
ABSTRACT
Heterozygous germline BRCA2 mutations predispose to breast, ovarian, pancreatic and other types of cancer. The presence of a pathogenic mutation in patients or their family members warrants close surveillance or prophylactic surgery. Besides clearly pathogenic mutations, variants leading only to a single amino acid substitution are often identified. The influence of such variants on cancer risk is often unknown, making their presence a major clinical problem. When genetic methods are insufficient to classify these variants, functional assays with various cellular models are performed. We developed and applied a new syngeneic model of human cancer cells to test all variants of unknown significance in exon 18 identified by genetic testing of high-risk cancer patients in the Czech Republic, via introduction of constructs containing each of these variants into the wild-type allele of BRCA2-heterozygous DLD1 cells (BRCA2wt/Δex11). We found unaffected DNA repair function of BRCA2 in cell lines BRCA27997G>C/Δex11, BRCA28111C>T/Δex11, BRCA28149G>T/Δex11, BRCA28182G>A/Δex11, and BRCA28182G>T/Δex11, whereas the cell line BRCA28168A>G/Δex11 and the nonsense mutation carrying line BRCA28305G>T/Δex11 did affect protein function. Targeting the BRCA2 wild-type allele with a construct carrying the variant c.7988A> G resulted in incorporation exclusively into the already defective allele in all viable clones, strongly suggesting a detrimental phenotype.
Our model thus offers a valuable tool for the functional evaluation of unclassified variants in the BRCA2 gene and provides a stable and distributable cellular resource for further research.
Introduction
The BRCA2 gene on chromosome 13q12-13 encodes a 3418 amino acid protein. Although the functions of this protein are well-known, its sequence reveals only some clues to its cellular function, and thus variations in this sequence cannot be easily interpreted. The protein’s role in DNA repair specifically involves homologous recombination, mediated by its interaction with RAD51.Citation1-Citation3 BRCA2 interacts with RAD51 through highly conserved BRC repeats in exon 11 and an interaction domain mapped to exon 27.Citation3,Citation4
The BRCA2 gene exhibits allelic differences in its sequence among members of the human population. Some of the variants are known to be deleterious, causing inherited susceptibility to breast, ovarian, pancreatic and other cancer types.Citation5,Citation6 Unfortunately, in many patients who underwent genetic testing, BRCA2 variants causing a single amino acid change are being foundCitation7. For many of these variants, it is not possible to use epidemiological data or laboratory studies of protein function to determine unambiguously or efficiently whether one should classify the variant as either a deleterious mutation or a polymorphism without functional significance. Furthermore, most functional studies of BRCA2 have been hampered by lack of well-controlled human cancer cellular models.Citation8 Such absence was in the past overcome by employing methods of considerable technical difficulty.Citation9
Cell lines with natural expression of homozygous variants are often unavailable and do not allow isogenic experimental controls. Artificial introduction of null states can lead to impaired survival. For some clinically important genes like BRCA2, even the exogenous expression of wild-type gene fragments can cause interference with endogenous gene function (squelching) unless assays are performed with extreme technical care and multiple controls.Citation9-Citation12
To accurately annotate sequence variations of the BRCA2 gene in an unambiguous and technically facile model, we previously performed targeted disruption of BRCA2 exon 11 by homologous recombination, yielding the first available syngeneic human cancer BRCA2 knock-out cell line. Using hemizygous BRCA2 cells, we constructed a library of syngeneic exon 27 genetic variant lines (SyVaL). By functional evaluation of multiple clones harboring individual variants, we were able to classify their effect as deleterious, hypomorphic or neutral, and to create a pseudo-phosphorylated BRCA2 variant replacing the normal gene.Citation13
The aim of our recent study was to apply the previously developed technique of functional evaluation of syngeneic cell lines harboring individual mutations to clinical practice. In collaboration with a clinical unit providing genetic testing in high-risk cancer individuals, we aimed to apply our technique to unclassified variants identified in Czech patients.
Results
Mutational and segregation analysis
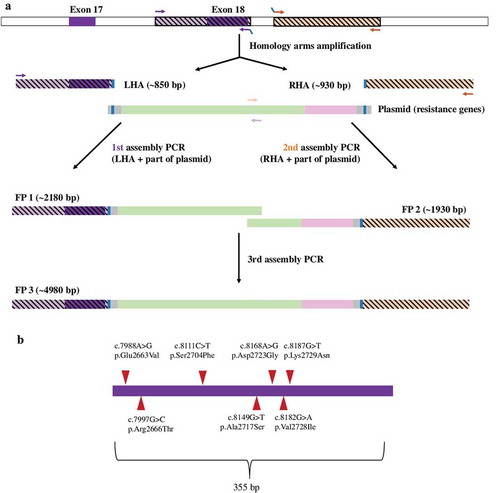
Mutational analysis of BRCA2 in high-risk individuals up to year 2011 revealed 78 BRCA2 variants; overall 71 of them were located in the coding region of gene. Of these 71 variants, three were classified as likely pathogenic, 17 as neutral and 54 as unclassified variants. In exon 18, seven variants were found in multiple families (). As the variants c.8149G> T (p.Ala2717Ser, rs28897747) and c.8182G> A (p.Val2728Ile, rs28897749) occurred at a high frequency in controls, we did not collect data for segregation analysis. For the remaining five variants, segregation analysis was possible in three families (). A unique variant c.8168A> G (p.Asp2723Gly) was found to segregate with the disease in two clinically unrelated families. In contrast, the variant c.7997G> C (p.Arg2666Thr, rs80359033) detected in the third family did not segregate with the disease (data not shown).
Table 1. Nomenclature and basic characteristics of variants selected for functional evaluation. BIC – Breast cancer Information Core; Align-GVGD – prediction based on calculation of Grantham Variation (GV) for positions in a PMSA and the Grantham Deviation (GD) for missense substitutions at those positions; PolyPhen2 (Polymorphism Phenotyping 2) – predicts possible impact of an amino acid substitution on the structure and function of a human protein using straightforward physical and comparative considerations; SIFT (Sorting Intolerant From Tolerant) – prediction based on the degree of conservation of amino acid residues in sequence alignments derived from closely related sequences; UMD – Universal Mutation Database, LOVD – Leiden Open Variation Database, IARC – International Agency for Research on Cancer, UV – unclassified variant. COSMIC – Catalogue Of Somatic Mutations In Cancer; FATHMM – Functional Analysis Through Hidden Markov Models.
Table 2. Segregation analysis of the tested variants. DX – cancer diagnosed at age, * year of born, † – deceased at age.
Introducing sequence variants into exon 18
For each mutation, 2–4 clones having homologously integrated the targeting construct with the desired mutation were identified by PCR. Clones with the correct phase (incorporation of the mutation in the wild-type allele) were used for subsequent experiments. We failed to obtain viable cell clones with the variant c.7988A> G in the active allele, probably due to their detrimental phenotype.Citation14 We identified two clones with the presence of the targeting construct in the wild-type allele by PCR (); however, all these clones were lost in early passage.
Figure 1. Preparation of the targeting construct (a) and the studied variants in exon 18 (b). Panel A: Two genomic sequences – the upstream sequence encompassing the wild type exon 18 (left homology arm, LHA) and the downstream sequence in intron 18 (right homology arm, RHA) were amplified in two separate assemply PCR reactions together with the pNeDaKO plasmid to produce two partially overlapping fragments FP1 and FP2 of the targeting construct. The targeting construct was obtained as a fusion product FP3 in the third assembly PCR reaction. Panel B: The depicted missense variations were introduced to the LHA of the targeting construct ligated to the pAAV-MCS vector by site-directed mutagenesis. Panel C: Presence of mutation 7988A> G (1) and determination of „phase“ (rs397729406, wild type allele) of the same clone (2).
Cell proliferation after drug treatment
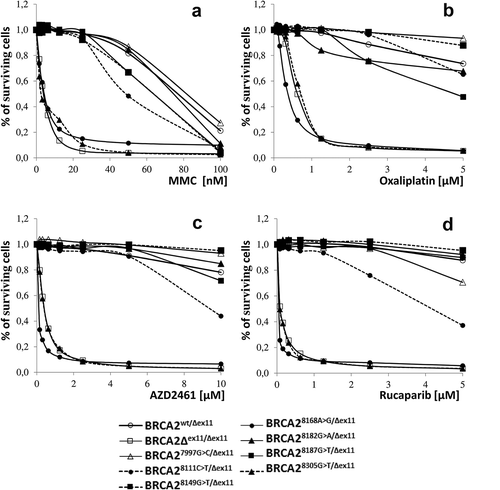
The cell line BRCA2wt/Δex11, used both as a positive control and the default cell line for targeted mutagenesis of exon 18 in the wild type allele, was verified to have intact BRCA2 protein function by demonstrating the absence of MMC hypersensitivity. Similarly, none of the tested cell lines BRCA27997G>C>/Δex11, BRCA28111C>T/Δex11, BRCA28149G>T/Δex11, BRCA28182G>A/Δex11, and BRCA28187G>T/Δex11 showed any differences in MMC sensitivity in comparison with the parental cell line BRCA2wt/Δex11 (). In contrast, the cell lines BRCA28168A>G/Δex11 and BRCA28305G>T/Δex11, the latter harboring a nonsense mutation p.Glu2769* in the active allele, were hypersensitive to MMC treatment (). Clinically more relevant drugs were also used. Similar results to MMC were seen upon exposure to oxaliplatin and PARP inhibitors AZD 2461 and rucaparib. In contrast, no difference in proliferation were found on treatment with gemcitabine ( and data not shown).
Table 3. Evaluation of variants in SyVaL. NA – not available.
Figure 2. Survival of BRCA2 syngeneic clones. Each panel compares survival of a clone carrying tested variant together with parental and knock-out cell lines (BRCA2wt/Δex11 and BRCA2Δex11/Δex11, respectively) in presence of a drug. Panel A: MMC. Panel B: oxaliplatin. Panel C: AZD 2461. Panel D: rucaparib. Values of surviving cell fraction were calculated as means of triplicates measured in different concentrations of MMC related to mean value of untreated cells (MMC = 0). Bars represent SEM.
RAD51 focus formation
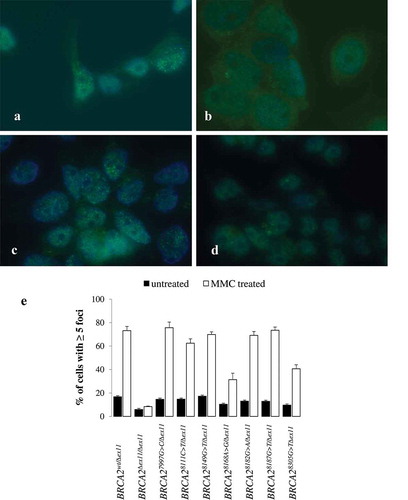
Treatment with MMC induced RAD51 foci (more than 5 foci/cell) in about 60–80% of BRCA2-proficient BRCA2wt/Δex11 cells. The BRCA2∆ex11/∆ex11 cells had considerably reduced levels of foci in the untreated state and no induction of foci was observed upon induction of DNA damage. The cell lines BRCA27997G>C/Δex11, BRCA28111C>T/Δex11, BRCA28149G>T/Δex11, BRCA28182G>A/Δex11, and BRCA28187G>T/Δex11 formed RAD51 foci upon MMC to a similar extent as did the parental cell line BRCA2wt/Δex11 (, ), whereas the cell lines BRCA28168A>G/Δex11 and BRCA28305G>T/Δex11 showed a significantly reduced amount of foci in comparison to the BRCA2wt/Δex11 cell line, although not to such an extent as the BRCA2∆ex11/∆ex11 negative control cell line.
Figure 3. Characterization of BRCA2 syngeneic clones by formation of RAD51 nuclear foci. Green immunofluorescence indicates RAD51 nuclear foci in MMC treated (2.4 μg/ml) parental cells (BRCA2wt/Δex11, panel a), BRCA2 knock-out cells (BRCA2Δex11/Δex11, panel b), cells with a functionally neutral variant in the active allele (BRCA28182A>G/Δex11, panel c) and cells carrying a nonsense variant in the active allele (BRCA28305G>T/Δex11. panel d). Relative counts of cells with nuclear RAD51 foci in untreated and MMC treated cells are shown in panel E (bars represent SEM).
Discussion
We previously developed a model in which human gene function could be evaluated based on comparative assessments of the functional impact of individual in-situ sequence alterations of one allele. Now we sought to apply this model to clinical practice. We constructed a syngeneic stable library of sequence variants of BRCA2 exon 18 identified in a genetic laboratory in Czech patients. This enabled us to functionally classify BRCA2 genomic sequence variants of unknown significance in a robust manner that may help to predict the cancer risk associated with presence of the same mutations in heterozygous patients.
The lack of understanding of the functional importance and associated cancer risk of sequence variants in the BRCA2 gene represents a serious clinical problem. The strongest interpretative evidence is inclined to come from segregation analyses and observed co-occurrence with known pathogenic mutations, both of which however require information that is often not availableCitation7. This is supported also by our data. Segregation analysis was possible or conclusive only in 3 out of 11 families. The most common reason for failure was impossibility of material collection. That was mostly caused by the fact that relatives who suffered from cancer were deceased at the time of analysis. Another important cause was unwillingness to provide genetic material for testing. Therefore, other methods of pathogenicity evaluation are urgently required. However, functional studies of human BRCA2 have proven very difficult. Previous cell-based in vitro assays depended upon expression of either partial or complete wild-type or mutant proteins. Testing of incomplete gene and protein sequences may not fully reveal the influence of the variant on the activity of the intact protein, and results coming from such experiments were not always confirmed using independent types of assayCitation15. The use of full-length wild-type or mutant constructs has been mostly limited to transient expression since it has proven difficult and at times impossible to generate stably expressing linesCitation11. Expression of exogenous BRCA2 including the BRC repeats may result in BRCA2 deficiency, perhaps due to squelching (titrating-out) of free RAD51Citation10-Citation12. Studies utilizing cells having endogenous wild-type BRCA2 alleles depend on the presumption that the co-dominant or dominant-negative effects of exogenous mutated constructs will govern the assay results. Thus, evaluating BRCA2 function utilizing exogenous expression constructs is of extreme technical and interpretational difficulty.
We present here a new approach to evaluate genomic variants by applying a homologous recombination replacement technique to produce syngeneic clones. This strategy was approximately as rapid as stable transfection of routine expression plasmids, a technique comprised of many invariant steps. Such approach allows an unambiguous read-out by generating multiple clones of stable cell lines having the endogenous gene altered. We applied a panel of functional assays to our syngeneic knock-out cell pair and selected two robust assays that we applied to our library of variants: RAD51 focus-formation and MMC sensitivity. The assays in combination provided a clear and unambiguous read-out of BRCA2 function.
Three of the seven tested variants we found in exon 18 – c.8182G> A, c.8187G> T, and c.8168A> G, were previously characterized by both computational methods and functional assays. Another three variants c.7997G> C, c.8111C> T and c.8149G> T, were as yet characterized only in-silico and one variant (c.7988A> G) has not been previously functionally characterized ().
In our functional assays, we showed no effect of MMC treatment on either survival or RAD51 focus formation in BRCA28182G>A/∆ex11, BRCA28187G>T/∆ex11, BRCA27997G>C/∆ex11, BRCA28111C>T/∆ex11, and BRCA28149G>T/∆ex11 cells and thus we classified them as neutral. In contrast, the cell line BRCA28168A>G/∆ex11 evinced impaired protein function in both our functional tests (). Our conclusion was further supported by obtaining similar proliferation differences upon treatment with oxaliplatin and PARP inhibitors, drugs that have been shown to induce selective hypersensitivity in BRCA2 mutated cells.Citation16
Our results do not fully match all previous predictions (). Interestingly, two of the variants that showed normal function in our assays had previously been predicted by some methods as damaging. The variant c.8149G> T was predicted as possibly damaging by the PolyPhen2, the VET (Variant Effect Predictor) and the FATHMM (Functional Analysis Through Hidden Markov Models) scores and the variant c.8187G> T was predicted harmful by the Align-Grantham Variation Grantham Deviation, PolyPhen2 and SIFT scores.Citation17-Citation19 We believe that our results, based on a human stable knock-out cancer cell model allowing for syngeneic controls and multiple tests evaluating various gene functions, are more reliable, relevant and feasible in clinical practice. The discrepancy highlights the importance of the use of multiple methods in evaluating mutation relevance, since neither method is fully reliable and infallible and only when combined, a more certain conclusion is possible.Citation20 Nonpathogenicity of these variants is further supported by the result of multifactorial likelihood ratio models that score these variants as as non-pathogenic.Citation21-Citation25
We failed in obtaining long-term viable cell lines carrying the unique variant c.7988A> G in the active (wild-type) allele, causing amino acid change from glutamic acid to glycine. This variant has never been characterized by computational or functional assays; however, according to prediction softwares (), this alteration seems to be important for the protein. Furthermore, a substitution for variant in the same position to valine (p.E2663V) is considered deleterious by both computer-based and functional tests.Citation23-Citation28
Inactivation of BRCA2 has been suggested to be predominantly selected against in cancer cells. Knock-out mice with BRCA2 null mutations are embryonic lethal and only some truncating mutations sparing BRC repeats have a viable phenotype. Lethality of BRCA2 bialelic inactivation is common in cell line models. Kuznetsov et al. (2008) performed their functional evaluation in a model, where bialelic BRCA2 inactivation was lethal and rescued only when a functional BRCA2 was expressed. Gallmeier et al. were unable to obtain a viable BRCA2 exon 11 knock-out clone, suggesting a detrimental phenotype.Citation14 Similarly to their study, all the viable clones that we obtained after several independent targeting rounds had integrated the targeting construct containing variant c.7899A> G in the already deleted allele. The only clones with the presence of the targeting construct in the wild-type allele identified by PCR were lost in early passage, supporting the assumption of a detrimental phenotype. Different severity of various pathogenic mutations has been shown in functional assays.Citation29 Murine and cancer risk models also suggest genotype-phenotype correlation.Citation30,Citation31 Thus, one possible explanation for the variant c.7899A> G causing cell lethality is that it stands at the more severe end of the mutation spectrum and does not allow cells to survive. Common artifacts such as sequence differences at the targeted locus or regional differences in chromatin that may be responsible for a failure in targeting the wild-type allele were excluded since we were able to obtain cells carrying other mutations including the truncating mutation c.8305G> T (p.Glu2769Ter) that leads to protein termination. The BRCA28305G>T/Δex11 cells showed increased sensitivity to MMC and impaired RAD51 focus formation similarly to the knock-out cell line BRCA2∆ex11/∆ex11 and served as a positive control.
In recent years, novel techniques of targeted genome editing have been developed. The CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) and CRISPR-associated (Cas) genes, which form an adaptive immune system in bacteria, have been modified for genome-editing protocols. Site-specific cleavage of double-stranded DNA results in activation of non-hemologous end joining, an error prone repair mechanism generating a diverse array of mutations, some of which disrupt the targeted gene.Citation32 Alternatively, if a homologous template is supplied, the break may be repaired by homology-directed repair, allowing for specific sequence variations to be made. The major limitation of our model, the low efficiency of homology directed repair, can be, in part, overcome with the CRISPR system. Multiple modifications, such as using mutaded Cas9 proteins having only nickase activity or the use of single stranded homology templates, greatly increase the efficiency and ease of the system,Citation33,Citation34 allowing for large scale missense variants generation. This has recently been shown for the BRCA1 gene.Citation35
In conclusion, our study introduces a novel method to predict the impact of BRCA2 variants of unknown significance via targeted variant insertion into the BRCA2 gene along with its first clinical application. We created a syngeneic variant library for naturally occurring BRCA2 variants found in real patients and evaluated them by functional assays. This method allowed a rapid assessment of pathogenicity of the variants and enabled in combination with other information patients’ counseling. It may serve as a method for investigating other human disease genes.
Materials and methods
Genotyping of high-risk individuals and segregation analysis
Genetic analysis of the BRCA2 gene in high-risk families was carried out by heteroduplex analysis (up to 2006) or High Resolution Melting analysis (from 2007) using LightScanner (Idaho Technology, Salt Lake City, UT) in the Masaryk Memorial Cancer Institute. Presence of mutation was confirmed by sequencing on 3130 Genetic Analyser (Thermo Fisher Scientific, Waltham, MA).
All sequence variants were named and are referred to in the text according to the nomenclature used by Human Genome Variation SocietyCitation36 recommendation guidelines, using the A of the ATG-translation initiation codon as nucleotide + 1Citation37 on the BRCA2 (Ref.Seq.: NM_000059.3). Variants listed in the dbSNP database are described using dbSNP identifiers whenever appropriate.
Selection of variants
For functional testing, we selected for testing variants in exon 18 (). The selected variants represented all mutations found in the Czech patients; however, their clinical impact would extend beyond the Czech Republic since most of them have been reported also from other regions of the world. In addition, a nonsense mutation c.8305G> T (p.Glu2769*) was introduced to serve as a positive control.
Cell lines
Cell lines with both heterozygous and homozygous deletion c.5324_5672del in exon 11 of BRCA2 were obtained from Scott E. Kern, Johns Hopkins University, Baltimore, MD. The AAV-293 cells were obtained as part of the AAV-helper free system kit (Agilent Technologies, cat. # 240073).
DNA isolation, PCR and sequencing
Genomic DNA from cells was isolated using QIAamp DNA Blood Mini Kit (Qiagen, cat. # 51304) following standard protocols. Sequencing was performed on 3130 Genetic Analyser (Thermo Fisher Scientific, Waltham, MA) using standard protocols.
Targeting construct preparation
The targeting construct (), containing the left homology arm (LHA), resistance genes for ZeocinCitation30 and neomycin,Citation38 and the right homology arm (RHA), was prepared according to Kohli et al.Citation39 with a few modifications.
The sequence of the LHA encompassed the wild type exon 18, whereas the sequence of the RHA was located approximately 80 bp downstream of the end of exon 18 (). The homology arms were amplified and assembled into the fusion product which was then ligated pAAV-MCS and further transformed as previously described.Citation39
Site-directed mutagenesis
Mutations were introduced into the targeting construct using the Platinum Taq DNA polymerase High Fidelity (Thermo Fisher Scientific, cat. # 11304011) or the QuickChange Site-Directed Mutagenesis Kit (Agilent Technologies, cat. # 200518). The PCR products were transformed colonies were screened for presence of the desired mutations.
Packaging of rAAV targeting construct, gene targeting of human cells and screening for targeting events
Equal amounts of the targeting construct, RC-plasmid, and pHelper (AAV-helper free system, cat. # 240071) were combined and transfected into AAV-293 cells at ~ 80% confluence in 75 cm2 flasks. Virus harvesting was performed after 48 hours according to Kohli et al.Citation39 and the virus was frozen at −80°C.
Heterozygous DLD-1 cells BRCA2wt/∆ex11 at 70–80% confluence were washed with PBS and infected as previously described.Citation39 After 48 hour incubation, the cells were trypsinized and transferred to geneticin-containing media (1 mg/ml, Sigma, cat. # G8168) and distributed to 96-well plates. Plates were wrapped with Saran foil and incubated for 2–3 weeks at 37°C. Only cells in wells containing single clones were trypsinized, transferred into new test plates and allowed to grow to full confluence for 4–5 days before DNA was obtained by cell lysis.Citation39
Locus-specific targeting events were screened by PCR using allele specific PCR (one primer situated within the targeting construct and the other outside of the targeted sequence). Presence of the correct size product on agarose electrophoresis was evaluated as a positive targeting event. Presence of the desired mutation in the allele containing the non-deleted exon 11 (active allele) was confirmed by sequencing.
Distinguishing the phase of the homologous recombination events relative to the allele inactivated by the deletion in exon 11
A previously described method for distinguishing the phase of the targeting consctruct and the deletion in exon 11 carrying allelesCitation13 was used. We found that the naturally occurring heterozygous polymorphism c.8331 + 1086_8331 + 1087insT (rs397729406) and the exon 11 deletion are located in cis whereas the rs397729406 wild-type allele is located in trans in the active allele. The phase of the recombined targeting construct was then determined by sequencing the PCR product encompassing the phase marker SNP rs397729406.
Functional analysis
Proliferation assays
Cells were seeded in density of ~ 1000 cells per well in 96-well plates, allowed to adhere and treated with mitomycin C (MMC, Sigma, cat. # M4287) in various concentration (0–100 nM). After 6 days, cells were washed with PBS and lysed with 100 µl of water. Then, 100 µl of 2x SYBR® Green I Nucleic Acid Gel Stain (Thermo Fisher Scientific, cat. # S7567) in TE buffer was added, cells were incubated in dark at 37°C for 10 min and then fluorescence at 485/520 nm was measured on Synergy 2 ™ Multi-Mode Microplate Reader (BioTek Instruments Inc., Winooski, VT).
Immunofluorescence
Cells were grown on cover slips in 6-well plates to a density of ~ 500 000 cells/well. After 24 h, cells were treated by MMC (2,4 µg/ml) or irradiation and were incubated at 37°C and 5% CO2 for the next 24 hours. Immunofluorescence staining of cells was performed according to Hucl et al.Citation13. Two hour incubation with primary Anti-Rad51 antibody (Santa Cruz Biotechnology, cat. # sc-8349,; diluted to 1:50 in blocking solution) followed by 1 hour staining with goat anti-rabbit IgG (H + L) cross-adsorbed secondary antibody, Alexa Fluor 488 (Thermo Fisher Scientific, cat. # A-11008; diluted to 1:300 in blocking solution) was used. Finally, cells were analyzed with a DM IRE2 fluorescent microscope (Leica Microcystems, Wetzlar, Germany).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The study was supported by IGA grant 10536-3.
Additional information
Funding
References
- Connor F, Bertwistle D, Mee PJ, Ross GM, Swift S, Grigorieva E, Tybulewicz VL, Ashworth A. Tumorigenesis and a DNA repair defect in mice with a truncating Brca2 mutation. Nat Genet. 1997;17:423–430. doi:10.1038/ng1297-423.
- Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell. 2001;7:263–272.
- Wong AK, Pero R, Ormonde PA, Tavtigian SV, Bartel PL. RAD51 interacts with the evolutionarily conserved BRC motifs in the human breast cancer susceptibility gene brca2. J Biol Chem. 1997;272:31941–31944.
- Sharan SK, Morimatsu M, Albrecht U, Lim DS, Regel E, Dinh C, Sands A, Eichele G, Hasty P, Bradley A. Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature. 1997;386:804–810. doi:10.1038/386804a0.
- Goggins M, Schutte M, Lu J, Moskaluk CA, Weinstein CL, Petersen GM, Yeo CJ, Jackson CE, Lynch HT, Hruban RH, et al. Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Res. 1996;56:5360–5364.
- King MC, Marks JH, Mandell JB. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science. 2003;302:643–646. doi:10.1126/science.1088759.
- Frank TS, Deffenbaugh AM, Reid JE, Hulick M, Ward BE, Lingenfelter B, Gumpper KL, Scholl T, Tavtigian SV, Pruss DR, et al. Clinical characteristics of individuals with germline mutations in BRCA1 and BRCA2: analysis of 10,000 individuals. J Clin Oncol. 2002;20:1480–1490. doi:10.1200/JCO.2002.20.6.1480.
- Gallmeier E, Kern SE. Targeting fanconi anemia/BRCA2 pathway defects in cancer: the significance of preclinical pharmacogenomic models. Clin Cancer Res. 2007;13:4–10. doi:10.1158/1078-0432.CCR-06-1637.
- Wu K, Hinson SR, Ohashi A, Farrugia D, Wendt P, Tavtigian SV, Deffenbaugh A, Goldgar D, Couch FJ. Functional evaluation and cancer risk assessment of BRCA2 unclassified variants. Cancer Res. 2005;65:417–426.
- Davies AA, Masson JY, McIlwraith MJ, Stasiak AZ, Stasiak A, Venkitaraman AR, West SC. Role of BRCA2 in control of the RAD51 recombination and DNA repair protein. Mol Cell. 2001;7:273–282.
- Esashi F, Christ N, Gannon J, Liu Y, Hunt T, Jasin M, West SC. CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature. 2005;434:598–604. doi:10.1038/nature03404.
- Yuan SS, Lee SY, Chen G, Song M, Tomlinson GE, Lee EY. BRCA2 is required for ionizing radiation-induced assembly of Rad51 complex in vivo. Cancer Res. 1999;59:3547–3551.
- Hucl T, Rago C, Gallmeier E, Brody JR, Gorospe M, Kern SE. A syngeneic variance library for functional annotation of human variation: application to BRCA2. Cancer Res. 2008;68:5023–5030. doi:10.1158/0008-5472.CAN-07-6189.
- Gallmeier E, Hucl T, Calhoun ES, Cunningham SC, Bunz F, Brody JR, Kern SE. Gene-specific selection against experimental fanconi anemia gene inactivation in human cancer. Cancer Biol Ther. 2007;6:654–660.
- Goldgar DE, Easton DF, Deffenbaugh AM, Monteiro AN, Tavtigian SV, Couch FJ. Integrated evaluation of DNA sequence variants of unknown clinical significance: application to BRCA1 and BRCA2. Am J Hum Genet. 2004;75:535–544. doi:10.1086/424388.
- Pishvaian MJ, Biankin AV, Bailey P, Chang DK, Laheru D, Wolfgang CL, Brody JR. BRCA2 secondary mutation-mediated resistance to platinum and PARP inhibitor-based therapy in pancreatic cancer. Br J Cancer. 2017;116:1021–1026. doi:10.1038/bjc.2017.40.
- Sorting intolerant from tolerant. 2. http://sift.bii.a-star.edu.sg.
- PolyPhen-2. 3. http://genetics.bwh.harvard.edu/pph2/.
- Align GVGD. 1. http://www.hgvd.genome.med.kyoto-u.ac.jp/.
- Lindor NM, Goldgar DE, Tavtigian SV, Plon SE, Couch FJ. BRCA1/2 sequence variants of uncertain significance: a primer for providers to assist in discussions and in medical management. Oncologist. 2013;18:518–524. doi:10.1634/theoncologist.2012-0452.
- Spurdle AB, Healey S, Devereau A, Hogervorst FB, Monteiro AN, Nathanson KL, Radice P, Stoppa-Lyonnet D, Tavtigian S, Wappenschmidt B, et al. ENIGMA–evidence-based network for the interpretation of germline mutant alleles: an international initiative to evaluate risk and clinical significance associated with sequence variation in BRCA1 and BRCA2 genes. Hum Mutat. 2012;33:2–7. doi:10.1002/humu.21628.
- Lindor NM, Guidugli L, Wang X, Vallee MP, Monteiro AN, Tavtigian S, Goldgar DE, Couch FJ. A review of a multifactorial probability-based model for classification of BRCA1 and BRCA2 variants of uncertain significance (VUS). Hum Mutat. 2012;33:8–21. doi:10.1002/humu.21627.
- Karchin R, Agarwal M, Sali A, Couch F, Beattie MS. Classifying variants of undetermined significance in BRCA2 with protein likelihood ratios. Cancer Inform. 2008;6:203–216.
- Farrugia DJ, Agarwal MK, Pankratz VS, Deffenbaugh AM, Pruss D, Frye C, Wadum L, Johnson K, Mentlick J, Tavtigian SV, et al. Functional assays for classification of BRCA2 variants of uncertain significance. Cancer Res. 2008;68:3523–3531. doi:10.1158/0008-5472.CAN-07-1587.
- Easton DF, Deffenbaugh AM, Pruss D, Frye C, Wenstrup RJ, Allen-Brady K, Tavtigian SV, Monteiro ANA, Iversen ES, Couch FJ, et al. A systematic genetic assessment of 1,433 sequence variants of unknown clinical significance in the BRCA1 and BRCA2 breast cancer-predisposition genes. Am J Hum Genet. 2007;81:873–883. doi:10.1086/521032.
- Chenevix-Trench G, Healey S, Lakhani S, Waring P, Cummings M, Brinkworth R, Deffenbaugh AM, Burbidge LA, Pruss D, Judkins T, et al. Genetic and histopathologic evaluation of BRCA1 and BRCA2 DNA sequence variants of unknown clinical significance. Cancer Res. 2006;66:2019–2027. doi:10.1158/0008-5472.CAN-05-3546.
- Kuznetsov SG, Liu P, Sharan SK. Mouse embryonic stem cell-based functional assay to evaluate mutations in BRCA2. Nat Med. 2008;14:875–881. doi:10.1038/nm.1719.
- Walker LC, Whiley PJ, Couch FJ, Farrugia DJ, Healey S, Eccles DM, Lin F, Butler SA, Goff SA, Thompson BA, et al. Detection of splicing aberrations caused by BRCA1 and BRCA2 sequence variants encoding missense substitutions: implications for prediction of pathogenicity. Hum Mutat. 2010;31:E1484–505. doi:10.1002/humu.21267.
- Guidugli L, Pankratz VS, Singh N, Thompson J, Erding CA, Engel C, Schmutzler R, Domchek S, Nathanson K, Radice P, et al. A classification model for BRCA2 DNA binding domain missense variants based on homology-directed repair activity. Cancer Res. 2013;73:265–275. doi:10.1158/0008-5472.CAN-12-2081.
- Rebbeck TR, Mitra N, Wan F, Sinilnikova OM, Healey S, McGuffog L, Mazoyer S, Chenevix-Trench G, Easton DF, Antoniou AC, et al. Association of type and location of BRCA1 and BRCA2 mutations with risk of breast and ovarian cancer. JAMA. 2015;313:1347–1361. doi:10.1001/jama.2014.5985.
- Evers B, Jonkers J. Mouse models of BRCA1 and BRCA2 deficiency: past lessons, current understanding and future prospects. Oncogene. 2006;25:5885–5897. doi:10.1038/sj.onc.1209871.
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi:10.1126/science.1225829.
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi:10.1126/science.1231143.
- Jacobi AM, Rettig GR, Turk R, Collingwood MA, Zeiner SA, Quadros RM, Harms DW, Bonthuis PJ, Gregg C, Ohtsuka M, et al. Simplified CRISPR tools for efficient genome editing and streamlined protocols for their delivery into mammalian cells and mouse zygotes. Methods. 2017;121-122:16–28. doi:10.1016/j.ymeth.2017.03.021.
- Findlay GM, Daza RM, Martin B, Zhang MD, Leith AP, Gasperini M, Janizek JD, Huang X, Starita LM, Shendure J. Accurate classification of BRCA1 variants with saturation genome editing. Nature. 2018;562:217–222. doi:10.1038/s41586-018-0461-z.
- Human Genom Variation Society. http://www.hgvs.org.
- Den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat. 2000;15:7–12. doi:10.1002/(SICI)1098-1004(200001)15:1<7::AID-HUMU4>3.0.CO;2-N.
- Nagasawa JY, Song J, Chen H, Kim HW, Blazel J, Ouk S, Groschel B, Borges V, Ong V, Yeh L-T, et al. 6-Benzylamino 4-oxo-1,4-dihydro-1,8-naphthyridines and 4-oxo-1,4-dihydroquinolines as HIV integrase inhibitors. Bioorg Med Chem Lett. 2011;21:760–763. doi:10.1016/j.bmcl.2010.11.108.
- Kohli M, Rago C, Lengauer C, Kinzler KW, Vogelstein B. Facile methods for generating human somatic cell gene knockouts using recombinant adeno-associated viruses. Nucleic Acids Res. 2004;32:e3. doi:10.1093/nar/gnh009.