
ABSTRACT
Background
Ixazomib (Ninlaro), a novel proteasome inhibitor, has been developed for the treatment of many cancers and has demonstrated anti-tumor efficacy against various malignancies. However, the mechanism of the anti-tumor effect of ixazomib in colorectal cancer (CRC) cells remains unclear.
Methods
MTS and flow cytometry were performed to determine the effect of ixazomib on CRC cells. Western blotting and real-time RT-PCR were performed to detect ixazomib-induced DR5 upregulation. ChIP was performed to detect CHOP binding to DR5 promoter. Finally, xenograft experiments were carried out to measure the antitumor effect of ixazomib in vivo.
Results
In this study, we revealed the mechanism by which ixazomib inhibits the growth of CRC cells. Our findings indicated that ixazomib treatment induces CHOP-dependent DR5 induction, irrespective of p53 status. Furthermore, DR5 is necessary for ixazomib-mediated apoptosis. Ixazomib also synergized with TRAIL to induce marked apoptosis via DR5 in CRC cells.
Conclusions
Our findings further suggested that ixazomib sensitizes TRAIL/death receptor signaling pathway-targeted CRC and suggested that DR5 induction could be a valuable indicator of ixazomib sensitivity.
Background
Colorectal cancer (CRC) is one of the most frequently diagnosed types of cancer in the US.Citation1 Globally, it is the third most common type of cancer.Citation2,Citation3 In CRC, conventional chemotherapy including DNA damaging drugs may be used in addition to surgery in certain cases to increase life expectancy. These chemotherapeutic drugs include combinations of irinotecan, oxaliplatin, 5-fluorouracil (5-FU) and capecitabine (FOLFIRI, FOLFOX or CAPOX).Citation4 Currently, targeted therapy has been identified as a promising cancer treatment strategy that can help overcome the adverse effects of conventional cancer therapies.Citation5,Citation6 Ixazomib, compared with bortezomib, is an effective oral 20S subunit-selective inhibitor with improved pharmacokinetics and pharmacodynamics.Citation7,Citation8 The proteasome half-time of ixazomib is six-fold greater, leading to more rapid tumor entry and faster red blood cell dissociation.Citation8 Moreover, compared with bortezomib, ixazomib also showed greater proteasome inhibition and more potent anticancer activity in cancer tissues.Citation9 Ixazomib citrate can be rapidly hydrolyzed in capsule form to ixazomib in physiological conditions.Citation10 Previously studies reported that ixazomib induces apoptosis in a verity of cancers including liver cance,Citation11 melanomCitation12 and CR.Citation13,Citation14 The above unique features of ixazomib may help to maintain optimal serum concentrations of ixazomib and improve treatment convenience for the patient.
Endoplasmic reticulum (ER) is the core of the synthesis, maturation and secretion of newly synthesized proteins.Citation15,Citation16 Proteins may increase ER stress by increasing translational dysregulation and protein quality control loading, including protein folding and degradation of misfolded proteins.Citation17 PERK-mediated phosphorylation of eIF2α is a key regulator of ER stress, which reduces general translation by inhibiting 43S PIC but increases ATF4 and its targets such as chaperone proteins, GADD34 and CHOP.Citation18,Citation19 However, prolonged ER stress induces apoptosis via CHOP-dependent or independent induction of DR5, Bim, Noxa and PUMA.Citation20,Citation21 Previous reports found that proteasome inhibitors such as MG132 and bortezomib induce apoptosis through ER stress-mediated DR5 induction.Citation22–Citation24
Apoptosis, a process of programmed cell death, occurs by the extrinsic apoptotic pathway (death receptor-mediated) and the intrinsic apoptotic pathway (mitochondria-mediated).Citation25 The intrinsic apoptotic pathway is activated by the members of the Bcl-2 family.Citation26 Stimulation activates the BH3-only protein, which inhibits the pro-survival Bcl-2 proteins, thereby activating the pro-apoptotic effectors BAX or BAK and leading to the disruption of the mitochondrial outer membrane permeabilization (MOMP).Citation27 The activation of extrinsic apoptotic pathway occurs by the binding of pro-apoptotic ligands to tumor necrosis factor (TNF) family receptors, which are negatively regulated by decoy receptors (DcRs).Citation28 Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL, also known as apo2 ligand) is a cytokine the has no effect on normal cells, but the ability of which to selectively induce apoptosis in cancer cells is well known.Citation29 TRAIL binds to cell surface death receptor 4 (DR4; TRAIL-R1) or death receptor 5 (DR5; TRAIL-R2), which further recruit the adaptor Fas-associated protein with death domain (FADD) and caspase 8, leading to the activation of caspase 3.Citation30,Citation31
In the current study, our findings demonstrated that ixazomib sensitizes TRAIL-induced apoptosis via CHOP-dependent DR5 induction. Our results highlight the newest biological functions of ixazomib in the regulation of apoptosis and the need to further investigate the potential treatment of CRC using a combination with ixazomib and TRAIL. Our results suggest that DR5 induction is a biomarker of the efficacy of ixazomib and other targeted chemotherapeutic drugs.
Materials and methods
Cell culture
CRC cell lines including HCT116, Lim1215, DLD1, LoVo, SW48, and HT29 were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). HCT116 p53-Knockout (p53-KO) cells were obtained from Horizon Discovery. All cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) medium with 10% heat-inactivated fetal bovine serum (FBS), 100 units/mL penicillin, and 100 µg/mL streptomycin (Invitrogen, Carlsbad, CA, USA). Ixazomib (Selleckchem, Houston, TX, USA) was diluted with DMSO. TRAIL (Sigma, Cleveland, OH, USA) was diluted with water.
Real-time reverse transcriptase PCR (RT-PCR)
Total RNA was extracted using the TRIzol RNA Kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instruction. Briefly, total RNA was used to generate cDNA with SuperScript II reverse transcriptase (Invitrogen, Carlsbad, CA, USA). PCR was performed in triplicate using the SsoFasrTM Probes Supermix (Bio-Rad) in a final reaction volume of 20 μL with gene-specific primer/probe sets and a standard thermal cycling procedure (35 cycles) on a Bio-Rad CFX96TM Real-Time PCR System. DR5 and β-actin levels were analyzed by TaqMan Gene Expression Real-Time PCR assays. The results were expressed as the threshold cycle (Ct). The relative quantification of the target transcripts was determined by the comparative Ct method (ΔΔCt) according to the manufacturer’s instruction. The 2−ΔΔCt method was used to analyze the relative changes in gene expression. Control experiments were conducted without reverse transcription to confirm that the total RNA was not contaminated with genomic DNA. The primers are as followed: DR5, Forward: 5′-CCAGCAAATGAAGGTGATCC-3′, Reverse: 5′-GCACCAAGTCTGCAAAGTCA-3′; DR4, Forward: 5′-TCCAGCAAATGGTGCTGAC-3′, Reverse: 5′-GAGTCAAAGGGCACGATGTT-3′; β-actin, Forward: 5′-TGGACATCCGCAAAGACCTG-3′, Reverse: 5′-CCGATCCACACGGAGTACTT-3′.
Western blotting
Western blotting was performed with antibodies against cleaved caspase-8, cleaved caspase-3, CHOP, FADD (Cell Signaling Technology, Danvers, MA, USA), DR5, HMGB1, β-actin, DR4 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), Mcl-1, FAS, Bcl-2 and Bcl-XL (BD, Franklin Lakes, NJ, USA).
ATP assay
ATP assay was performed using ATP assay kit (Abcam) according to the manufacturer’s instructions.
Apoptosis assays
Apoptosis was analyzed by nuclear staining with Hoechst 33258 (Invitrogen, Carlsbad, CA, USA). Annexin V/propidium iodide (PI) staining was performed using annexin-Alexa 488 (Invitrogen, Carlsbad, CA, USA) and PI. Caspase activity was measured using the SensoLyte Homogeneous AMC Caspase-3/7 Assay Kit (Anaspec, Fremont, CA, USA).
MTS assay
The indicated cell lines were seeded in 96-well plates at a density of 5 × 103 cells/well. After 24 hours of incubation, the cells were treated with ixazomib for an additional 72 hours. The 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay was performed using the MTS assay kit (Promega, Fitchburg, WI, USA) according to the manufacturer’s instructions. Absorbance was measured with a Wallac Victor 1420 Multilabel Counter at 490nm (Perkin Elmer, Bridgeville, PA, USA). Each assay was conducted in triplicate and repeated three times.
Sirna/shrna knockdown
The indicated cells were transfected with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Knockdown experiments were performed 24 hours prior to ixazomib treatment using 300 pmol of siRNA. The control scrambled siRNA and the siRNA for human CHOP (AAGACCCGCGCCGAGGUGAAG) were from Invitrogen. For stable transfection, a shRNA-expressing plasmid that containing the FADD-targeting sequence (GCAGUCCUCUUAUUCCUAA), DR5-targeting sequence (AAGACCCUUGUGCUCGUUG UC), caspase 8-targeting sequence (AACCUCGGGAUACUGUCUGA), or a vector containing a scrambled sequence was transfected into HCT116 and DLD1 cells. Followed transfection, the cells were plated in 96-well plates in the presence of 5 µg/mL puromycin for 2 weeks. The protein expression of the puromycin-resistant clones was then analyzed by western blotting.
Chromatin immunoprecipitation (chip)
ChIP with CHOP antibody (Cell Signaling Technology, Danvers, MA, USA) was performed using the Chromatin Immunoprecipitation Assay Kit (Millipore, Billerica, MA, USA) according to the manufacturer’s instructions. The precipitates were analyzed by PCR using the primers 5ʹ- AGGTTAGTTCCGGTCCCTTC −3ʹ and 5ʹ- CAACTGCAAATTCCACCACA −3ʹ to amplify a DR5 promoter fragment containing CHOP binding sites.
Statistical analysis
Statistical analyses were carried out using GraphPad Prism V software. P values were calculated by Student’s t-test and were considered significant if p < 0.05. The means ± one standard deviation (SD) is displayed in the figures.
Results
Ixazomib inhibits cell growth and promotes apoptosis in CRC
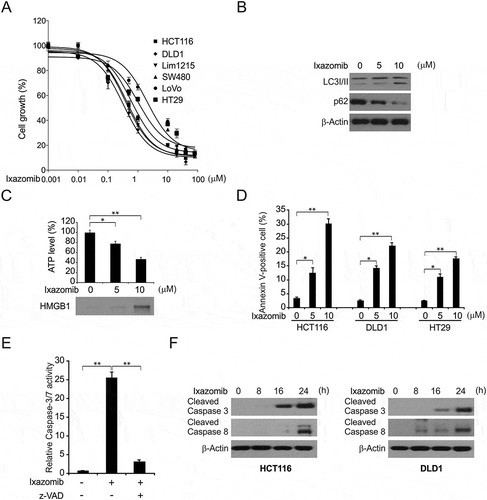
To determine the effect of ixazomib on CRC cells, 6 CRC cell lines were treated with increasing concentrations of ixazomib for 72 hours, and cell growth was examined by MTS assay. Based on the MTS assay results, we found that ixazomib significantly suppressed the growth of those cell lines (). In consistence with previously study, we found ixazomib induced autophagy in HCT116 cells (). Previously study has shown that MG132 promotes RIPK3 and RHIM dependent necroptosis,Citation32 we also detected whether ixazomib induced necroptosis in HT29 cells. We found ixazomib increased the intracellular ATP and extracellular release of nuclear High-Mobility Group Box 1 (HMGB1), which indicated ixazomab induced necroptosis in HT29 cells (). Next, we examined whether apoptotic cell death was involved in ixazomib-induced CRC growth inhibition. In the apoptotic assays, HCT116, DLD1 and HT29 cells treated with ixazomib at 5–10 µM induced Annexin V-positive cell populations (). In this study, we focused on the mechanisms of ixazomib induced apoptosis in CRC. Following ixazomib treatment, the activation of caspases 3/7 was strongly induced in HCT116 cells (). Moreover, the apoptotic response was also found to be blocked by z-VAD-fmk, a pan-caspase inhibitor (), indicating an ixazomib-mediated caspase-dependent apoptotic response. Then, we observed the cleaved forms of caspase 3 and 8 in cells with different p53 statuses, including p53-WT HCT116 and p53-mutant DLD1 with ixazomib treatment for 24 hours, and found that ixazomib activates caspase 3 and 8 in both of these cells (). In summary, these data indicate that ixazomib decrease cell proliferation and induced caspase-dependent but p53-independent apoptosis in CRC.
Figure 1. Ixazomib inhibits cell proliferation and induces apoptosis in CRC. (A) The indicated cell lines were treated with increasing concentrations of ixazomib for 72 hours. Cell proliferation was analyzed by MTS assay. (B) HCT116 cells were treated with ixazomib for 24 hours. Indicated protein level was analyzed by Western blotting. (C) HT29 cells were treated with ixazomib for 24 hours. ATP level (upper) and HMGB1 release (lower) were analyzed. (D) Indicated cell lines were treated with ixazomib for 24 hours at indicated concentrations. Apoptosis was analyzed by Annexin V/PI staining followed by flow cytometry. (E) HCT116 cells were treated with ixazomib with or without z-VAD-fmk, caspase 3/7 activity was determined by fluorogenic analysis. (F) The indicated cell lines were treated with 10 μmol/L ixazomib at indicated time point. Cleaved caspase 3 and 8 were analyzed by western blotting. Results in (A), (D) and (F) were expressed as means ± SD of 3 independent experiments. **, P < 0.01; *, P < 0.05.
DR5 is required for ixazomib-induced apoptosis
The above results demonstrated that ixazomib enhances caspase 8 activation, which is a well-known initiator caspase in the extrinsic apoptotic pathway.Citation33 We then analyzed the mechanism by which ixazomib activates the extrinsic apoptosis pathway. It well known that both DR5 and DR4 are important cell surface death receptors, which activate extrinsic apoptotic signaling by recruiting FADD.Citation34 To determine whether ixazomib can directly activate extrinsic apoptotic signaling through death receptors, we detected DR5 and DR4 expression in HCT116 cells treated with ixazomib. Ixazomib markedly induced the mRNA and protein expression levels of DR5, but not DR4, in a time-dependent manner in HCT116 cells (-). The above results indicate that ixazomib strongly increases DR5 expression in HCT116 cells.
Figure 2. DR5 is required for ixazomib-induced apoptosis. (A) HCT116 cells were treated with 10μmol/L ixazomib at indicated time point. Indicated protein levels were analyzed by western blotting. (B) HCT116 cells were treated with ixazomib at indicated concentration. DR5 expression was analyzed by western blotting. (C) HCT116 cells were treated with 10μmol/L ixazomib at indicated time points. DR5 mRNA level was analyzed by gel electrophoresis. β-Actin was used as a control for normalization. (D) HCT116 cells were treated with ixazomib for 24 hours at indicated concentration. mRNA expression of DR5 and DR4 were analyzed by semiquantitive reverse transcription PCR (RT-PCR). (E) Parental and DR5-KD HCT116 cells were treated with 10μmol/L ixazomib for 24 hours. DR5 expression was analyzed by western blotting. (F) Parental and DR5-KD HCT116 cells were treated with 10μmol/L ixazomib for 24 hours. Apoptosis was analyzed by flow cytometry. (G) Parental and DR5-KD HCT116 or DLD1 cells were treated with 10μmol/L ixazomib 24 hours. Cleaved caspase 3 and 8 were analyzed by western blotting. Results in (D) and (F) were expressed as means ± SD of 3 independent experiments. **, P < 0.01; *, P < 0.05.
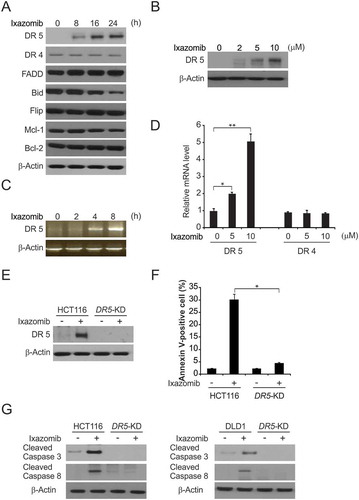
Next, we investigated whether DR5 is required for ixazomib-induced apoptosis in CRC. The effects of ixazomib on apoptosis and caspase activation in parental and DR5 stable knockdown (DR5-KD) HCT116 cells were compared. It was revealed that the apoptosis induced by ixazomib is significantly reduced in DR5-KD cells ( and ). Moreover, apoptosis and the cleavage of caspases 3 and 8 were markedly enhanced by ixazomib treatment in parental HCT116 and DLD1 cells, but not in DR5-KD cells (). Together, these finding suggest that DR5 upregulation is required for ixazomib-induced apoptosis.
FADD and caspase 8 are required for ixazomib induced apoptosis
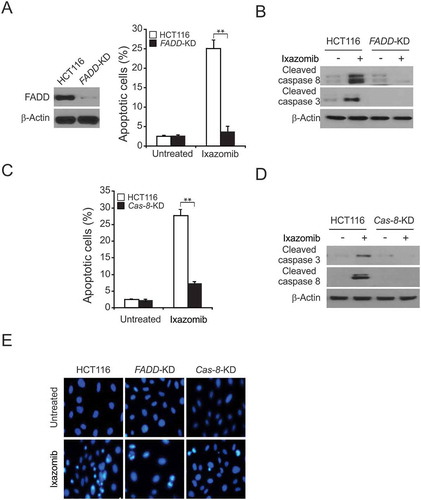
Next, we compared the effects of ixazomib on the induction of apoptosis in parental and FADD stable knockdown (FADD-KD) HCT116 cells, and we found that apoptosis was markedly reduced in FADD-KD cells in response to ixazomib treatment (, and ). We further examined whether caspase-8 is necessary for ixazomib-induced apoptosis. We treated parental and caspase 8 stable knockdown (Casp 8-KD) HCT116 cells with ixazomib and analyzed apoptosis by nuclear staining. In line with the FADD results, compared to parental cells, casp 8-KD cells displayed significantly reduced apoptosis induced by ixazomib (, and ). The above results indicated that caspase 8 mediates the effects of ixazomib-induced apoptosis. Together, these results demonstrate that FADD is an essential component in the mediation of the extrinsic apoptotic pathway, and the depletion of FADD abrogated the anticancer activity of ixazomib, suggesting that the activation of the extrinsic apoptotic pathway plays an important role in ixazomib-induced apoptosis.
Figure 3. Caspase 8 and FADD mediated ixazomib-induced apoptosis. (A) Parental and FADD-KD HCT116 cells were treated with 10 μmol/L ixazomib for 24 hours. Apoptosis was analyzed by a nuclear fragmentation assay. (B) Parental and FADD-KD HCT116 cells were treated with 10 μmol/L ixazomib for 24 hours. Cleaved caspase 3 and 8 were analyzed by western blotting. (C) Parental and Cas 8-KD HCT116 cells were treated with 10 μmol/L ixazomib for 24 hours. Apoptosis was analyzed by a nuclear fragmentation assay. (D) Parental and Cas 8-KD HCT116 cells were treated with 10 μmol/L ixazomib for 24 hours. Cleaved caspase 3 and 8 were analyzed by Western blotting. (E) The images of representative fields of nuclear staining as treatment in (A) and (C). Results in (A) and (C) were expressed as means ± SD of 3 independent experiments. **, P < 0.01.
Ixazomib induces chop-mediated DR5 upregulation
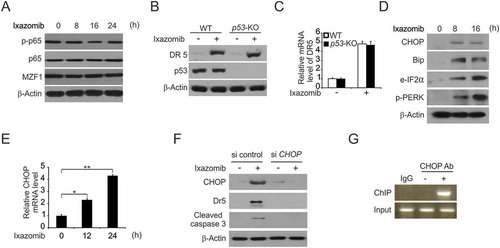
Then, we investigated the mechanism of p53-independent DR5 induction by ixazomib in CRC. Several transcription factors that can mediate DR5 induction were examined by western blotting. P65 is not involved because phosphorylation did not occur in the response to ixazomib treatment. Another transcription factor, MZF1 was also ruled out because of lack of induction after ixazomib treatment (). To confirm p53-independent DR5 induction by ixazomib, we treated WT and p53-KO HCT116 cells with ixazomib, and found the protein and mRNA level of DR5 are equally in the WT and p53-KO HCT116 cells ( and ). Previously, studies suggested that CHOP is involved in DR5 induction and that is contributes to the sensitization of TRAIL-mediated apoptosis.Citation35,Citation36 Thus, we investigated whether CHOP is required for DR5 induction and apoptosis in ixazomib-treated CRC. We determined the effects of ixazomib on the expression of CHOP in addition to other ER stress markers. Importantly, we found that ixazomib-induced apoptosis correlated with various markers of ER stress activation apart from CHOP upregulation, such as Bip, e-IF2α, p-PERK, in a time-dependent manner (). Further-more, the CHOP mRNA level was upregulated by ixazomib (). We also found that the knockdown of CHOP by siRNA obviously suppressed the induction of DR5 and the cleavage of caspase-3 by ixazomib (). To investigate whether CHOP can activate DR5 transcription directly, we performed the chromatin immunoprecipitation (ChIP) experiment. The ChIP results showed that CHOP was recruited to the genomic region containing the CHOP binding sites following ixazomib treatment (). These findings indicate that CHOP directly binds to the DR5 promoter region to drive its transcriptional activation and is required for the induction of DR5 and apoptosis, as well as the activation of ER stress in response to ixazomib treatment.
Figure 4. CHOP-mediated ixazomib-induced DR5 expression. (A) HCT116 cells were treated with 10 μmol/L ixazomib at indicated time point. Indicated protein expression was analyzed by western blotting. (B) WT and p53-KO HCT116 cells were treated with 10 μmol/L ixazomib for 24 hours. DR5 and p53 were analyzed by Western blotting. (C) WT and p53-KO HCT116 cells were treated with 10 μmol/L ixazomib for 24 hours. DR5 mRNA level was analyzed by Real-time PCR. (D) HCT116 cells were treated with 10 μmol/L ixazomib at indicated time point. Indicated protein expression was analyzed by western blotting. (E) HCT116 cells were treated with 10μmol/L ixazomib at indicated time point. CHOP mRNA level was analyzed by real-time PCR. (F) HCT116 cells transfected with si control or si CHOP were treated with 10 μmol/L ixazomib for 24 hours. CHOP, DR5 and cleaved caspase 3 were analyzed by western blotting. (G) Chromatin immunoprecipitation (ChIP) was performed using anti-CHOP antibody on HCT116 cells following ixazomib treatment for 12 hours. ChIP with the control IgG was used as a control. PCR was carried out using primers surrounding the CHOP binding sites in the DR5 promoter. Results in (C) were expressed as means ± SD of 3 independent experiments. **, P < 0.01; *, P < 0.05.
Ixazomib promotes trail-induced apoptosis via DR5 upregulation
We further hypothesized that ixazomib sensitizes CRC to TRAIL-mediated apoptosis in CRC. As expected, we found that a combination treatment with ixazomib and TRAIL induced more apoptosis compared to treatment with either single agent (). In addition, it was also found that the apoptotic response was attenuated after pretreatment with z-VAD-fmk (a pan-caspase inhibitor) (). Furthermore, combination treatment with ixazomib and TRAIL was also much more effective than single treatment in terms of increasing cleaved caspases 3 and 8 in HCT116 (). The combination index was calculated using the Chou-Talalay method to assess the synergy (CI < 1) or antagonism (CI> 1) for each drug combinatio.Citation37 Our results showed that the co-treatment with ixazomib and TRAIL resulted in a synergistic effect on the cell viability of HCT116 cells using a combination of 5 μM ixazomib with 10 ng/mL TRAIL.
Figure 5. Ixazomib sensitizes TRAIL-mediated apoptosis. (A) HCT116 cells were treated with 5 μmol/L or 10 μmol/L ixazomib, 10 ng/mL TRAIL or their combination with or without 10 μmol/L z-VAD-fmk for 24 hours. Apoptosis was analyzed by flow cytometry. (B) HCT116 cells were treated with 5 μmol/L ixazomib, 10 ng/mL TRAIL or their combination for 24 hours. Cleaved caspase 3 and 8 were analyzed by western blotting. (C) HCT116 cells were treated with the combination of 5 μmol/L ixazomib and 10 ng/mL TRAIL with or without 10 μmol/L z-VAD for 24 hours. Cleaved caspase 3 was analyzed by western blotting. (D) Parental and DR5-KD HCT116 cells were treated with 5 μmol/L ixazomib, 10ng/mL TRAIL or their combination for 24 hours. Apoptosis was analyzed by a nuclear fragmentation assay. (E) Parental and DR5-KD HCT116 cells were treated with 5 μmol/L ixazomib, 10 ng/mL TRAIL or their combination for 24 hours. Cleaved caspase 3 and 8 were analyzed by western blotting. (F) Parental and DR5-KD DLD1 cells were treated with 5 μmol/L ixazomib, 10 ng/mL TRAIL or their combination for 24 hours. Cleaved caspase 3 and 8 were analyzed by western blotting. Results in (A) and (C) were expressed as means ± SD of three independent experiments. ***, P < 0.001; **, P < 0.01; *, P < 0.05.
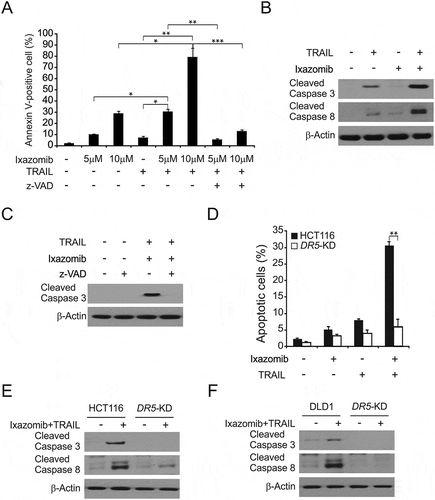
Apoptosis induction and caspase 3 activation by the combination treatment were largely blocked by pretreatment with z-VAD-fmk (), indicating that the combination of ixazomib and TRAIL induced caspase-dependent apoptosis in CRC.
This is consistent with the fact that the apoptosis induced by the combination of ixazomib and TRAIL was significantly reduced in DR5-KD cells (). Moreover, apoptosis and the cleavage of caspase 3 and 8 were enhanced by ixazomib in parental HCT116 and DLD1 cells, but not in DR5-KD cells ( and ). The above results suggest that DR5 mediates the combined effects of ixazomib and TRAIL in vitro.
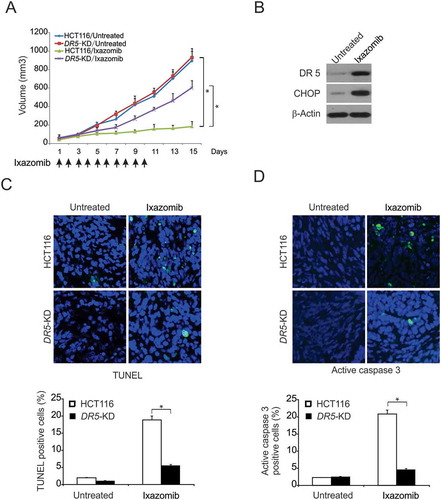
DR5 mediates antitumor effect of ixazomib in vivo
To determine if DR5 mediates tumor suppression by ixazomib, we treated nude mice bearing parental and DR5-KD HCT116 xenografts daily for 10 consecutive days by oral gavage with 20 mg/kg ixazomib or the vehicle. There was no significant different in the growth of parental and DR5-KD tumors in the control group (). Ixazomib treatment caused significant reductions in the tumor growth of the parental tumors (). In contrast, DR5-KD tumors were significantly less sensitive to ixazomib treatment compared to the parental tumors (), suggesting that the loss of DR5 abrogated the antitumor activity of ixazomib. Consistent with the observations in vitro, CHOP and DR5 expression were markedly increased in ixazomib-treated parental tumors (). TUNEL staining showed that ixazomib-treated WT tumors had marked apoptosis, which was significantly blocked in DR5-KD tumors (). Active caspase 3 staining confirmed ixazomib-induced and DR5-dependent apoptosis in tumors (). Therefore, these data suggest a pivotal role of the extrinsic apoptotic pathway in the antitumor activities of ixazomib in vivo.
Figure 6. DR5-mediated antitumor effect of ixazomib in vivo. (A) Nude mice were injected s.c. with 4 × 106 parental or DR5-KD HCT116 cells. After 1 week, mice were oral gavaged with 20 mg/kg ixazomib or the vehicle control for 10 consecutive days. Tumor volume at indicated time points after treatment was calculated and plotted with p values, n = 6 in each group. Arrows indicate ixazomib injection. (B) Mice with parental HCT116 xenograft tumors were treated with 20 mg/kg ixazomib or the vehicle as in (A) for 4 consecutive days. The levels of CHOP and DR5 in randomly selected tumors were analyzed by western blotting. (C) Paraffin-embedded sections of parental or DR5-KD tumor tissues from mice treated as in (B) were analyzed by TUNEL staining. Upper, representative TUNEL staining pictures; Lower, TUNEL-positive cells were counted and plotted. (D) Tissue sections from (C) were analyzed by active caspase 3 staining. Upper, representative staining pictures; Lower, active caspase 3-positive cells were counted and plotted. In (C)–(D), results were expressed as means ± SD of 3 independent experiments. *, P < 0.05.
Discussion
Ixazomib is a novel, 20S proteasome subunit selective inhibitor that can be administered orally and intravenously.Citation38,Citation39 The distribution characteristics of ixazomib are similar after intravenous and oral administration.Citation40 The oral bioavailability of ixazomib is approximately 60%, according to the pooled population pharmacokinetic data analysis of intravenous and oral regimens.Citation38 Preclinical studies demonstrated that the antitumor activity of ixazomib is similar to that of bortezomib.Citation41 The study also showed greater proteasome inhibition and better pharmacodynamic and pharmacokinetic parameters in ixazomib.Citation7,Citation41 In addition, compared to bortezomib, ixazomib has been shown to have a 6-fold faster proteasome disassociation half-life, leading to a faster tumor entry and faster red blood cell dissociation.Citation1,Citation42 In this study, our findings indicated that ixazomib effectively reduces cell growth and induces apoptosis in CRC. In addition, our findings suggested that ixazomib induces the extrinsic apoptotic pathway because the absence of FADD or caspase 8 protected cells against ixazomib-induced apoptosis and caspase cleavage. In addition, our results showed that ixazomib sensitizes TRAIL-mediated apoptosis, as evidenced by the synergistic induction of DR5-dependent apoptosis by the combination of ixazomib and TRAIL. Ixazomib-induced CHOP-mediated upregulation of DR5 is required for the antitumor effects of ixazomib. This is the first report to demonstrate that ixazomib induces the DR5-dependent extrinsic apoptosis pathway in CRC.
TRAIL is an alternative antitumor agent that induces apoptosis in a variety of cancer cell types without cytotoxic effect in normal cells and therefore represents a promising novel target for antitumor therapeutics.Citation29 However, many studies indicated that many types of cancer are resistant to the antitumor effects of TRAIL.Citation43 Therefore, developing drugs that sensitize cancer cells to TRAIL-mediated antitumor activity is important to improve the therapeutic efficacy of TRAIL. TRAIL mediates apoptosis by binding to the cell surface receptors DR4 and DR5, which leads to the formation of the death-inducing signaling complex (DISC) with the subsequent binding of caspase 8.Citation44 In addition, the expression of DR4 and DR5 is also crucial for the sensitization of cells to TRAIL.Citation45 In the current study, we found that DR5 expression was induced after treatment with ixazomib in CRC. Thus, the present study focused on the role of DR5 in mediating the potentiation of ixazomib-induced apoptosis and TRAIL-induced apoptosis as well as the mechanism of ixazomib-induced DR5 upregulation.
CHOP is upregulated by endoplasmic reticulum (ER) stress and is involved in ER-mediated apoptosis.Citation46 Moreover, CHOP, which belongs to the C/EBP family, is a transcription factor responsible for DR5 induction.Citation47 Here, our results showed that ixazomib-mediated upregulation of CHOP is a key regulator of DR5 expression and that CHOP contributes to ixazomib-induced apoptosis. Moreover, CHOP is known as an apoptotic inducer through ER stress, and the depletion of CHOP prevents the apoptosis induced by various antitumor agents.Citation46 ER stress also increases the expression of CHOP, thereby promoting the ER stress-mediated expression of apoptosis molecules, such as DR5.
In summary, our results suggest that ixazomib exerts potent inhibitory and cytotoxic effects on CRC, regardless of p53 status. In addition, ixazomib sensitizes TRAIL-mediated apoptosis through CHOP-dependent upregulation of DR5. Our findings provide a novel therapeutic approach to overcome TRAIL-resistant cancer therapy.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Authors’s contributions
DY performed the experiments; DY and XS made the figures and wrote the manuscript.
Ethics approval and consent to participate
All animal experiments used for this study were performed according to the animal use guideline (Directive 2010/63/EU in Europe). The China Medical University research ethics committee approved the current study, which was performed in accordance with the principles expressed in the Declaration of Helsinki.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank our lab members for critical reading of the manuscript.
References
- De Rosa M, Pace U, Rega D, Costabile V, Duraturo F Izzo P and Delrio P. Genetics, diagnosis and management of colorectal cancer (Review). Oncol Rep. 2015;34:1087–1096. doi:10.3892/or.2015.4108.
- Lynch HT, Lynch JF Lynch PM and Attard T. Hereditary colorectal cancer syndromes: molecular genetics, genetic counseling, diagnosis and management. Fam Cancer. 2008;7:27–39. doi:10.1007/s10689-007-9165-5.
- Tariq K, Ghias K. Colorectal cancer carcinogenesis: a review of mechanisms. Cancer Biol Med. 2016;13:120–135. doi:10.28092/j.issn.2095-3941.2015.0103.
- Gustavsson B, Carlsson G, Machover D, Petrelli N, Roth A, Schmoll HJ Tveit KM and Gibson F. A review of the evolution of systemic chemotherapy in the management of colorectal cancer. Clin Colorectal Cancer. 2015;14:1–10. doi:10.1016/j.clcc.2014.11.002.
- El Zouhairi M Charabaty A and Pishvaian MJ. Molecularly targeted therapy for metastatic colon cancer: proven treatments and promising new agents. Gastrointest Cancer Res.2011;4:15–21.
- Tabernero J, Salazar R, Casado E, Martinelli E. Gomez P and Baselga J. Targeted therapy in advanced colon cancer: the role of new therapies. Ann Oncol. 2004;15(Suppl 4):iv55–62. doi:10.1093/annonc/mdh905.
- Al-Salama ZT Garnock-Jones KP and Scott LJ. Ixazomib: A Review in Relapsed and/or Refractory Multiple Myeloma. Target Oncol. 2017;12:535–542. doi:10.1007/s11523-017-0504-7.
- Offidani M, Corvatta L, Gentili S Maracci L and Leoni P. Oral ixazomib maintenance therapy in multiple myeloma. Expert Rev Anticancer Ther. 2016;16:21–32. doi:10.1586/14737140.2016.1123627.
- Salvini M, Troia R, Giudice D, Pautasso C Boccadoro M and Larocca A. Pharmacokinetic drug evaluation of ixazomib citrate for the treatment of multiple myeloma. Expert Opin Drug Metab Toxicol. 2018;14:91–99. doi:10.1080/17425255.2018.1417388.
- Richardson PG, Kumar S, Laubach JP, Paba-Prada C, Gupta N, Berg D van de Velde H and Moreau P. New developments in the management of relapsed/refractory multiple myeloma - the role of ixazomib. J Blood Med. 2017;8:107–121. doi:10.2147/JBM.S102328.
- Augello G, Modica M, Azzolina A, Puleio R, Cassata G, Emma MR, Di Sano C, Cusimano AMontalto G and Cervello M. Preclinical evaluation of antitumor activity of the proteasome inhibitor MLN2238 (ixazomib) in hepatocellular carcinoma cells. Cell Death Dis. 2018;9:28. doi:10.1038/s41419-017-0195-0.
- Suarez-Kelly LP, Kemper GM, Duggan MC, Stiff A, Noel TC, Markowitz J, Luedke EA, Yildiz VO, Yu L, Jaime-Ramirez AC, et al.. The combination of MLN2238 (ixazomib) with interferon-alpha results in enhanced cell death in melanoma. Oncotarget. 2016;7:81172–81186. doi:10.18632/oncotarget.12791.
- Engur S, Dikmen M. The evaluation of the anti-cancer activity of ixazomib on Caco2 colon solid tumor cells, comparison with bortezomib. Acta Clin Belg. 2017;72:391–398. doi:10.1080/17843286.2017.1302623.
- Fan Q, Liu B. Identification of the anticancer effects of a novel proteasome inhibitor, ixazomib, on colorectal cancer using a combined method of microarray and bioinformatics analysis. Onco Targets Ther. 2017;10:3591–3606. doi:10.2147/OTT.S139686.
- Schwarz DS, Blower MD. The endoplasmic reticulum: structure, function and response to cellular signaling. Cell Mol Life Sci. 2016;73:79–94. doi:10.1007/s00018-015-2052-6.
- Phillips MJ, Voeltz GK. Structure and function of ER membrane contact sites with other organelles. Nat Rev Mol Cell Biol. 2016;17:69–82. doi:10.1038/nrm.2015.8.
- Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol. 2015;10:173–194. doi:10.1146/annurev-pathol-012513-104649.
- Cnop M, Toivonen S Igoillo-Esteve M and Salpea P. Endoplasmic reticulum stress and eIF2alpha phosphorylation: the Achilles heel of pancreatic beta cells. Mol Metab. 2017;6:1024–1039. doi:10.1016/j.molmet.2017.06.001.
- Leitman J, Barak B, Benyair R, Shenkman M, Ashery U Hartl FU and Lederkremer GZ. ER stress-induced eIF2-alpha phosphorylation underlies sensitivity of striatal neurons to pathogenic huntingtin. PLoS One. 2014;9:e90803. doi:10.1371/journal.pone.0090803.
- Iurlaro R, Munoz-Pinedo C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016;283:2640–2652. doi:10.1111/febs.13598.
- Anania VG, Yu K, Gnad F, Pferdehirt RR, Li H, Ma TP, Jeon D, Fortelny N, Forrest W, Ashkenazi A Overall CM and Lill JR. Uncovering a Dual Regulatory Role for Caspases During Endoplasmic Reticulum Stress-induced Cell Death. Mol Cell Proteomics. 2016;15:2293–2307. doi:10.1074/mcp.M115.055376.
- Zhu W, Zhan D, Wang L, Ma D, Cheng M, Wang H, Zhao J Cai Y and Cheng Z. Proteasome inhibitor MG132 potentiates TRAIL-induced apoptosis in gallbladder carcinoma GBC-SD cells via DR5-dependent pathway. Oncol Rep. 2016;36:845–852. doi:10.3892/or.2016.4839.
- Yoshida T, Shiraishi T, Nakata S, Horinaka M, Wakada M, Mizutani Y Miki T and Sakai T. Proteasome inhibitor MG132 induces death receptor 5 through CCAAT/enhancer-binding protein homologous protein. Cancer Res. 2005;65:5662–5667. doi:10.1158/0008-5472.CAN-05-0693.
- Liu X, Yue P, Chen S, Hu L, Lonial S Khuri FR and Sun SY. The proteasome inhibitor PS-341 (bortezomib) up-regulates DR5 expression leading to induction of apoptosis and enhancement of TRAIL-induced apoptosis despite up-regulation of c-FLIP and survivin expression in human NSCLC cells. Cancer Res. 2007;67:4981–4988. doi:10.1158/0008-5472.CAN-06-4274.
- Fulda S, Debatin KM. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene. 2006;25:4798–4811. doi:10.1038/sj.onc.1209608.
- Brunelle JK, Letai A. Control of mitochondrial apoptosis by the Bcl-2 family. J Cell Sci. 2009;122:437–441. doi:10.1242/jcs.031682.
- Martinou JC, Youle RJ. Mitochondria in apoptosis: bcl-2 family members and mitochondrial dynamics. Dev Cell. 2011;21:92–101. doi:10.1016/j.devcel.2011.06.017.
- Sayers TJ. Targeting the extrinsic apoptosis signaling pathway for cancer therapy. Cancer Immunol Immunother. 2011;60:1173–1180. doi:10.1007/s00262-011-1008-4.
- Naimi A, Movassaghpour AA, Hagh MF, Talebi M, Entezari A Jadidi-Niaragh F and Solali S. TNF-related apoptosis-inducing ligand (TRAIL) as the potential therapeutic target in hematological malignancies. Biomed Pharmacother. 2017;98:566–576. doi:10.1016/j.biopha.2017.12.082.
- Siegmund D Lang I and Wajant H. Cell death-independent activities of the death receptors CD95, TRAILR1, and TRAILR2. FEBS J. 2017;284:1131–1159. doi:10.1111/febs.13968.
- Bertsch U, Roder C Kalthoff H and Trauzold A. Compartmentalization of TNF-related apoptosis-inducing ligand (TRAIL) death receptor functions: emerging role of nuclear TRAIL-R2. Cell Death Dis. 2014;5:e1390. doi:10.1038/cddis.2014.351.
- Moriwaki K, Chan FK. Regulation of RIPK3- and RHIM-dependent Necroptosis by the Proteasome. J Biol Chem. 2016;291:5948–5959. doi:10.1074/jbc.M115.700997.
- Svandova EB, Vesela B, Lesot H Poliard A and Matalova E. Expression of Fas, FasL, caspase-8 and other factors of the extrinsic apoptotic pathway during the onset of interdigital tissue elimination. Histochem Cell Biol. 2017;147:497–510. doi:10.1007/s00418-016-1508-6.
- Pobezinskaya YL, Liu Z. The role of TRADD in death receptor signaling. Cell Cycle. 2012;11:871–876. doi:10.4161/cc.11.5.19300.
- Yamaguchi H, Wang HG. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J Biol Chem. 2004;279:45495–45502. doi:10.1074/jbc.M406933200.
- Tiwary R, Yu W, Li J, Park SK Sanders BG and Kline K. Role of endoplasmic reticulum stress in alpha-TEA mediated TRAIL/DR5 death receptor dependent apoptosis. PLoS One. 2010;5:e11865. doi:10.1371/journal.pone.0011865.
- Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–446. doi:10.1158/0008-5472.CAN-09-1947.
- Moreau P, Masszi T, Grzasko N, Bahlis NJ, Hansson M, Pour L, Sandhu I, Ganly P, Baker BW, Jackson SR, Stoppa AM, Simpson DR, Gimsing P, Palumbo A, Garderet L, Cavo M, Kumar S, Touzeau C, Buadi FK, Laubach JP, Berg DT, Lin J, Di Bacco A, Hui AM, van de Velde H, Richardson PG. Group T-MS,Oral Ixazomib, Lenalidomide, and Dexamethasone for Multiple Myeloma. N Engl J Med. 2016;374:1621–1634. doi:10.1056/NEJMoa1516282.
- Kumar SK, Berdeja JG, Niesvizky R, Lonial S, Laubach JP, Hamadani M, Stewart AK, Hari P, Roy V, Vescio R, et al.. Safety and tolerability of ixazomib, an oral proteasome inhibitor, in combination with lenalidomide and dexamethasone in patients with previously untreated multiple myeloma: an open-label phase 1/2 study. Lancet Oncol. 2014;15:1503–1512. doi:10.1016/S1470-2045(14)71125-8.
- Mateos MV, Masszi T, Grzasko N, Hansson M, Sandhu I, Pour L, Viterbo L, Jackson SR, Stoppa AM, Gimsing P, et al.. Impact of prior therapy on the efficacy and safety of oral ixazomib-lenalidomide-dexamethasone vs. Placebo-Lenalidomide-Dexamethasone in Patients with Relapsed/Refractory Multiple Myeloma in TOURMALINE-MM1. Haematologica. 2017;102:1767–1775.
- Brayer J, Baz R. The potential of ixazomib, a second-generation proteasome inhibitor, in the treatment of multiple myeloma. Ther Adv Hematol. 2017;8:209–220. doi:10.1177/2040620717710171.
- Bonnet A, Moreau P. Safety of ixazomib for the treatment of multiple myeloma. Expert Opin Drug Saf. 2017;16:973–980. doi:10.1080/14740338.2017.1344212.
- Wang F Lin J and Xu R. The molecular mechanisms of TRAIL resistance in cancer cells: help in designing new drugs. Curr Pharm Des. 20;2014:6714–6722.
- Sessler T, Healy S. Samali A and Szegezdi E. Structural determinants of DISC function: new insights into death receptor-mediated apoptosis signalling. Pharmacol Ther. 2013;140:186–199. doi:10.1016/j.pharmthera.2013.06.009.
- Shlyakhtina YPavet V and Gronemeyer H. Dual role of DR5 in death and survival signaling leads to TRAIL resistance in cancer cells. Cell Death Dis. 2017;8:e3025. doi:10.1038/cddis.2017.518.
- Lei Y, Wang S, Ren B, Wang J, Chen J, Lu J, Zhan S, Fu YHuang L and Tan J. CHOP favors endoplasmic reticulum stress-induced apoptosis in hepatocellular carcinoma cells via inhibition of autophagy. PLoS One. 2017;12:e0183680. doi:10.1371/journal.pone.0183680.
- Kim IY, Kang YJ, Yoon MJ, Kim EH, Kim SU, Kwon TKKim IA and Choi KS. Amiodarone sensitizes human glioma cells but not astrocytes to TRAIL-induced apoptosis via CHOP-mediated DR5 upregulation. Neuro Oncol. 2011;13:267–279. doi:10.1093/neuonc/noq195.