
ABSTRACT
Objectives: Distant metastasis is a critical factor for cancer-associated death. Our previous studies identified collapsin response mediator protein 4a (CRMP4a) as a metastasis suppressor in prostate cancers. Enhancing CRMP4 expression by promoter-targeted small activating RNAs reduced cell migration in vitro and abolished distal metastasis in mouse xenograft models. In this study, we investigated the mechanism for CRMP4a-mediated suppression of cell migration.
Methods: PC-3 cells were stably infected with lentiviruses expressing CRMP4a cDNA or a shRNA sequence. Cytoskeletal organization was analyzed by measuring cellular focal adhesion area and number, percentage of cell area and lamellipodia numbers after phalloidin staining or anti-vinculin immunocytofluorescent staining. Cell migration was evaluated with TranswellTM chambers coated with MatriGel. RhoA activation was determined with a Rhotekin RBD agarose bead-based assay kit. Lentiviruses harboring RhoA-Q63L or RhoA-T19N mutant constructs were used to overexpress mutant RhoA proteins.
Results: CRMP4a overexpression largely reduced while CRMP4a knockdown remarkably increased cytoskeletal organization in PC-3 cells. CRMP4a immunoprecipitation pulled down RhoA but not cdc42 or Rac1 proteins. Manipulating CRMP4a expression levels reversely altered active RhoA levels. Overexpression of RhoA active (Q63L) but not inactive (T19N) mutants reversed CRMP4a-mediated reduction of cancer cell migration while RhoA inhibitor Rhosin diminished CRMP4a shRNA-induced increase of cancer cell migration. CRMP4a overexpression also largely reduced cell spreading that was abolished by overexpressing RhoA active mutant.
Conclusion: Our data demonstrated that CRMP4a interacts with RhoA and sequesters its activity, resulting in suppression of cytoskeletal organization, cell migration and spreading.
Keywords:
Introduction
Distant metastasis is a primary cause of cancer-associated mortalityCitation1. There are multiple cellular processes involved in distant metastasis of cancer, including local invasion from the primary sites, entering into bloodstream or lymphatic system, circulation through the bloodstream to land in distant organs or tissuesCitation2. During all these processes, cell motility or migration is essential and the small GTPase Rho family is a critical regulatory factorCitation3.
Collapsin response mediator protein 4 (CRMP4), also called dihydropyrimidinase-like protein 3 (DPYSL3), belongs to a cytosolic phospho-protein family of five isoforms and their functions are mainly associated with the assembly of cytoskeletal proteins and neural axonal growthCitation4. Among the CRMP family proteins, CRMP4 was found to directly bind to F-actin in modulating cytoskeletal organizationCitation5 and is the only one that has been shown to interact with RhoA, the key regulator of actin cytoskeletonCitation6.
Recently, altered expression levels of CRMP family proteins were reported in multiple human cancersCitation7. Our studies demonstrated that expression of CRMP4a, the shorter splicing variant, is significantly lower in metastatic prostate cancers than that in primary cancers at the mRNA and protein levels and that CRMP4a overexpression drastically suppressed cell migration and tumor metastasis in prostate cancer modelsCitation8. Additionally, using the small activating RNA (saRNA) technique, we demonstrated that enhancement of CRMP4a expression completely blocked distant metastases in mouse xenograft models of prostate cancerCitation9, which has been supported by others utilizing different approachesCitation10,Citation11.
In this study, we continued studying the mechanism involved in CRMP4a-mediated suppression of cell migration/invasion in prostate cancer cells. Our data revealed that CRMP4a interacts with RhoA protein but not with other two Rho family members cdc42 and Rac1. Consistent with previous reportsCitation5,Citation6,Citation12, CRMP4a interaction suppressed RhoA activity, leading to reduced cytoskeletal reorganization and cell motility.
Materials and methods
Cell lines, antibodies and reagents
Human prostate cancer PC-3 cells were obtained from ATCC (Manassas, VA) and were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) plus 100 μg/ml streptomycin and 100 IU/ml penicillin at 37°C and 5% CO2 as described in our previous study Citation9. RhoA antibody (clone 67B9) and RIPA Buffer (#9806) were obtained from Cell Signaling Tech (Danvers, MA). Antibodies for Rac1, cdc42, mouse IgG-AlexaFluor-488 and AlexaFluor-594, phalloidin-iFluor-555 were purchased from Abcam (Cambridge, MA). Actin antibody was obtained from Sigma-Aldrich (St. Louis, MO). Antibodies for CRMP4 and RhoA (sc-418), as well as protein A/G PLUS-Agarose (sc-2003) were obtained from Santa Cruz Biotech (Santa Cruz, CA). Antibodies for Vinculin (66305–1-Ig), CRMP4 (13661–1-AP), HRP-conjugated goat anti-mouse IgG (SA00001-1) and goat anti-rabbit IgG (SA00001-2) were obtained from Proteintech Group (Chicago, IL). Hoechst33342 (HY-15559A) and RhoA inhibitor Rhosin hydrochloride (HY-12646) were obtained from MCE (New Jersey, NJ). MatriGel (catalog #35623) and Transwell (Catalog #353097) were obtained from Corning Life Sciences (Corning, NY).
Plasmid constructs, lentivirus package and cell infection
To establish stable expression sublines, DPYSL3v2 cDNA sequence (CRMP4a) was cloned into pLVX-IRES-Neo construct (Clontech Laboratories, Mountain View, CA) with the primer pairs as follow: forward 5ʹ-CCGCTCGAG ATG TCC TAC CAA GGC AAG AAG AA-3ʹ and reverse 5ʹ- CTAGTCTAGA TTA ACT CAG AGA TGT GAT ATT AGA ACG G-3ʹ from the expression construct as described previouslyCitation9. RhoA-Q63L and RhoA-T19N mutants were generated using a PCR-based three-step mutagenesis approach as described in our recent publication Citation9 with the primers as follow: RhoA63L-a: 5ʹ-CCGCTCGAG ATG GCT GCC ATC CGG AA-‘3 and 5ʹ-CTTC aag CC CAG CTG TGT CC-3ʹ; RhoA63L-b: 5ʹ-AGC TGG G ctt GA AGA TTA TGA TC-3ʹ and 5ʹ-CGCGGATCC TCA CAA GAC AAG GCA CCC AGA T-3ʹ; RhoA19N-1: 5ʹ-CCGCTCGAG ATG GCT GCC ATC CGG AA-3ʹ and 5ʹ-CAA GCA gtt CTT TCC ACA GGC-3ʹ; RhoA19N-2: 5ʹ-GGA AAG aac TGC TTG CTC ATA GTC T-3ʹ and 5ʹ-CGCGGATCC TCA CAA GAC AAG GCA CCC AGA T-3ʹ. The completed mutants were sub-cloned into pLVX-Puro (Clontech). The lower-case letters indicate mutation sites for RhoA. The small hairpin interfering RNAs against DPSL3v2 (shCRMP4a) were designed and cloned into pGLVU6/Puro (GenePharma, catalog #C06002, Shanghai, China) with the sequences as follow: 5ʹ-GTC CTA CCA AGG CA AGA AGA A-3ʹ and 5ʹ-TTC TTC TTG CCT TGG TAG GAC-3ʹ. The scramble control shRNAs (shControl) were also purchased from GenePharma.
Lentivirus were produced in 293T cells using psPAX2/pMD2.G system obtained from Dr Didier Trono as a gift (Addgene plasmid #12259–12260). Lentivirus-containing supernatants were clarified via filtration through a 0.45 μm filter and stored at −80°C before use. PC-3 cells were infected with lentiviruses encoding the indicated genes for 24 h respectively. Stable expression clones were selected with puromycin (2 μg/ml) or G418 (500 μg/ml). Monoclonal stable subline cells were maintained in RPMI 1640 supplemented 10% FBS. Overexpression or knockdown efficacy was examined by western blot assays.
Western blotting and immunoprecipitation
Western blot was performed as previously described in our recent publicationsCitation9. Total cellular proteins were extracted from cells with RIPA buffer containing protease inhibitors, and then subjected to SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto PVDF membrane. The membrane was blocked for 1 h in 5% non-fat milk solution and incubated with indicated primary antibody overnight at 4°C, followed by peroxidase-conjugated secondary antibody incubation at room temperature for 1 h. Immunoblot bands were visualized using ECL reagent obtained from Santa Cruz Biotech. Actin blot was included as an endogenous protein loading control.
For immunoprecipitation, cells were lysed in 500 μl cold NP-40 lysis buffer (50 mM Tris pH 7.4, 50 mM NaCl, 1% NP-40, 1x Complete Protease Inhibitors, 10 mM NaF, 1 mM Na2VO3, 2 mM sodium pyrophosphate and 2 mM β-glycerophosphate)Citation13. 2 μg of antibodies were mixed with 25 μl Protein A/G-Agarose and incubated overnight at 4C. The antibodies A/G-Agarose complexes were collected and incubated with protein lysate for 8 h at 4C with rotation. The immunoprecipitants were eluted for western blot assays with anti-antibodies as indicated in the figures.
Immunofluorescence staining and cytoskeleton visualization
Immunocytofluorescent staining was performed as previously describedCitation14. Briefly, cells grown on coverslips were fixed in 4% paraformaldehyde for 10 min and then permeabilized in 0.1% TritonX-100 for 5 min. Following the blocking with 5% normal goat serum, the coverslips were incubated with indicated primary antibodies for 2 h at room temperature and then incubated with indicated fluorescent labeled secondary antibodies. Nuclei were counterstained with Hoechst33342.
For visualizing cytoskeleton reorganization, F-actin was detected with Phalloidin-iFluor555 staining according to the manufacturer’s protocol. Focal adhesion (FA) was stained with anti-vinculin antibody conjugated with AlexaFluor-488. Nuclei were visualized with Hoechst33342 staining. The microscopic images were taken using a confocal microscope LSM 800 Zeiss (Carl Zeiss Micro-Imaging, Inc.). Quantification of focal adhesion and lamellipodia formation were determined with ImageJ soft (NIH, Rockville Pike, MD, USA). Briefly, after global background staining was removed from the images, the pictures were inverted to black-and-white image. Focal adhesion number was obtained using “Analyze Particles” feature of ImageJ. Focal adhesion and cells’ area were calculated with the measurement function of ImageJ. Lamellipodia formation was manually measured using ImageJ software as describedCitation15. A total of 30 ~ 100 cells per each condition from three independent experiments were analyzed.
Transwell migration assay
The transwell migration assay was conducted using the 8.0 μm pore size membranes transwells (Corning catalog #353097) as previously describedCitation9. In brief, 2 × 10Citation4 cells (in 200 μl medium) were plated into the upper chamber coated with MatriGel (5 μg/ml) (Corning catalog #354263) in serum-free RPMI 1640 media. The lower chamber contained 300 μl of RPMI 1640 media supplemented with 10% FBS. Following incubation at 37C for 24 h, cells migrated into the bottom chamber were fixed and stained with crystal violet. The number of cells from three different fields was counted per filter for quantification. The number of migration cells in the control group was assigned a relative value of 100%.
Active rhoa pull-down assays
The RhoA activation assays were performed with the RhoA activation assay kit (Abcam catalog #ab211164) according to the manufacture’s instruction. Briefly, equal amount of total proteins from cell lysis was incubated with Rhotekin RBD agarose beads at 4C for 1 h. The beads were pelleted and washed three times with 0.5 mL of 1X assay buffer. Following the last wash, the beads were resuspended in 40 µL of 2X reducing SDS sample buffer and then boiled for 5 minutes. Finally, the precipitated GTP-Rho was detected by western blot assays using an anti-RhoA specific monoclonal antibody (Abcam catalog #ab211164).
Live-cell imaging
PC-3 cells were plated onto 2-cm glass bottom culture dish in RPMI1640 medium at 37C and 5% CO2 inside a Live-cell images system. Time-lapse images were continually captured at intervals of 5 min for 3 h using the IX2-ZDC2 laser-based autofocusing system. Cell migrate trajectories and length were determined using the centroid of the nuclei with the ImageJ plugin “MTrackJ”Citation15.
Statistical analysis
Data are presented as mean ± SEM. Statistically significant was analyzed using the statistical software SPSS 20.0 software (SPSS, Inc., Chicago, IL, USA).
Results
CRMP4a regulates cytoskeleton reorganization
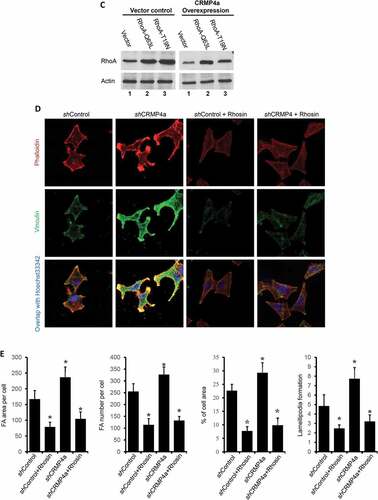
In our previous studies, CRMP4a was identified as a tumor metastasis suppressor in human prostate cancersCitation8 and enhancing CRMP4a expression with a small activating RNA approach suppressed cancer cell migration in vitro and distant tumor metastases in vivo in prostate cancer modelsCitation9. In this study, we investigated the mechanism underlying CRMP4a-induced suppression of cancer cell migration in vitro. Cytoskeleton reorganization is a major process during cell movementCitation16,Citation17, thus, we first examined whether manipulating CRMP4a expression results in pattern changes of cytoskeleton reorganization. Cytoskeleton reorganization was evaluated by detecting focal adhesion (FA) and F-actin organization with anti-vinculin immunocytofluorescent approach and iFluor 555-conjugated phalloidin staining, respectively. To increase CRMP4a expression, prostate cancer PC-3 cells were stably infected with lentiviruses harboring CRMP4a expression constructs, as shown in . To reduce CRMP4a expression, the small interfering RNA approach was utilized and CRMP4a knockdown was confirmed (). As expected, overexpressing CRMP4a protein in PC-3 cells largely reduced cellular focal adhesion area/numbers, as well as lamellipodia numbers per cell compared to the empty vector control cells ( & ). Conversely, CRMP4a knockdown in PC-3 cells significantly increased cellular focal adhesion area/numbers and lamellipodia numbers per cell compared to the control shRNA ( & ). These results suggest that CRMP4a is involved in modulating cytoskeleton reorganization.
Figure 1. CRMP4a overexpression reduces cytoskeleton organization in prostate cancer cells. A. PC-3 cells stably infected with lentiviruses harboring the control empty vector or CRMP4a expression constructs were harvested for western blot with the antibodies as indicated. Actin blot served as protein loading control. B. PC-3 stable subline cells as indicated (empty vector or CRMP4a) were seeded on cover glass in full culture media for 24 h and then stained with iFluor555-conjugated phalloidin (red) and Hoechst33342 (blue). Cells were also subjected to immunocytofluorescent staining with anti-vinculin antibodies (green) followed by visualization with AlexaFluor®488-labeld secondary antibodies. The representative microscopic images were shown from four independent experiments. C. Quantitative data for focal adhesion area or number per cell, percentage of cell area, and lamellipodia numbers per cell formation were shown as mean ± SEM. The asterisk indicates a statistical significance compared to the control (student’s t-test, p < 0.01).
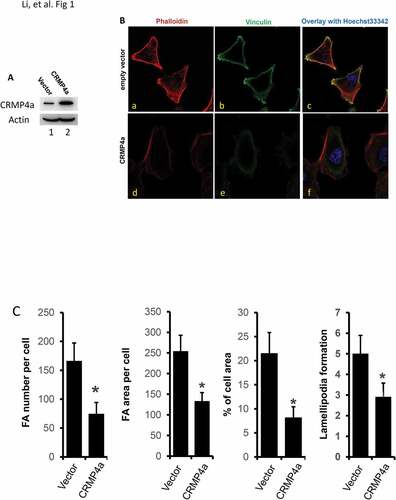
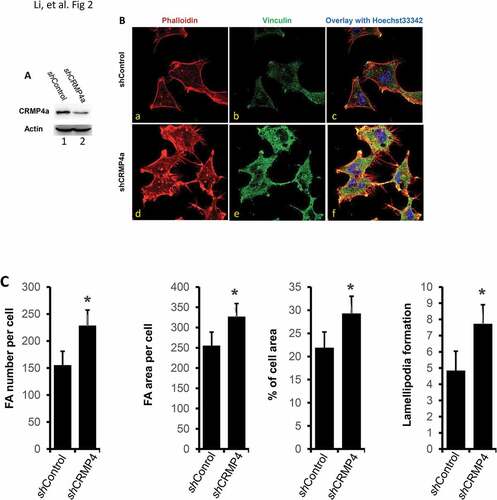
Figure 2. CRMP4a knockdown increases cytoskeleton organization in prostate cancer cells. A. PC-3 cells stably infected with lentiviruses harboring the control shRNA (shControl) or CRMP4a shRNA (shCRMP4a) constructs were harvested for western blot with the antibodies as indicated. Actin blot served as protein loading control. B. PC-3 stable subline cells as indicated (shControl or shCRMP4a) were seeded on cover glass in full culture media for 24 h and then stained with iFluor555-conjugated phalloidin (red) and Hoechst33342 (blue). Vinculin proteins were visualized by immunocytofluorescent staining with anti-vinculin antibodies (green) coupled with AlexaFluor®488-labeled secondary antibodies. The representative microscopic images were shown from four independent experiments. C. Quantitative data for focal adhesion area or number per cell, percentage of cell area, and lamellipodia numbers per cell formation were shown as mean ± SEM. The asterisk indicates a statistical significance compared to the control (student’s t-test, p < 0.01).
CRMP4a interacts with rhoa but not cdc42 and rac1
Rho family proteins are the key mediators of cytoskeleton reorganization and cell motilityCitation18-Citation20. To understand how CRMP4a mediates cytoskeleton reorganization, we investigated whether CRMP4a mediates cytoskeleton reorganization via Rho pathway. We determined whether CRMP4a interacts with Rho family proteins RhoA, cdc42 and Rac1, the predominant members in Rho pathway. Immunoprecipitation assays were conducted with anti-CRMP4a antibody, followed by immunoblotting with RhoA, cdc42 or Rac1. As show in , anti-CRMP4a immunoprecipitation pulled down RhoA but not cdc42 or Rac1 proteins. Consistently, anti-RhoA immunoprecipitation pulled down CRMP4a protein (). To verify the interaction between RhoA and CRMP4a, we examined if CRMP4a co-localizes with RhoA by immunocytofluorescent staining. As shown in , co-localization signals of CRMP4a and RhoA were visualized (yellow spots) within the cytoplasm compartment. These data suggest that CRMP4a protein interacts with RhoA protein to modulate cytoskeletal organization.
Figure 3. CRMP4a interacts with RhoA but not cdc42 or Rac1. A. Exponentially grown PC-3 stable subline cells as indicated were harvested for immunoprecipitation assay with anti-CRMP4a antibody (sc-100323) and the elutes were subjected to western blot with Rho family proteins as indicated. Whole cell protein lysates from CRMP4a overexpressing subline cells were used as protein input control. B. Similarly, immunoprecipitation was conducted with anti-RhoA antibody (sc-418) and the elutes were subjected to western blot with anti-CRMP4a antibody. C. PC-3 cells plated on coverslips were used for immunocytofluorescent staining with anti-CRMP4a antibody (Proteintech #13661–1-AP) or anti-RhoA (sc-418) antibody. Antibody visualization was carried out using AlexaFluor®488 or AlexaFluor®488, respectively, respectively. Cell nuclei were counterstained with Hoechst33342. Yellow arrows indicate spots with co-localization of CRMP4a and RhoA. D. PC-3 cells stably infected with lentiviruses harboring control vector, CRMP4a expression constructs or shRNA constructs were harvested for confirming the efficiency of CRMP4a expression by western blot assay. Actin blot served as protein loading control. E. PC-3 cells stably infected with lentiviruses as indicated were harvested for RhoA activation assay with Rhotekin RBD agarose beads. The eluted RhoA proteins were evaluated in western blot assay. Equal amount of whole cellular proteins from each subline was used as protein loading control. F. Exponentially grown PC-3 cells stably infected with lentiviruses as indicated were lysed in RIPA buffer for western blots. Expression of Rho family proteins were analyzed with anti-antibodies as indicated. Actin blot was included as an endogenous protein loading control.
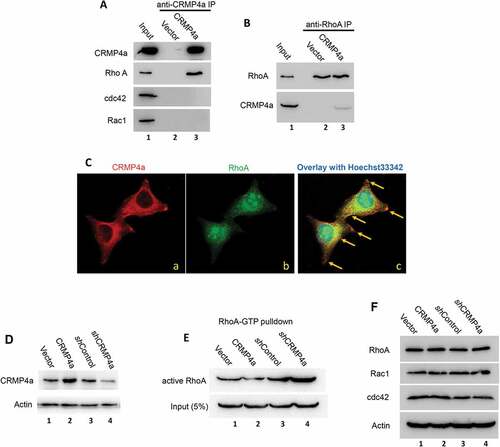
We then investigated whether CRMP4a modulates RhoA activity. RhoA activation was determined using a kit based on Rhotekin RBD agarose beads that binds to active RhoA protein. As shown in , manipulation of CRMP4a expression was successfully confirmed. CRMP4a overexpression largely reduced the level of active RhoA protein compared to the vector control, while knocking down of CRMP4a expression led to a drastic increase of active RhoA level in PC-3 cells (). However, altering CRMP4a expression did not affect the total levels of Rho family proteins (). These data suggest that CRMP4a interacts with RhoA but not cdc42 or Rac1 to suppress RhoA activation.
CRMP4a suppresses rhoa-dependent cytoskeleton reorganization
We then investigated whether CRMP4a-mediated RhoA suppression leads to reduced cytoskeleton organization. Similar as shown in , CRMP4a overexpression in PC-3 cells reduced cytoskeleton organization as measured by FA area/number per cell, percentage of cell area and lamellipodia numbers per cell ( & ). We introduced two mutant RhoA proteins, active RhoA-Q63L and inactive RhoA-T19N to counter-act on CRMP4a-mediated RhoA suppression (). Overexpression of active RhoA-Q63L mutant protein not only enhanced cytoskeleton re-organization but also reversed CRMP4a-mediated reduction of cytoskeleton organization. However, inactive RhoA-T19N mutant protein had no obvious effect on CRMP4a-mediated reduction of cytoskeletal organization ( & ). Consistently, knocking down CRMP4a expression resulted in a significant increase of FA area and numbers per cell, percentage of cell area and lamellipodia numbers per cell, which were almost abolished by co-treatment with a RhoA inhibitor Rhosin ( & ). These data indicate that CRMP4a reduces cytoskeleton organization by suppressing RhoA activation.
Figure 4. RhoA antagonizes CRMP4a-modulated cytoskeletal organization. A. PC-3 cells stably infected with lentiviruses as indicated were left untreated or re-infected with RhoA-Q63L or RhoA-T19N lentiviruses for 24 h. Cells were then stained with iFluor555-conjugated phalloidin, immunocytofluorescent stained with anti-vinculin antibodies. Cell nuclear were counter-stained with Hoechst33342. The representative microscopic images from three independent experiments were shown. B. Quantitative data for focal adhesion area & numbers per cell, percentage of average cell area, and lamellipodia numbers per cell were measured under fluorescent microscope with ImageJ soft. Data are shown as mean ± SEM from three independent experiments. The asterisk indicates a statistical significance compared to the vector control (student’s t-test, p < 0.05). C. PC-3 cells stably infected with CRMP4a and RhoA mutants were harvested for western blot assays with the antibodies as indicated. Actin blot served as protein loading control. D. PC-3 cells stably infected with lentiviruses as indicated were left untreated or treated with Rhosin (30 μM). Cytoskeletal organization was evaluated with iFluor555-conjugated phalloidin or anti-vinculin immunocytofluorescent staining. Cell nuclei were counter-stained with Hoechst33342. E. Quantitative data in focal adhesion & numbers per cell, the percentage of cell area and lamellipodia numbers per cell were measured under fluorescent microscope with ImageJ software. Data are shown as mean ± SEM from three independent experiments. The asterisk indicates a significant difference compared to the shControl (student’s t-test, p < 0.05).
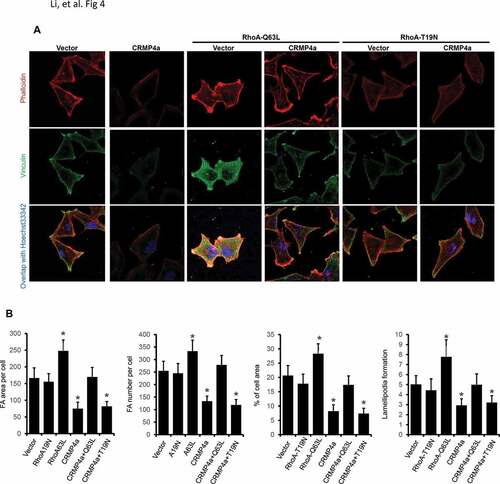
Figure 4. RhoA antagonizes CRMP4a-modulated cytoskeletal organization. A. PC-3 cells stably infected with lentiviruses as indicated were left untreated or re-infected with RhoA-Q63L or RhoA-T19N lentiviruses for 24 h. Cells were then stained with iFluor555-conjugated phalloidin, immunocytofluorescent stained with anti-vinculin antibodies. Cell nuclear were counter-stained with Hoechst33342. The representative microscopic images from three independent experiments were shown. B. Quantitative data for focal adhesion area & numbers per cell, percentage of average cell area, and lamellipodia numbers per cell were measured under fluorescent microscope with ImageJ soft. Data are shown as mean ± SEM from three independent experiments. The asterisk indicates a statistical significance compared to the vector control (student’s t-test, p < 0.05). C. PC-3 cells stably infected with CRMP4a and RhoA mutants were harvested for western blot assays with the antibodies as indicated. Actin blot served as protein loading control. D. PC-3 cells stably infected with lentiviruses as indicated were left untreated or treated with Rhosin (30 μM). Cytoskeletal organization was evaluated with iFluor555-conjugated phalloidin or anti-vinculin immunocytofluorescent staining. Cell nuclei were counter-stained with Hoechst33342. E. Quantitative data in focal adhesion & numbers per cell, the percentage of cell area and lamellipodia numbers per cell were measured under fluorescent microscope with ImageJ software. Data are shown as mean ± SEM from three independent experiments. The asterisk indicates a significant difference compared to the shControl (student’s t-test, p < 0.05).
CRMP4a inhibits rhoa-dependent cell motility
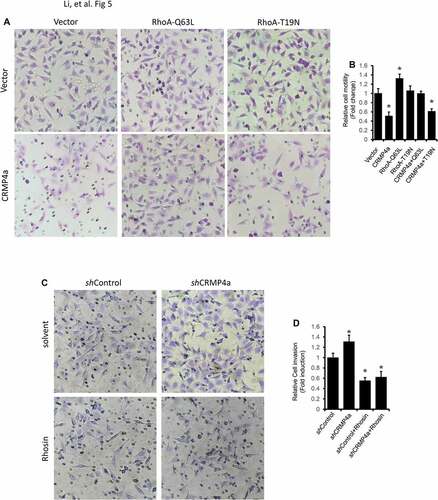
We utilized the Boyden chamber assay to determine if CRMP4a-mediated reduction of cytoskeleton organization is associated with cell migration. Similar as reported in our previous publicationCitation9, CRMP4a overexpression significantly reduced PC-3 cells migration to the bottom chambers compared to the vector control ( & ). Co-transfection of the cells with RhoA-Q63L active mutant but not RhoA-T19N inactive mutant reversed CRMP4-mediated reduction of cell migration. On the other hand, knocking down CRMP4a expression increased PC-3 cell migration, which was reversed by co-treatment with the RhoA inhibitor Rhosin ( & ). These data indicate that CRMP4a modulates cell migration via antagonizing RhoA activity.
Figure 5. RhoA activation reverses CRMP4a-mediated reduction of cell migration. A. PC-3 stable subline cells were infected with lentiviruses of active RhoA (Q63L) or inactive RhoA (T19N) and then plated in the upper chamber TranswellTM coated with MatriGel. After a 48-h incubation, migrated cells into the bottom chambers were stained with crystal violet. Microscopic images were taken, and the representative images were shown from three independent experiments. B. Quantitative data of migrated cells per condition from panel A experiments were summarized as the mean ± SEM. The asterisk indicates a significant difference compared to the vector control (student’s t-test, p < 0.05). C. PC-3 subline cells as indicated were plated in the upper chamber TranswellTM coated with MatriGel were left untreated or treated Rhosin (30 μM). After 48 h incubation, migrated cells into the bottom chamber were stained with crystal violet. Microscopic images were taken, and the representative images were shown from three independent experiments. D. Quantitative data for migrated cells per condition from panel C experiments were summarized as the mean ± SEM. The asterisk indicates a significant difference compared to the vector control (student’s t-test, p < 0.05).
CRMP4 inhibits rhoa-dependent cell spreading capability
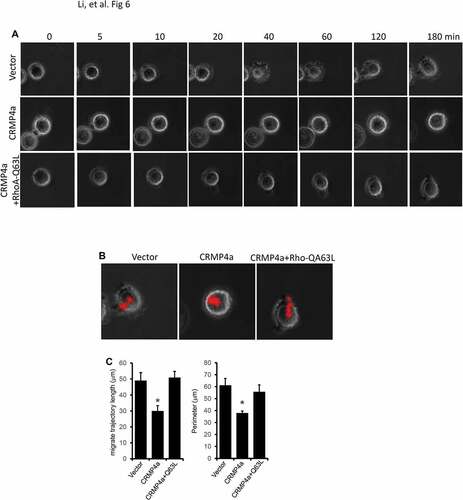
Cell motility strongly depends on cell adhesion and spreadingCitation21. Thus, we investigated the effect of CRMP4a expression on cell spreading using a Live-Cell Imaging System. For the control PC-3 subline cells, cell attachment was observed within 5 min after platted on plastic surface and it only took about 40 min for cell to spread and 180 min to a full adhesion (). In contrast, CRMP4a overexpression dramatically delayed the spreading process. However, co-expression with the active RhoA-Q63L mutant eliminated the suppressive effect of CRMP4a overexpression on cell spreading and adhesion ( & Supplementary Movie S1-S3). In addition, CRMP4a overexpression significantly reduced migrate trajectory length and cell perimeter but active RhoA-Q63L reversed this reduction (-). These data indicate that CRMP4a suppresses RhoA-dependent cell spreading and adhesion.
Figure 6. RhoA activation blocks CRMP4a-mediated suppression of cell spreading. A. PC-3 cells stably infected with empty vector, CRMP4a or CRMP4a plus RhoA-Q63L lentiviruses were plated in glass-bottomed culture dish for living cell system. Time-lapse sequential images were captured at intervals of 5 min for 3 h using the IX2-ZDC2 laser-based autofocusing system. B. Cell migration trajectories were determined using the ImageJ software. The movie files were opened in ImageJ program and cell migrate trajectories were tracked along the centrality of cells with the “add” button in plugin ‘MTrackJ’. The lengths of cell migrate trajectories were calculated using the “measure” button in plugin ‘MTrackJ’. C. Quantitative data for trajectory length and perimeter were shown as mean ± SEM from three independent microscopic fields. The asterisk indicates a significant difference compared to the vector control (student’s t-test, p < 0.05).
Discussion
In this study, we provided the novel insights into the mechanism for CRMP4a modulation of cancer cell migration. We determined that manipulating CRMP4a gene expression attenuated the pattern of cytoskeletal organization in parallel with altered RhoA activation. We also demonstrate that CRMP4a interacted with RhoA but not cdc42 or Rac1 and suppressed RhoA-dependent cell migration and spreading. Consistent with previous reportsCitation5,Citation6,Citation12, our data demonstrated that CRMP4a mediates tumor metastasis suppression via sequestering RhoA activity.
CRMP4a is the shorter splicing variant compared with CRMP4b and has been identified as a tumor metastasis suppressor in prostate cancersCitation8. Proteomic and genomic analyses revealed that CRMP4a expression was significantly reduced in metastatic tumor tissues mainly due to promoter methylationCitation22-Citation24. CRMP4 expression could be enhanced at the transcriptional level by altering its promoter status with the small activating RNA (saRNA) approach or the transcription activator-like effector (TALE)-guided DNA demethylase approachCitation9,Citation10, leading to reduced cancer cell migration and tumor metastasis in prostate cancer models. In this study, our data indicated that CRMP4a is involved in modulating cytoskeletal organization such as focal adhesion and cell spreading in prostate cancer cells. These data were supported by previous reports that CRMP4 directly binds to F-actin in neural cellsCitation5. Our data also showed that CRMP4 interacted with RhoA but not cdc42 or Rac1 and reduces RhoA activity. Indeed, CRMP4 was found as the only CRMP family proteins to interact with RhoA protein in neural cellsCitation6,Citation12. Therefore, it is postulated that CRMP4 suppresses cancer cell migration and tumor metastasis by interacting with and sequestering RhoA activity.
Rho family proteins belong to small GTPases consisting of more than 60 members and there are three major Rho sub-families, Rho (RhoA, RhoB & RhoC), Rac (Rac1-3 & RhoG) and cdc42 (cdc42m RhoQ & RhoJ) based on their protein structure and functional specificityCitation3. Rho family protein activity is determined by its GTP-bound status and is regulated by numerous factors including guanine nucleotide activating proteins (GAP), guanine nucleotide dissociation inhibitors (GDI) and guanine nucleotide exchange factors (GEF). Currently, it is not clear how CRMP4a sequesters RhoA activity by interaction. It was shown that CRMP2 interacts with Rho downstream protein kinase ROCK2 to reduce its function in modulating neural cell motility and cancer cell migrationCitation25-Citation27. On the other hand, CRMP2 was shown to be phosphorylated at residue threonine-555 by ROCK2 in brain cellsCitation28, leading to a functional reduction of microtubule assemblyCitation29. Interestingly, in our studies, we also showed that active RhoA counter-acted CRMP4-mediated modulation of cytoskeletal organization and cell migration. Therefore, we hypothesize that CRMP4a might trap RhoA or its effecter kinase ROCK1 or ROCK2, or vice versa, to modulate cancer cell migration or invasion.
In conclusion, in this study, we determined the mechanistic actions of CRMP4a in modulating cancer cell motility. We demonstrated that CRMP4a interacts with RhoA and sequesters its activation to suppress cytoskeletal organization. Further investigation is ongoing to analyze the mode of CRMP4-RhoA interaction.
Acknowledgments
This study was supported mainly by a grant from Shandong Provincial Natural Science Foundation of China (ZR2016HL25) and partially by an internal fund of Jining Medical University (grant #JY2017KJ030).
Additional information
Funding
References
- Steeg PS. 2016. Targeting metastasis. Nat Rev Cancer. 16:201–218. doi:10.1038/nrc.2016.25.
- Peinado H, Zhang H, Matei IR, Costa-Silva B, Hoshino A, Rodrigues G, Psaila B, Kaplan RN, Bromberg JF, Kang Y, et al. Pre-metastatic niches: organ-specific homes for metastases. Nat Rev Cancer. 2017;17:302–317. doi:10.1038/nrc.2017.6.
- Jansen S, Gosens R, Wieland T, Schmidt M. 2018. Paving the Rho in cancer metastasis: rho GTPases and beyond. Pharmacol Ther. 183:1–21. doi:10.1016/j.pharmthera.2017.09.002.
- Nagai J, Baba R, Ohshima T. 2017. CRMPs Function in Neurons and Glial Cells: potential Therapeutic Targets for Neurodegenerative Diseases and CNS Injury. Mol Neurobiol. 54:4243–4256. doi:10.1007/s12035-016-0005-1.
- Rosslenbroich V, Dai L, Baader SL, Noegel AA, Gieselmann V, Kappler J. 2005. Collapsin response mediator protein-4 regulates F-actin bundling. Exp Cell Res. 310:434–444. doi:10.1016/j.yexcr.2005.08.005.
- Alabed YZ, Pool M, Ong Tone S, Fournier AE. 2007. Identification of CRMP4 as a convergent regulator of axon outgrowth inhibition. J Neurosci. 27:1702–1711. doi:10.1523/JNEUROSCI.5055-06.2007.
- Tan F, Thiele CJ, Li Z. 2014. Collapsin response mediator proteins: potential diagnostic and prognostic biomarkers in cancers (Review). Oncol Lett. 7:1333–1340. doi:10.3892/ol.2014.1909.
- Gao X, Pang J, Li LY, Liu WP, Di JM, Sun QP, Fang YQ, Liu XP, Pu XY, He D, et al. Expression profiling identifies new function of collapsin response mediator protein 4 as a metastasis-suppressor in prostate cancer. Oncogene. 2010;29:4555–4566. doi:10.1038/onc.2010.213.
- Li C, Jiang W, Hu Q, Li LC, Dong L, Chen R, Zhang Y, Tang Y, Thrasher JB, Liu CB, et al. Enhancing DPYSL3 gene expression via a promoter-targeted small activating RNA approach suppresses cancer cell motility and metastasis. Oncotarget. 2016;7:22893–22910. doi:10.18632/oncotarget.8290.
- Li K, Pang J, Cheng H, Liu WP, Di JM, Xiao HJ, Luo Y, Zhang H, Huang WT, Chen MK, et al. Manipulation of prostate cancer metastasis by locus-specific modification of the CRMP4 promoter region using chimeric TALE DNA methyltransferase and demethylase. Oncotarget. 2015;6:10030–10044. doi:10.18632/oncotarget.3192.
- Zhou W, Xie P, Pang M, Yang B, Fang Y, Shu T, Liu C, Wang X, Zhang L, et al. Upregulation of CRMP4, a new prostate cancer metastasis suppressor gene, inhibits tumor growth in a nude mouse intratibial injection model. Int J Oncol. 2015;46:290–298. doi:10.3892/ijo.2014.2705.
- Khazaei MR, Girouard MP, Alchini R, Ong Tone S, Shimada T, Bechstedt S, Cowan M, Guillet D, Wiseman PW, Brouhard G, et al. Collapsin response mediator protein 4 regulates growth cone dynamics through the actin and microtubule cytoskeleton. J Biol Chem. 2014;289:30133–30143. doi:10.1074/jbc.M114.570440.
- Havel LS, Kline ER, Salgueiro AM, Marcus AI. 2015. Vimentin regulates lung cancer cell adhesion through a VAV2-Rac1 pathway to control focal adhesion kinase activity. Oncogene. 34:1979–1990. doi:10.1038/onc.2014.123.
- Liao X, Thrasher JB, Holzbeierlein J, Stanley S, Li B. 2004. Glycogen synthase kinase-3beta activity is required for androgen-stimulated gene expression in prostate cancer. Endocrinology. 145:2941–2949. doi:10.1210/en.2003-1519.
- Pellegrino L, Stebbing J, Braga VM, Frampton AE, Jacob J, Buluwela L, Jiao LR, Periyasamy M, Madsen CD, Caley MP, et al. miR-23b regulates cytoskeletal remodeling, motility and metastasis by directly targeting multiple transcripts. Nucleic Acids Res. 2013;41:5400–5412. doi:10.1093/nar/gkt245.
- Steffen A, Stradal TE, Rottner K. 2017. Signalling Pathways Controlling Cellular Actin Organization. Handb Exp Pharmacol. 235:153–178. doi:10.1007/164_2016_35.
- Rottner K, Faix J, Bogdan S, Linder S, Kerkhoff E. 2017. Actin assembly mechanisms at a glance. J Cell Sci. 130:3427–3435. doi:10.1242/jcs.206433.
- Narumiya S, Tanji M, Ishizaki T. 2009. Rho signaling, ROCK and mDia1, in transformation, metastasis and invasion. Cancer Metastasis Rev. 28:65–76. doi:10.1007/s10555-008-9170-7.
- Haga RB, Ridley AJ. 2016. Rho GTPases: regulation and roles in cancer cell biology. Small GTPases. 7:207–221. doi:10.1080/21541248.2016.1232583.
- Ridley AJ. 2015. Rho GTPase signalling in cell migration. Curr Opin Cell Biol. 36:103–112. doi:10.1016/j.ceb.2015.08.005.
- Cuvelier D, Thery M, Chu YS, Dufour S, Thiery JP, Bornens M, Nassoy P, Mahadevan L. The universal dynamics of cell spreading. Curr Biol. 2007;17:694–699. doi:10.1016/j.cub.2007.02.058.
- Gao X, Li LY, Rassler J, Pang J, Chen MK, Liu WP, Chen Z, Ren SC, Zhou FJ, Xie KJ, et al. Prospective Study of CRMP4 Promoter Methylation in Prostate Biopsies as a Predictor For Lymph Node Metastases. J Natl Cancer Inst. 2017;109(6):djw282.
- Huang QX, Xiao CT, Chen Z, Lu MH, Pang J, Di JM, Luo ZH, Gao X. Combined analysis of CRMP4 methylation levels and CAPRA-S score predicts metastasis and outcomes in prostate cancer patients. Asian J Androl. 2018;20:56–61. doi:10.4103/aja.aja_3_17.
- Gao X, Mao YH, Xiao C, Li K, Liu W, Li LY, Pang J. Calpain-2 triggers prostate cancer metastasis via enhancing CRMP4 promoter methylation through NF-kappaB/DNMT1 signaling pathway. Prostate. 2018. doi:10.1002/pros.23512.
- Yoneda A, Morgan-Fisher M, Wait R, Couchman JR, Wewer UM. 2012. A collapsin response mediator protein 2 isoform controls myosin II-mediated cell migration and matrix assembly by trapping ROCK II. Mol Cell Biol. 32:1788–1804. doi:10.1128/MCB.06235-11.
- Leung T, Ng Y, Cheong A, Ng CH, Tan I, Hall C, Lim L. p80 ROKalpha binding protein is a novel splice variant of CRMP-1 which associates with CRMP-2 and modulates RhoA-induced neuronal morphology. FEBS Lett. 2002;532:445–449.
- Morgan-Fisher M, Couchman JR, Yoneda A. 2013. Phosphorylation and mRNA splicing of collapsin response mediator protein-2 determine inhibition of rho-associated protein kinase (ROCK) II function in carcinoma cell migration and invasion. J Biol Chem. 288:31229–31240. doi:10.1074/jbc.M113.505602.
- Arimura N, Inagaki N, Chihara K, Menager C, Nakamura N, Amano M, Iwamatsu A, Goshima Y, Kaibuchi K. Phosphorylation of collapsin response mediator protein-2 by Rho-kinase. Evidence for Two Separate Signaling Pathways for Growth Cone Collapse. J Biol Chem. 2000;275:23973–23980.
- Arimura N, Menager C, Kawano Y, Yoshimura T, Kawabata S, Hattori A, Fukata Y, Amano M, Goshima Y, Inagaki M, et al. Phosphorylation by Rho kinase regulates CRMP-2 activity in growth cones. Mol Cell Biol. 2005;25:9973–9984. doi:10.1128/MCB.25.22.9973-9984.2005.