistent gradient exposure to cisplatin. The GC cell lines were grown according to the recommended guidelines and were tested negative for mycoplasma contamination. All these cells were passaged fewer than 6 months after obtaining for all the experiments. These GC cells lines were cultured in RPMI 1640 medium (Hyclone, SH30809.01) supplemented with 10% fetal bovine serum (Gibco, 15140–122), 100 U/ml of penicillin, and 100 μ g/ml of streptomycin (Gibco, 15070063). Cells were grown in a 5% CO2 humidified incubator at 37°C. For cell transfection, AGS and SGC7901 cells were transfected with pcDNA6.0-RNF138 or pcDNA6.0 vector (negative control). Stably transfected cells expressing RNF138 were selected by culturing in the presence of G418 (Invivogen, ant-gn-1).
Chemicals and reagents
Cisplatin (Biovalley, 20043889) was obtained from Inner Mongolia medical university, and crystal violet was purchased from Beyotime Biotechnology (C0121). Cycloheximide (CHX) (C7698-1G), RNase A (R-4875) and propidium iodide (PI) (P4170) were obtained from Sigma-Aldrich.
Plasmid, constructs and sirna
The human full-length RNF138 cDNA was cloned into a modified pcDNA6.0 vector containing a Flag tag vector to generate pcDNA6.0-RNF138. DNA sequencing was performed to ensure that the coding region of all plasmids used in this study contained no undesired mutations. Three small interfering RNA (siRNA) oligonucleotides against RNF138 were designed and synthesized by GenePharma with sequences as follows, siRNA1: 5ʹ-CCUGUGUCAAGAAUCAAAU-3ʹ, siRNA2: 5ʹ-GGAUCACUGUAACAGUAAU-3ʹ, siRNA3: 5ʹ-CUUCAGCUAGAUGAAGAAA-3ʹ, and the siRNAs were mixed as a RNAi pool for gene silencing experiments. A negative control siRNA was also purchased from GenePharma. Plasmid transfection was carried out using Lipofectamine 3000 (Invitrogen, L3000008) and the siRNA transfection was carried out using Lipofectamine RNAi MAX (Invitrogen, 13778150) following the protocol suggested by the manufacturer’ instruction.
Real-time quantitative PCR (real-time qpcr)
Total RNAs from human GC cell lines (AGS, AGS/DDP, SGC7901, SGC7901/DDP) were extracted using TRIzol reagent (Invitrogen, 15596018). 2µg of isolated total RNA was converted to cDNA using the TransScript® First-Strand cDNA Synthesis (TransGen biotech, AT301-03). The TransStart Green qPCR SuperMix (TransGen biotech, AQ131–02) was added to the cDNA samples, and a real-time qPCR was performed using the Step One Plus (Applied Bio-systems, California, USA) Real-Time Detection System and following the manufacturer’s instructions. The PCR primers were designed using Primer 3, and their specificity was verified using BLAST (NCBI). Primers used in this study are as follows: The relative levels of target gene mRNAs were normalized against that of β-actin. RNF138: F: CAGACAGCGTTTACTGGATCAC R: TGGTAATCTGGCTAGGATCTCC, β-ACTIN: F: CATGTACGTTGCTATCCAGGC R: CTCCTTAATGTCACGCACGAT.
Immunoblotting analysis
Cells were harvested and lysed in SDS lysis buffer [50 mM Tris–HCl (pH 6.8), 10% glycerol, and 2% SDS], and the protein concentration was quantified using the BCA protein assay reagent (Pierce, 23225). Cell lysates were loaded onto a 10%-15% SDS-polyacrylamide gel electrophoresis (PAGE) gel and then electrophoretically transferred to PVDF membrane (GE Healthcare, RPN303F). Membranes were blocked in 5% bovine serum albumin (BSA) or skim milk for 1 hour at room temperature and then incubated overnight with the indicated antibodies at 4°C. Antibodies against γ-H2AX (#9718), PARP (#9532), P-Chk1 (#2348), P-Cdc25C (#4901), Chk1 (#2360) and Cdc25C (#4688) were purchased from Cell Signaling Technology. Antibody against GAPDH (#60004–1-Ig) was purchased from Proteintech, and α-Tubulin (#T6199) was from Sigma -Aldrich. An antibody against RNF138 was raised by our lab. The PVDF membrane was then incubated with secondary antibody (Zhongshan Golden Bridge Biotechnology, ZB-2301, ZB-2305, ZB-2307) for 1 hour at room temperature. Protein of interest was finally detected using an ECL blot detection system (Millipore, WBULS0500).
Cytotoxicity assay
First, AGS and SGC7901 cells, stably transfected with RNF138 or the vector alone, and AGS/DDP and SGC7901/DDP cells transiently transfected with siRNF138 or siCON were plated at a density of 10,000 cells per well in 96-well plates (Corning-Costar, 3599). Cells were then treated with cisplatin of various concentrations. After 48 hours, cell viability was determined using the Cell Counting Kit-8 (Dojindo, CK04), following the manufacturer’s instruction. The cell variability curves were plotted and IC50 values were determined through non-linear regression analysis using Graph Pad Prism software.
Clonogenic assay
Prior to separation, AGS and SGC7901 cells transfected with RNF138 or the vector alone, and AGS/DDP and SGC7901/DDP cells transfected with siRNF138 or siCON were treated with cisplatin at the indicated concentrations for 2 hours. Cells were then trypsinized, seeded 1000 cells per well into six-well plates (Corning-Costar, 3516) and cultured for additional 2 weeks in the case of AGS and SGC7901 cells or 3 weeks in the case of AGS/DDP and SGC7901/DDP cells, until visible colonies were formed. Cells were then stained with crystal violet, imaged, and the numbers of colonies were counted. Each assay was carried out in triplicate.
Cycloheximide (CHX) chase assay
GC cells were treated with 10 μM CHX and then harvested at 0 min, 20 min, 40 min, 1 h, 2 h and 4 h. Proteins were extracted using SDS lysis buffer and cell lysates were resolved by SDS-PAGE, and analyzed by immunoblotting.
Flow cytometry
For cell cycle analysis, cells were harvested and immobilized by 70% ethanol for at least two hours. RNase A (100 μg/ml) was then added for 10 min to inactivate RNase, and finally stained with PI. Apoptosis was detected by Annexin V/PI double staining (Bimake, B32115) following the manufacturer’s instructions. Cell cycle and apoptotic cells were measured using a flow cytometer (BD, New Jersey, USA).
Immunofluorescence
Cells were fixed for 10 min in PBS containing 4% paraformaldehyde and then permeabilized with 0.2% Triton X-100. Then, the cells were blocked in PBS containing 5% BSA for 30 min at room temperature. Afterwards, the cells were incubated with primary antibodies at 4℃ overnight. Antibodies against γ-H2AX (#9718) was purchased from Cell Signaling Technology. Antibody against RNF138 was obtained from our lab. The cells were washed 3 times in PBS, then incubated with fluorescein isothiocyanate (FITC)-conjugated or tetramethylrhodamine β-isothiocyanate (TRITC)-labeled secondary antibodies (Invitrogen, R37117, A-11006) for 1 h at room temperature. The nuclei were counterstained with DAPI. Immunofluorescent images were captured by a confocal microscope FV1200 (Olympus, Tokyo, Japan).
Statistical analysis
Experiments were repeated for at least three times and data were presented as mean + or ± standard deviation (SD). The two-tailed student’s t-test was performed. A value of *, p ≤ 0.05 and **, p ≤ 0.01, ***, p ≤ 0.001 was considered statistically significant. All statistical analyses were carried out using GraphPad Prism 6.0 and SPSS 20.0.
Conflict of interest
All authors that are listed in this manuscript have not any editorial or financial conflict of interest, and they have contributed to, read and approved to submit to your journal, I declare that all the experiments involved in this manuscript meet ethical requirements.
Abbreviations
Acknowledgments
This work was supported by grants from the National Natural Sciences Foundation of China (81672472 and 81372661), CAMS Innovation Fund for Medical Sciences (CIFMS, 2016-12M-1-001, 2017-12M-3-009), the State Key Laboratory Special fund from the Ministry of Science (2060204), grants from the National Key Basic Research Program of China [2015CB943001] and the Natural Sciences Foundation of Inner Mongolia Autonomous Region (2014MS0871). In addition, deeply thank Prof. Jianwei Zhou (Nanjing Medical University) and Prof. Chengchao Shou (Peking University Cancer Hospital & Institute) for kindly providing paired sensitive and resistant gastric cancer cell lines. We also thank the colleagues who involve in this current study for technical guidance at the State Key Laboratory of Medical Molecular Biology, Peking Union Medical College.
Correction Statement
This article was originally published with errors, which have now been corrected in the online version. Please see Correction (http://dx.doi.org/10.1080/15384047.2024.2375109)
References
- Zhao L, Pan Y, Gang Y, Wang H, Jin H, Tie J, Xia L, Zhang Y, He L, Yao L, et al. Identification of GAS1 as an epirubicin resistance-related gene in human gastric cancer cells with a partially randomized small interfering RNA library. J Biol Chem. 2009; 284(39): 26273–26285.doi:10.1074/jbc.M109.028068.
- Zhang Y, Xu W, Ni P, Li A, Zhou J, Xu S. MiR-99a and MiR-491 Regulate Cisplatin Resistance in Human Gastric Cancer Cells by Targeting CAPNS1. Int J Biol Sci. 2016;12(12):1437–1447. doi:10.7150/ijbs.16529.
- Abuzeid WM, Jiang X, Shi G, Wang H, Paulson D, Araki K, Jungreis D, Carney J, O’Malley BW Jr., Li D. Molecular disruption of RAD50 sensitizes human tumor cells to cisplatin-based chemotherapy. J Clin Invest. 2009;119(7):1974–1985. doi:10.1172/JCI33816.
- Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014; 513(7517): 202–209. doi: 10.1038/nature13480.
- Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22(47):7265–7279. doi:10.1038/sj.onc.1206933.
- Mo D, Fang H, Niu K, Liu J, Wu M, Li S, Zhu T, Aleskandarany MA, Arora A, Lobo DN, et al. Human Helicase RECQL4 Drives Cisplatin Resistance in Gastric Cancer by Activating an AKT-YB1-MDR1. Signaling Pathway Cancer Res. 2016; 76(10): 3057–3066.doi:10.1158/0008-5472.CAN-15-2361.
- Xu W, Wang S, Chen Q, Zhang Y, Ni P, Wu X, Zhang J, Qiang F, Li A, Roe OD, et al. TXNL1-XRCC1 pathway regulates cisplatin-induced cell death and contributes to resistance in human gastric cancer. Cell Death Dis. 2014;5:(e1055. doi:10.1038/cddis.2014.27.
- Aranda F, Bloy N, Pesquet J, Petit B, Chaba K, Sauvat A, Kepp O, Khadra N, Enot D, Pfirschke C, et al. Immune-dependent antineoplastic effects of cisplatin plus pyridoxine in non-small-cell. Lung Cancer Oncogene. 2015; 34(23): 3053–3062.doi:10.1038/onc.2014.234.
- Lipinska N, Romaniuk A, Paszel-Jaworska A, Toton E, Kopczynski P, Rubis B. Telomerase and drug resistance in cancer Cell. Mol Life Sci. 2017. doi:10.1007/s00018-017-2573-2.
- Wilk A, Waligorska A, Waligorski P, Ochoa A, Reiss K. Inhibition of ERbeta induces resistance to cisplatin by enhancing Rad51-mediated DNA repair in human medulloblastoma cell lines. PLoS One. 2012;7(3):e33867. doi:10.1371/journal.pone.0033867.
- Pouliot LM, Chen YC, Bai J, Guha R, Martin SE, Gottesman MM, Hall MD. Cisplatin sensitivity mediated by WEE1 and CHK1 is mediated by miR-155 and the miR-15 family. Cancer Res. 2012;72(22):5945–5955. doi:10.1158/0008-5472.CAN-12-1400.
- Duan L, Perez RE, Hansen M, Gitelis S, Maki CG. Increasing cisplatin sensitivity by schedule-dependent inhibition of AKT and Chk1 Cancer. Biol Ther. 2014;15(12):1600–1612. doi:10.4161/15384047.2014.961876.
- Zhang D, Piao HL, Li YH, Qiu Q, Li DJ, Du MR, Tsang BK. Inhibition of AKT sensitizes chemoresistant ovarian cancer cells to cisplatin by abrogating S and G2/M arrest. Exp Mol Pathol. 2016;100(3):506–513. doi:10.1016/j.yexmp.2016.05.003.
- Yarden RI, Metsuyanim S, Pickholtz I, Shabbeer S, Tellio H, Papa MZ. BRCA1-dependent Chk1 phosphorylation triggers partial chromatin disassociation of phosphorylated Chk1 and facilitates S-phase cell cycle arrest. Int J Biochem Cell Biol. 2012;44(11):1761–1769. doi:10.1016/j.biocel.2012.06.026.
- Chen S, Chen X, Xie G, He Y, Yan D, Zheng D, Li S, Fu X, Li Y, Cdc6 PX. contributes to cisplatin-resistance by activation of ATR-Chk1 pathway in bladder cancer cells. Oncotarget. 2016;7(26):40362–40376. doi:10.18632/oncotarget.9616.
- Sen T, Tong P, Stewart CA, Cristea S, Valliani A, Shames DS, Redwood AB, Fan YH, Li L, Glisson BS, et al. CHK1 Inhibition in Small-Cell Lung Cancer Produces Single-Agent Activity in Biomarker-Defined Disease Subsets and Combination Activity with Cisplatin or Olaparib. Cancer Res. 2017; 77(14): 3870–3884.doi:10.1158/0008-5472.CAN-16-3409.
- Feng Z, Xu S, Liu M, Zeng YX, Chk1 KT. inhibitor Go6976 enhances the sensitivity of nasopharyngeal carcinoma cells to radiotherapy and chemotherapy in vitro and in vivo. Cancer Lett. 2010;297(2):190–197. doi:10.1016/j.canlet.2010.05.011.
- Yamada M, Ohnishi J, Ohkawara B, Iemura S, Satoh K, Hyodo-Miura J, Kawachi K, Natsume T, Shibuya H. NARF, an nemo-like kinase (NLK)-associated ring finger protein regulates the ubiquitylation and degradation of T cell factor/lymphoid enhancer factor (TCF/LEF). J Biol Chem. 2006;281(30):20749–20760. doi:10.1074/jbc.M602089200.
- Xu L, Lu Y, Han D, Yao R, Wang H, Zhong S, Luo Y, Han R, Li K, Fu J, et al. Rnf138 deficiency promotes apoptosis of spermatogonia in juvenile male mice. Cell Death Dis. 2017; 8(5): e2795.doi:10.1038/cddis.2017.110.
- Han D, Liang J, Lu Y, Xu L, Miao S, Lu LY, Song W, Wang L. Ubiquitylation of Rad51d Mediated by E3 Ligase Rnf138 Promotes the Homologous Recombination Repair Pathway. PLoS One. 2016;11(5):e0155476. doi:10.1371/journal.pone.0155476.
- Giannini AL, Gao Y, Bijlmakers MJ. T-cell regulator RNF125/TRAC-1 belongs to a novel family of ubiquitin ligases with zinc fingers and a ubiquitin-binding domain. Biochem J. 2008;410(1):101–111. doi:10.1042/BJ20070995.
- Zhou YX, Chen SS, Wu TF, Ding DD, Chen XH, Chen JM, Su ZP, Li B, Chen GL, Xie XS, et al. A novel gene RNF138 expressed in human gliomas and its function in the glioma cell line U251. Anal Cell Pathol (Amst). 2012; 35(3): 167–178.doi:10.3233/ACP-2011-0051.
- Ismail IH, Gagne JP, Genois MM, Strickfaden H, McDonald D, Xu Z, Poirier GG, Masson JY, Hendzel MJ. The RNF138 E3 ligase displaces Ku to promote DNA end resection and regulate DNA repair pathway choice. Nat Cell Biol. 2015;17(11):1446–1457. doi:10.1038/ncb3259.
- Yard BD, Reilly NM, Bedenbaugh MK, Pittman DL. RNF138 interacts with RAD51D and is required for DNA interstrand crosslink repair and maintaining chromosome integrity DNA Repair. Amst. 2016;42:.82–93. doi:10.1016/j.dnarep.2016.04.006.
- Schmidt CK, Galanty Y, Sczaniecka-Clift M, Coates J, Jhujh S, Demir M, Cornwell M, Beli P, Jackson SP. Systematic E2 screening reveals a UBE2D-RNF138-CtIP axis promoting DNA repair. Nat Cell Biol. 2015;17(11):1458–1470. doi:10.1038/ncb3260.
- Matsuoka S, Ballif BA, Smogorzewska A, Rd MDE, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316(5828):1160–1166. doi:10.1126/science.1140321.
- Huang D, Duan H, Huang H, Tong X, Han Y, Ru G, Qu L, Shou C, Zhao Z. Cisplatin resistance in gastric cancer cells is associated with HER2 upregulation-induced epithelial-mesenchymal transition. Sci Rep. 2016;6(20502). doi: 10.1038/srep20502.
- Wu Q, Vasquez KM. Human MLH1 protein participates in genomic damage checkpoint signaling in response to DNA interstrand crosslinks, while MSH2 functions in DNA repair. PLoS Genet. 2008;4(9):e1000189. doi:10.1371/journal.pgen.1000189.
- Zhang YH, Wu Q, Xiao XY, Li DW, Wang XP. Silencing MRP4 by small interfering RNA reverses acquired DDP resistance of gastric cancer cell. Cancer Lett. 2010;291(1):76–82. doi:10.1016/j.canlet.2009.10.003.
- Marechal A, Li JM, Ji XY, Wu CS, Yazinski SA, Nguyen HD, Liu S, Jimenez AE, Jin J, Zou L. PRP19 transforms into a sensor of RPA-ssDNA after DNA damage and drives ATR activation via a ubiquitin-mediated circuitry . Mol Cell. 2014;53(2):235–246. doi:10.1016/j.molcel.2013.11.002.
- Ivanova T, Zouridis H, Wu Y, Cheng LL, Tan IB, Gopalakrishnan V, Ooi CH, Lee J, Qin L, Wu J, et al. Integrated epigenomics identifies BMP4 as a modulator of cisplatin sensitivity in gastric cancer. Gut. 2013; 62(1): 22–33. doi:10.1136/gutjnl-2011-301113.
- Xie X, Lozano G, Siddik ZH. Heterozygous p53(V172F) mutation in cisplatin-resistant human tumor cells promotes MDM4 recruitment and decreases stability and transactivity of p53. Oncogene. 2016;35(36):4798–4806. doi:10.1038/onc.2016.12.
- Wang Q, Chen Q, Zhu L, Chen M, Xu W, Panday S, Wang Z, Li A, Røe OD, Chen R, et al. JWA regulates TRAIL-induced apoptosis via MARCH8-mediated DR4 ubiquitination in cisplatin-resistant gastric cancer cells. Oncogenesis. 2017; 6(7): e353.doi:10.1038/oncsis.2017.57.