
ABSTRACT
Glioblastoma is a highly malignant and typically fatal tumor of the central nervous system. The tumor is characterized by marked cellular and molecular heterogeneity, including a subpopulation of brain tumor initiating cells (BTICs) that are highly resistant to radiation and chemotherapy. We previously reported that the RNA-binding protein HuR is: (1) overexpressed in glioblastoma, (2) necessary for tumor growth in vivo, and (3) a positive regulator of tumor-promoting genes in glioblastoma. These findings provide strong evidence that HuR might be a viable therapeutic target in glioblastoma. In this report, we investigated the effects of MS-444, a small molecule inhibitor of HuR, in xenograft-derived human glioblastoma cells and BTICs. We found that MS-444 treatment of glioblastoma cells resulted in loss of viability and induction of apoptosis, with evidence implicating death receptor 5. BTICs were particularly sensitive to MS-444. At sub-lethal doses, MS-444 attenuated invasion of glioblastoma cells and BTICs in a transwell model. At the molecular level, MS-444 treatment led to an attenuation of mRNAs in different tumor promoting pathways including angiogenesis, immune evasion and suppression of apoptosis. Although cytoplasmic HuR was reduced with MS-444 treatment, the attenuation of mRNAs could not be explained by RNA destabilization. In summary, this report provides proof of concept that small molecule inhibition of HuR could be a viable approach for treatment of glioblastoma.
Introduction
Glioblastoma is the most common primary malignant brain tumor and continues to carry a very poor prognosis with a median survival of ~20 monthsCitation1,Citation2 Challenges in treating glioblastoma center around its molecular and cellular heterogeneity, including the presence of small populations of brain tumor initiating cells (BTICs) that are typically resistant to standard treatment regimens.Citation3,Citation4 This complexity necessitates the development of therapies for targets that intersect multiple tumor-promoting pathways.Citation5 HuR, an RNA binding protein, is one potential target as it controls the expression of many genes regulating these pathways including anti-apoptotic (e.g. Bcl2, Bcl-xL, and cIAP2), pro-angiogenic (e.g. VEGF and COX-2), and pro-proliferation (e.g. cyclins D1, E and A2) factors.Citation6-Citation10 Previously, we showed that HuR is overexpressed in glioblastoma and its silencing promotes cytotoxicity invitro and suppresses tumor growth invivo.Citation9,Citation11 On the other hand, overexpression of HuR in gliomas enhances expression of VEGF, TNF-α, and IL-8 and promotes chemoresistance invitro.Citation9,Citation10 This background builds a strong case for developing small molecule inhibitors of HuR as a therapeutic approach to glioblastoma and other cancers. MS-444 is a small molecule inhibitor of HuR first discovered in a high throughput screen using a fluorescence-based RNA binding assay.Citation12 MS-444 inhibits HuR dimerization, a process necessary for its RNA binding and cytoplasmic translocation.Citation12 In colon and pancreatic cancer models, MS-444 produces a cytotoxic effect.Citation13,Citation14 Here we investigated the effect of MS-444 on human glioblastoma cells in vitro and found a cytotoxic effect on the whole cell population as well as BTICs. There was induction of death receptor 5 (DR5) and cleavage of caspase -8 and downstream caspase -3. Tumor cell invasion was significantly inhibited by MS-444. At the molecular level, there was a significant suppression of key genes that promote glioma progression through different pathways. RNA stability did not appear to be the mechanism for this attenuation. Our findings should prompt further investigation of MS-444 as a potential therapeutic agent in this devastating cancer.
Results
MS-444 is cytotoxic to glioblastoma cells
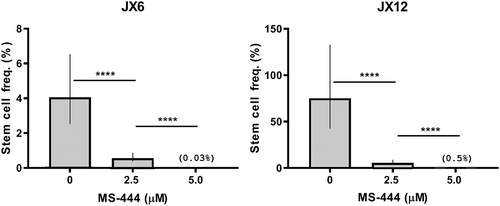
We have previously shown that HuR is strongly expressed in glioblastoma and that genetic silencing leads to attenuation of cell growth and sensitization to standard chemotherapies.Citation9,Citation11 Based on these findings, we hypothesized that chemical inhibition of HuR would produce an anti-cancer effect in glioblastoma. We focused on MS-444, a small molecule that inhibits HuR by preventing its exit from the nucleus and binding to target mRNAs.Citation12 We performed cell viability assays using three human-derived glioblastoma xenolines, JX6, JX12 and X1066, and observed significant toxicity, with an IC50 of ~31 to 63 µM in bulk tumor cells (–C). Primary astrocytes showed no toxicity in the same dose range (), indicating the potential for a large therapeutic window for HuR inhibition. Knockdown of HuR by siRNA also produced a significant attenuation of proliferation in both xenolines (Supplemental Fig, 1). In contrast, cells enriched for BTICs via CD133 sorting had substantially lower IC50 values (3–13 µM; –C), suggesting that BTICs would not be more resistant to HuR-based inhibitory approaches than the majority of glioblastoma cells. Untreated BTICS sorted from JX6 and JX12 cells demonstrated robust proliferative patterns (Supplemental ). There was a modest increase in proliferation with JX12 cells (BTICs and unsorted cells) compared to JX6. Using neurosphere formation as an assessment of a functional phenotype of BTICs, we confirmed by a limiting dilution assay that 2.5 µM was sufficient to attenuate neurosphere formation in both xenolines while 5 µM essentially abrogated it (). There was a marked increase in the estimated stem cell frequency in JX12 versus JX6 cells which may have contributed to differences in proliferation observed in Supplemental .
Figure 1. MS-444 produces cytotoxicity in glioblastoma xenolines and the subpopulation of CD133+ cells. (A–C) glioblastoma xenolines (parent and the CD133+ fraction) were plated and treated with different doses of MS-444 or vehicle for 24 h. Viability was determined by a luciferase assay as described in the Methods section. (D) Primary cortical astrocytes were treated with MS-444 and viability was similarly determined. Data points represent the mean ± SD of three independent tests.
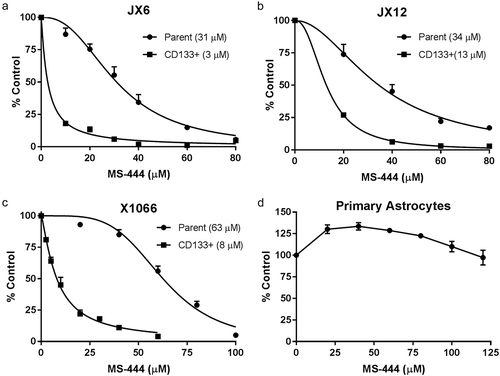
Figure 2. MS-444 inhibits neurosphere formation. Single cell suspensions of glioblastoma xenolines were plated at different densities and treated with MS-444. After 14 days, sphere formation was quantitated. Log-fraction curves were generated from 10 replicate wells per dose of MS-444 to estimate stem cell frequency using the Extreme Limiting Dilution Analysis tool.Citation15 Confidence intervals (95%) are shown. ****P < 1e-8.
MS-444 induces apoptosis in glioblastoma cells
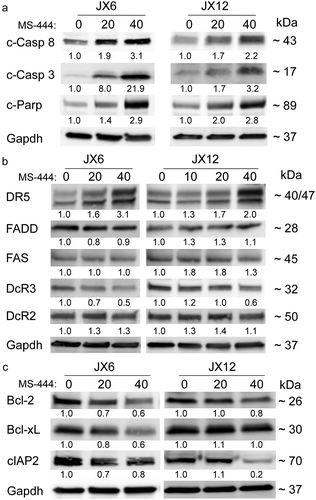
We next determined the underlying basis for the loss of viability with MS-444 treatment, in glioblastoma cells. After 24 h of treatment, we observed a strong dose-dependent increase in activated caspase -8 for both glioblastoma lines tested (). These findings are consistent with activation of the extrinsic apoptotic pathway. MS-444 treatment of BTICs induced activation of caspase 8 at lower doses compared to unsorted cells as determined by ELISA (Supplemental ). Cleaved caspase -3 and cPARP increased in a similar pattern, confirming apoptosis activation in glioblastoma cells upon MS-444 treatment (). To further characterize activation of the extrinsic pathway, we assessed key components including death receptor 5 (DR5) and decoy receptor 3 (DcR3), which are predominantly expressed in most glioblastomas ().Citation16-Citation18 We observed a dose-dependent increase in DR5 in both glioblastoma xenolines, whereas DcR3 was attenuated at similar doses. There was no clear change in FADD, Fas or DcR2. The anti-apoptotic factors Bcl-2 and Bcl-xL were attenuated inJX6 cells whereas in JX12 cells they were unaltered (). On the other hand, cIAP2 was markedly diminished in JX12 cells but only minimally decreased in JX6 cells. Taken together, these data indicate that MS-444 produces a dose-dependent cytotoxic effect on glioblastoma cells with evidence of apoptosis, in part through activation of caspases 3 and 8.
Figure 3. MS-444 induces apoptosis and suppression of anti-apoptotic factors in glioblastoma xenolines. (A–C) western blots of glioblastoma xenolines after treatment with MS-444 (24 h) at the doses shown. Antibodies are shown to the left and approximate band size to the right. Band intensity was determined by densitometry, adjusting for GAPDH intensity. Values are shown below each band relative to untreated cells (set at a value of 1).
Silencing DR5 partially rescues the cytotoxicity of MS-444
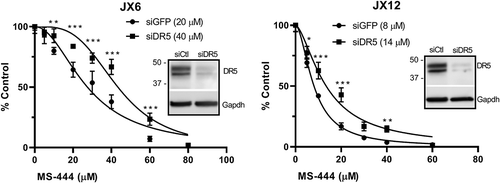
To determine the impact of DR5 on MS-444 induced cytotoxicity, we silenced DR5 with siRNA in the glioblastoma xenolines and then treated them with MS-444 (). We observed excellent suppression of DR5 protein compared to control siRNA-transfected cells. Loss of DR5 led to an increase in the IC50 for both xenolines (JX6, 20 to 40 µM; Jx12, 8 to 14 µM) indicating a partial rescue effect at each dose of MS-444. A second set of DR5 siRNAs showed a similar rescue effect (Supplemental ). Overall, our findings indicate that DR5 participates in the cytotoxic effects of MS-444.
Figure 4. MS-444 induced cytotoxicity is partially reversed with DR5 knockdown. Glioblastoma xenolines were transfected with siRNA targeting DR5 or a control siRNA followed by treatment with MS-444 for 24 h. Viability was determined by the Vialight luciferase assay. Data points represent three independent tests ± SD. *P < 0.05; **P < 0.01; ***P < 0.001.
MS-444 impairs glioblastoma cell invasion
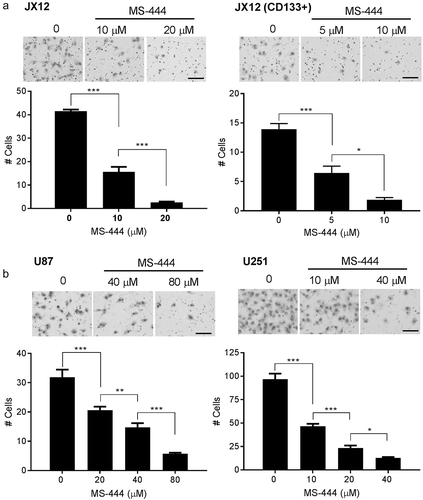
We previously observed that shRNA-induced silencing of HuR in glioblastoma cells attenuated invasive properties of the tumor in vivo, prompting us to examine the effects of MS-444 on invasion.Citation9 Using a matrigel-based transwell model, we assessed invasion by seeding glioblastoma cells in the top well which was pre-coated with matrigel. Serum was used as a chemoattractant in the bottom well. We observed robust invasion of JX12 cells at baseline, but this was significantly inhibited by MS-444 at doses of 10 and 20 µm which are well below the IC50 of 34 µm (). There was a 95% reduction in cells crossing into the lower chamber at a dose of 20 µM. Invasive properties of the CD133+ subpopulation were more sensitive to MS-444, showing nearly an 80% reduction at 10 μM . To mitigate further against the possibility that loss of invasive properties was related to cell death, we performed the assay at shorter time intervals of treatment (6 and 12 hours) and observed a similar attenuation of invasion (Supplemental ). We tested two additional malignant glioma cell lines, U87 and U251 (), and detected a similar dose-dependent attenuation of invasion using sublethal doses of MS-444 (the IC50s for these lines were substantially higher than the xenolines – data not shown). At the highest dose, there was an 80–90% reduction in invading cells. Migration of primary astrocytes, on the other hand, was unaffected by MS-444 at similar doses (Supplemental ). Taken together, these data indicate that MS-444 suppressed the invasive properties of glioblastoma cells.
Figure 5. MS-444 inhibits glioblastoma cell invasion. (A and B) glioblastoma cells were seeded in the top well of a matrigel-based transwell system with MS-444 at the dose indicated. Media supplemented 10% fetal calf serum was added to the bottom well as a chemoattractant. At 24 h, migrated cells were stained and quantitated using a microscope. Size bar = 100 µm. All data points are the mean ± SD of 3 independent tests. *P < 0.05; **P < 0.01; *** P < 0.001.
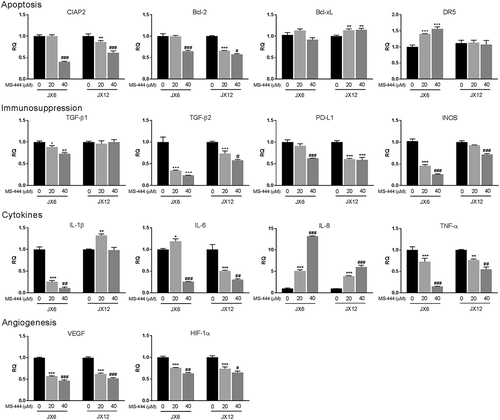
MS4-444 reduces expression of genes known to impact glioblastoma growth
To address potential mechanisms for MS-444-induced glioma toxicity, we evaluated expression levels of select mRNAs that are relevant to glioblastoma or known to be regulated by HuR in glioblastoma and other cancer cells ().Citation8-Citation10,Citation19-Citation23 Many of these mRNAs are also regulated by HuR in primary cells.Citation19,Citation24,Citation25 Using doses up to the IC50, we found that anti-apoptotic genes Bcl-2 and cIAP2 decreased significantly in a dose-dependent manner whereas Bcl-xL was either not affected (JX6) or modestly increased (JX12). DR5 mRNA was upregulated in JX6 but not JX12 cells even though DR5 protein increased in JX12 at similar doses of MS-444 (). Angiogenic factors VEGF and HIF1A decreased by ~40% at 40 µM. Immunosuppressive factors PD-L1, INOS, TGF-β1 (JX6 only), TGF-β2, and INOS, were all significantly suppressed, particularly in JX6 cells by as much as 75%. IL-1β (JX6 only), IL-6 and TNF-α, key cytokines supporting many aspects of glioma progression also significantly declined up to 70–80%. Surprisingly, IL-8 increased by nearly 13-fold at a dose of 40 µM in both lines. Although in general MS-444 produced consistent effects on gene expression, the responses of several mRNA targets including Bcl-xL, IL-1β, TGF-β1, and DR5 were different in the two xenolines. This discrepancy likely reflects the heterogeneity of glioblastoma and the variable dependence of these mRNAs on HuR.
Figure 6. MS-444 suppresses expression of genes promoting glioblastoma progression. Glioblastoma xenolines were treated for 24 h with MS-444 at the doses indicated. RNA levels were assessed by qPCR. All data points are the mean ± SD of three independent tests. Comparing 20 µM versus vehicle treated cells, P values are: * < 0.05; **, <0.01; ***, <0.001. Comparing 40 µM versus 20 µM, P values are: # < 0.05; ##, <0.01; ###, <0.001.
MS-444 attenuates cytoplasmic HuR but not mRNA stability
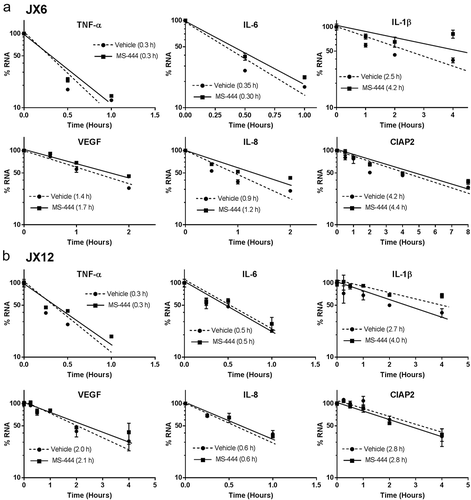
To confirm that MS-444 inhibits HuR translocation to the cytoplasm as previously determined in other cell types,Citation12 glioblastoma cells were treated with MS-444 for 24 h followed by preparation of nuclear and cytoplasmic lysates. We observed an attenuation of cytoplasmic HuR in both glioblastoma xenolines (). Nuclear and cytoplasmic controls (lamin and GAPDH respectively) showed equal loading and no cross-contamination of cellular compartments. Since one of the roles of cytoplasmic HuR is generally to promote mRNA stability, we determined whether the half-life was altered in a subset of mRNAs altered by MS-444. Cells were pulsed with actinomycin D and mRNA half-lives were estimated after treatment with MS-444 (20 µM). Degradation curves for nearly all mRNAs tested completely overlapped with vehicle-treated cells indicating no change in half-life (). This pattern included mRNAs that were significantly reduced (TNF-α, IL-6, VEGF and cIAP2) or IL-8 which was increased. For IL-1β and IL-8 (JX6 cells), there was even a prolongation of half-life with MS-444 treatment). Taken together, these findings suggest that changes in mRNA expression following MS-444 treatment were due to transcriptional effects as seen previously with siRNA-mediated suppression of HuR.Citation24,Citation25
Figure 7. MS-444 attenuates cytoplasmic HuR. Western blot analysis of nuclear and cytoplasmic extracts from glioblastoma xenolines treated with MS-444 for 24 h at the doses shown. Antibodies are shown to the left. This experiment was repeated one time with similar results.
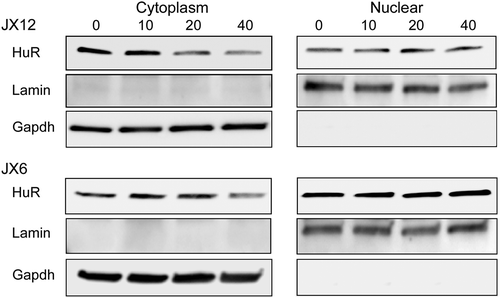
Figure 8. MS-444 does not affect mRNA half-life. Glioblastoma xenolines were treated with MS-444 for 24 h at 20 µM and then pulsed with actinomycin D. Target mRNAs were quantified at different time intervals to determine decay curves as shown. The RNA half-life was estimated from the point where 50% of the mRNA was remaining. Data points represent the mean ± SD of three independent samples.
Discussion
In this report, we demonstrate that the HuR inhibitor, MS-444, is cytotoxic to glioblastoma cells and especially BTICs, the subpopulation that drives tumor maintenance and treatment resistance in glioblastoma. We show evidence that apoptosis is induced by MS-444 treatment and that the invasive properties of glioblastoma cells and BTICs are inhibited. MS-444 reduces cytoplasmic HuR and leads to the suppression of mRNAs promoting glioma progression. Surprisingly this suppression is not linked to RNA destabilization and suggests an effect on transcription.
This work underscores our previous observations that HuR plays a key role in glioblastoma progression and demonstrates that HuR is a potentially druggable target in this devastating cancer.Citation9-Citation11,Citation26,Citation27 The much greater sensitivity of BTICs to MS-444, showing total abrogation of neurosphere formation at 5 µM (), was surprising and bolsters the rationale for developing this compound or other HuR inhibitors for glioblastoma treatment. BTICs represent a relatively small subpopulation of glioma cells that are important drivers of tumor progression and treatment resistance.Citation28-Citation30 The reason for enhanced sensitivity of BTICs to MS-444 is unclear but may be related to several key mRNA targets attenuated by MS-444 treatment. IL-6, for example, was suppressed by up to 75%. Acting via autocrine and paracrine pathways, this cytokine plays an integral role in activation of signal transducer and activator of transcription 3 (STAT3) which is required for maintenance of BTICs.Citation31-Citation33 Through p38 MAPK, HuR plays an important role in augmenting IL-6 production in glioblastoma cells.Citation34 Inducible nitric oxide synthase (INOS), hypoxia inducible factor 1α (HIF1α), TGF-β1 and TGF-β2, all of which were suppressed with MS-444 treatment (), participate in BTIC maintenance as well.Citation35-Citation37 TGF-β2 is the highest expressing TGF isoform in glioblastoma, particularly BTICs, and its level correlates with pathological grade.Citation36,Citation38 Its positive effect on maintaining stemness occurs through an autocrine signaling pathway.
Cytotoxicity with induction of apoptosis was a major effect of MS-444 similar to what we observed with shRNA-based HuR silencing.Citation9 Our finding in this report of cleaved caspase-8 implicates the extrinsic apoptotic pathway as part of the cytotoxic effect of MS-444. In this pathway, DR5, a key component, associates with Fas-associated death domain (FADD) adapter protein which binds to pro-caspase-8 to form the death-inducing signaling complex (DISC) which cleaves and activates caspase-8.Citation17,Citation39 DR5 is the predominant death receptor in glioblastoma where it is expressed consistently in primary tumors and cultured cells.Citation16,Citation17 Apoptosis is then initiated by downstream caspases activated by caspase -8 (e.g. caspase -3). This pathway was likely accentuated by the upregulation of DR5 after MS-444 treatment ( and ). In pancreatic cancer cells, HuR was shown to bind the 5ʹ untranslated region of DR5 mRNA and repress its translation.Citation23 MS-444 also promotes TRAIL sensitivity in these cancer cells by increasing DR4 and DR5 cell surface levels.Citation14 Since MS-444 reduced cytoplasmic HuR in glioblastoma cells () and is known to inhibit HuR-mRNA target binding,Citation12 the increase in DR5 protein expression may stem from translational de-repression, especially in JX12 cells where mRNA levels were unaffected by MS-444 (). It is also possible that this effect was indirect through altered expression of another HuR-regulated factor or pathway. On the other hand, decoy receptor 3 (DcR3), which is amplified and expressed in the majority of glioblastomas, was attenuated with MS-444 treatment ().Citation18,Citation40 Since DcR3 antagonizes Fas-mediated apoptosis, this attenuation might lead to an increase in apoptosis through that pathway. Activated caspase-8 can also promote mitochondrial (intrinsic pathway) contributions to apoptosis through activation of BID which binds and neutralizes anti-apoptotic factors Bcl-2 and Bcl-xL.Citation41,Citation42 This pathway would be accentuated in JX6 cells because these anti-apoptotic factors were attenuated by MS-444 (), which is consistent with our prior observations using shRNA to silence HuR.Citation9 Survival signaling in JX12 cells may additionally be hampered through loss of cIAP2 following MS-444 treatment (). cIAP2 is frequently amplified in glioblastoma and promotes apoptosis resistance in glioblastoma through NF-ĸB mediated survival pathways.Citation42,Citation43 Lastly, MS-444 attenuated key factors that enable glioblastoma to evade the cytotoxic effects of the immune system which could potentiate an anti-tumor effect in vivo. PD-L1, attenuated by ~40% after MS-444 treatment, suppresses T cell activation and reduces immune cell infiltration into the tumor.Citation44 The mRNA of this gene is regulated posttranscriptionally through AU-rich RNA binding motifs in the 3ʹ UTR and thus may be a direct target of HuR.Citation45 TGF-β, particularly the β2 isoform which was reduced by 40–75% at the mRNA level, suppresses cytotoxic T cell activation and recognition of glioblastoma cells while at the same time promoting immunosuppressive IL-10 expression by macrophages in the tumor microenvironment.Citation36 INOS, which was downregulated in both xenolines lines, has also been linked to immunosuppression in glioblastoma.Citation35
The other prominent cellular phenotype observed with MS-444 treatment, was decreased invasion. This effect was observed by our group with shRNA-induced HuR silencing in glioblastoma using an intracranial model.Citation9 In that report, there was a marked loss of tumor invasion and migration into local structures and the contralateral hemisphere. Glioblastoma progression is linked to migration of cancer cells, particularly BTICs, into the surrounding environment and along blood vessels.Citation37,Citation46 BTIC invasion was especially sensitive to MS-444 (), underscoring the therapeutic potential of this inhibitor. Defects in invasion have been observed in other cancer cells, including lung, breast and melanoma, when HuR was either chemically inhibited for silenced by interfering RNAs.Citation47-Citation49 The underlying basis for these defects could be multi-factorial in light of the broad number of mRNA targets that were attenuated. HIF1α, INOS, TNF-α and IL-6, for example, all play a key role in glioma invasion and migration, including BTICs and glioma tumor cells.Citation34,Citation35,Citation37,Citation50
The molecular response to MS-444 treatment was mainly suppression of HuR-regulated targets. Bcl-xL mRNA, however, did not change and actually increased in JX12 cells whereas Bcl-2 mRNA was significantly suppressed. This pattern is similar to what we reported in shRNA-induced silencing in glioblastoma cells.Citation9 The loss of Bcl-xL protein expression in JX6 cells () therefore suggests a translational effect of MS-444. Interestingly, we observed a 5–12 fold induction of IL-8 in both glioblastoma xenolines following MS-444 treatment (). Since HuR is a positive regulator of IL-8 expression in glioblastoma and colorectal cancer cells,Citation8,Citation10 this finding was unexpected. A prior report found a robust induction of IL-8 following ligation of DR5 receptor and caspase -8 activation.Citation51 This response was mediated by activation of AP-1 and NF-ĸB. Our finding that the IL-8 mRNA half-life did not change with MS-444 treatment is consistent with a transcriptional basis for this induction. Many mRNAs, however, including IL-1β, IL-6, INOS, and TNF-α, are induced by similar transcription factors but were suppressed here by MS-444 and did not have a decrease in RNA stability. We previously observed a similar pattern of mRNA suppression without change in half-life when HuR was knocked down in primary microglia and astrocytes.Citation24,Citation25 In those reports, a negative effect on transcription was demonstrated using reporter assays, with NF-ĸB elements possibly being involved. The effect on transcription may stem from altered transcription factor expression or localization. HuR, for example, has been shown to modulate transcriptional activity of NF-ĸB and retinoic acid receptor by regulating nuclear import of proteins.Citation52 Inhibition of HuR may also affect lncRNAs that are required for gene-specific transcription.Citation53 Studies are currently underway to identify the mechanism(s) of mRNA suppression induced by MS-444.
In conclusion, we have shown proof-of-concept that inhibition of HuR with MS-444 exerts anti-cancer effects in glioblastoma and merits further investigation with in vivo models. Little progress has been made in advancing therapies in glioblastoma as the median survival remains poor at ~20 months.Citation1,Citation2,Citation54 Treatment resistance results from the molecular and cellular heterogeneity of glioblastoma tumors, including BTICs, with multiple different survival pathways and their respective molecular drivers at play. Targeting HuR therapeutically in glioblastoma would represent a multi-pronged approach as it positively regulates many of these drivers.Citation55 The impact on multiple pathways is clearly reflected by the mRNA targets that were suppressed, including angiogenesis, stem cell maintenance, tumor cell migration/invasion and immunosuppression. It remains to be determined whether the extent of gene suppression observed in vitro will translate into phenotypic effects on these pathways in vivo. Our top priority is first to determine whether MS-444 crosses the blood brain barrier, and if so, to test it in xenograft models of glioblastoma.
Materials and methods
Cell culture and glioma stem-like cell isolation
JX6, JX12 and X1066 patient-derived glioblastoma xenografts (PDX) were acquired from the UAB Brain Tumor Core Facility, and propagated in the flanks of nude mice as described previously.Citation56 All animal procedures were reviewed and approved by the UAB Institutional Animal Care and Use Committee in compliance with the National Research Council Guide for the Care and Use of Laboratory Animals. Xenografts were dissociated into single cells using the Tumor Dissociation Kit (Miltenyi Biotec) according to manufacturer’s protocol. For cell culture, cells were grown in stem cell medium [Neurobasal™-A medium supplemented with 1% B-27™ minus vitamin A, 1% CTS™ N-2, 2 mM L-glutamine, 2.5 μg/mL Amphotericin B, 50 μg/mL Gentamicin, 10 ng/ml FGF, and 10 ng/ml EGF (Gibco)]. CD133+ BTICs were enriched using the CD133 MicroBead Kit (Miltenyi Biotec) and cultured in stem cell medium. Primary mouse astrocytes were isolated from C57BL/6J mice as described previously.Citation57
MS444 treatment and viability assay
MS-444 was kindly provided by Dr. Meisner-Kober (Novartis, Basel, Switzerland). MS-444 was reconstituted in DMSO at a concentration of 20 mM. Cells were seeded in 96-well plates at a density of 5,000 cells per well, and treated with different doses of MS-444 or DMSO control for 24 h. Cell viability was determined using the ViaLight™ Plus Cell Proliferation/Cytotoxicity Kit (Lonza).
RNA isolation, qRT-PCR and kinetics
Total RNA was extracted from cells using the illustra RNAspin Kit (GE Healthcare), and quantified by Nanodrop (Thermo Fisher Scientific). One microgram of RNA was reverse transcribed using the High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). Expression levels of mRNAs were measured by real time qPCR using predesigned TaqMan Gene Expression Assays (Thermo Fisher Scientific). For RNA kinetics, the cells were pretreated with MS444 or DMSO control for 24 hours. After adding Actinomycin D (MilliporeSigma) to the culture medium (10 μg/mL), cells were harvested at different intervals and target mRNA levels were quantitated by qPCR.
Protein extraction and western blot
Whole cell lysates were prepared using M-PER Mammalian Protein Extraction Reagent (Thermo Fisher Scientific) and quantified using the Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific). Cytoplasmic and Nuclear protein was extracted using the NE-PER™ Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific) according to the manufacturer’s protocol. Fifty micrograms of protein extract was denatured with Laemmli Sample Buffer (Bio-Rad Laboratories) at 95°C for 10 minutes and then resolved by electrophoresis using 4–20% Mini-PROTEAN® TGX™ Precast Protein Gels (Bio-Rad Laboratories), transferred to nitrocellulose membrane and probed with the following antibodies: Fas, DR5, FADD, Bcl-2, Bcl-xL, cleaved PARP, cleaved caspase-3, cleaved caspase-8, GAPDH (Cell Signaling Technology), HuR, Lamin A/C (Santa Cruz Biotechnology), cIAP2 (Abcam).
Limiting dilution assay
Single cell suspension of JX6 and JX12 cells were stained with the SYTOX™ Blue Dead Cell Stain (Thermo Fisher Scientific), and live cells were sorted using the BD FACS Aria cell sorter (Becton, Dickinson and Company) and seeded into 96-well plates. JX12 cells were seeded at densities of 100, 50, 20, 5, 1 cells per well, at 10 replicates for each density. JX6 cells were seeded at a higher density of 500, 250, 100, 50, 5 cells per well due to a lower percentage of BTICs. Cells were treated with MS-444 at different doses (0–20 μM) for 14 d, and tumor sphere formation was quantitated. BTIC frequency was calculated using the online Extreme Limiting Dilution Analysis tool.Citation15
Transwell invasion assay
The transwell invasion assay was performed using Corning® BioCoat™ Matrigel® Invasion Chambers with 8.0 μm membrane pores (Corning). A single cell suspension of 2.5 × 104 cells in stem cell medium were added to the top chamber and treated with different doses of MS-444 or DMSO control. Fetal bovine serum (10%) in DMEM/F-12 was added to the bottom chamber as a chemoattractant. Cells were allowed to migrate for 24 hours in the cell culture incubator, and then fixed and stained using the Hema 3™ Manual Staining System and Stat Pack (Thermo Fisher Scientific). The inserts were removed and mounted on microscope slides and stained cells. For quantitation, 30 images were captured (throughout the filter) for each well and stained cells were counted.
DR5 knockdown and rescue study
JX6 or JX12 cells at a density of 1 × 106 were transfected with 300 pmol of 4 pooled DR5 or HuR siRNAs from Dharmacon (ON-TARGETplus), siGFP control using Neon Transfection System (Thermo Fisher Scientific). A second set of 4 pooled DR5 siRNAs targeting different regions of the mRNA was purchased from Qiagen (GeneSolution siRNA). Seventy two hours after transfection, cells were seeded in 96-well plates and treated with different doses of MS-444 or DMSO control. Cell viability was measured using ViaLight™ kit as described previously.
Caspase-8 activity assay
caspase-8 activity was measured using the Caspase-Glo® 8 Assay System (Promega). Cells were seeded in 96-well plate and treated with different doses of MS444 or DMSO control, after 24 hours, cells were lysed and caspase-8 activity was measured by the luminescent signal generated by the cleavage of substrates.
Statistics
Statistical analyses were performed using Graphpad Prism 7 (Graphpad Software, Inc) except for the extreme limiting dilution assay for neurosphere frequencies where the significance between MS-444 dosing groups was determined by a Chi-squared test using an online analysis tool (http://bioinf.wehi.edu.au/software/elda/). Invasion, qRT-PCR, and ELISA assays were analyzed by one-way ANOVA with a Tukey’s multiple comparison test. Rescue studies were analyzed by a multiple t test comparison at different doses of MS-444 with significance determined using the Holm-Sidak method with alpha = 0.05.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplemental Material
Download MS Word (2.1 MB)Acknowledgments
We wish to acknowledge the UAB Comprehensive Flow Cytometry Facility (https://www.uab.edu/medicine/cfar/core-facilities/basic-research-core/flow-cytometry-core). We thank Dr. Yancey Gillespie and Ms. Catherine Langford for providing guidance on maintaining and propagating the glioma xenografts.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
Related Research Data
References
- van Den Bent MJ, Klein M, Smits M, Reijneveld JC, French PJ, Clement P, Fyf DV, Wick A, Mulholland PJ, Taphoorn MJB, et al. 2018. Bevacizumab and temozolomide in patients with first recurrence of WHO grade II and III glioma, without 1p/19q co-deletion (TAVAREC): a randomised controlled phase 2 EORTC trial. Lancet Oncol. 19:1170–1179. doi:10.1016/S1470-2045(18)30362-0.
- Desjardins A, Gromeier M, Herndon JE, Beaubier N, Bolognesi DP, Friedman AH, Friedman HS, McSherry F, Muscat AM, Nair S, et al. 2018. Recurrent glioblastoma treated with recombinant poliovirus. N Engl J Med. 379:150–161. doi:10.1056/NEJMoa1716435.
- Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN. 2015. Cancer stem cells in glioblastoma. Genes Dev. 29:1203–1217. doi:10.1101/gad.261982.115.
- Cai X, Sughrue ME. 2018. Glioblastoma: new therapeutic strategies to address cellular and genomic complexity. Oncotarget. 9:9540–9554. doi:10.18632/oncotarget.23476.
- Maeda H, Kuwahara H, Ichimura Y, Ohtsuki M, Kurakata S, Shiraishi A. TGF-beta enhances macrophage ability to produce IL-10 in normal and tumor-bearing mice. J Immunol. 1995;155:4926–4932.
- Markovic DS, Vinnakota K, Chirasani S, Synowitz M, Raguet H, Stock K, Sliwa M, Lehmann S, Kälin R, van Rooijen N, et al. 2009. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc Natl Acad Sci USA. 106:12530–12535. doi:10.1073/pnas.0804273106.
- Abdelmohsen K, Gorospe M. 2010. Posttranscriptional regulation of cancer traits by HuR. Wiley Interdiscip Rev RNA. 1:214–229. doi:10.1002/wrna.4.
- Dixon DA, Tolley ND, King PH, Nabors LB, McIntyre TM, Zimmerman GA, Prescott SM. 2001. Altered expression of the mRNA stability factor HuR promotes cyclooxygenase-2 expression in colon cancer cells. J Clin Invest. 108:1657–1665. doi:10.1172/JCI12973.
- Filippova N, Yang X, Wang Y, Gillespie GY, Langford C, King PH, Wheeler C, Nabors LB. 2011. The RNA-binding protein HuR promotes glioma growth and treatment resistance. Mol Cancer Res. 9:648–659. doi:10.1158/1541-7786.MCR-10-0325.
- Nabors LB, Suswam E, Huang Y, Yang X, Johnson MJ, King PH. Tumor necrosis factor alpha induces angiogenic factor up-regulation in malignant glioma cells: a role for RNA stabilization and HuR. Cancer Res. 2003;63:4181–4187.
- Nabors LB, Gillespie GY, Harkins L, King PH. HuR, an RNA stability factor, is expressed in malignant brain tumors and binds to adenine and uridine-rich elements within the 3ʹ untranslated regions of cytokine and angiogenic factor mRNAs. Cancer Res. 2001;61:2154–2161.
- Meisner N-C, Hintersteiner M, Mueller K, Bauer R, Seifert J-M, Naegeli H-U, Ottl J, Oberer L, Guenat C, Moss S, et al. Identification and mechanistic characterization of low-molecular-weight inhibitors for HuR. Nat Chem Biol. 2007;3:508–515. doi:10.1038/nchembio.2007.14.
- Blanco FF, Preet R, Aguado A, Vishwakarma V, Stevens LE, Vyas A, Padhye S, Xu L, Weir SJ, Anant S, et al. 2016. Impact of HuR inhibition by the small molecule MS-444 on colorectal cancer cell tumorigenesis. Oncotarget. 7:74043–74058. doi:10.18632/oncotarget.12189.
- Romeo C, Weber MC, Zarei M, DeCicco D, Chand SN, Lobo AD, Winter JM, Sawicki JA, Sachs JN, Meisner-Kober N, et al. 2016. HuR contributes to TRAIL resistance by restricting death receptor 4 expression in pancreatic cancer cells. Mol Cancer Res. 14:599–611. doi:10.1158/1541-7786.mcr-15-0448.
- Hu Y, Smyth GK. 2009. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 347:70–78. doi:10.1016/j.jim.2009.06.008.
- Bellail AC, Tse MC, Song JH, Phuphanich S, Olson JJ, Sun SY, Hao C. 2010. DR5-mediated DISC controls caspase-8 cleavage and initiation of apoptosis in human glioblastomas. J Cell Mol Med. 14:1303–1317. doi:10.1111/j.1582-4934.2009.00777.x.
- Song JH, Song DK, Pyrzynska B, Petruk KC, Van Meir EG, Hao C. TRAIL triggers apoptosis in human malignant glioma cells through extrinsic and intrinsic pathways. Brain Pathol. 2003;13:539–553. doi:10.1111/j.1750-3639.2003.tb00484.x.
- Roth W, Isenmann S, Nakamura M, Platten M, Wick W, Kleihues P, Bahr M, Ohgaki H, Ashkenazi A, Weller M. Soluble decoy receptor 3 is expressed by malignant gliomas and suppresses CD95 ligand-induced apoptosis and chemotaxis. Cancer Res. 2001;61:2759–2765.
- Srikantan S, Gorospe M. HuR function in disease. Front Biosci (Landmark Ed). 2012;17:189–205. doi:10.2741/3921.
- Galban S, Kuwano Y, Pullmann R Jr., Martindale JL, Kim HH, Lal A, Abdelmohsen K, Yang X, Dang Y, Liu JO, et al. 2008. RNA-binding proteins HuR and PTB promote the translation of hypoxia-inducible factor-1α. Mol Cell Biol. 28:93–107. doi:10.1128/mcb.00973-07.
- Rodriguez-Pascual F, Hausding M, Ihrig-Biedert I, Furneaux H, Levy AP, Forstermann U, Kleinert H. Complex contribution of the 3ʹ-untranslated region to the expressional regulation of the human inducible nitric-oxide synthase gene. Involvement of the RNA-binding protein HuR. J Biol Chem. 2000;275:26040–26049. doi:10.1074/jbc.M910460199.
- Levy NS, Chung S, Furneaux H, Levy AP. Hypoxic stabilization of vascular endothelial growth factor mRNA by the RNA-binding protein HuR. J Biol Chem. 1998;273:6417–6423. doi:10.1074/jbc.273.11.6417.
- Pineda DM, Rittenhouse DW, Valley CC, Cozzitorto JA, Burkhart RA, Leiby B, Winter JM, Weber MC, Londin ER, Rigoutsos I, et al. HuR’s post-transcriptional regulation of death receptor 5 in pancreatic cancer cells. Cancer Biol Ther. 2012;13:1–10.
- Kwan T, Floyd CL, Kim S, King PH. 2017. RNA binding protein human antigen R is translocated in astrocytes following spinal cord injury and promotes the inflammatory response. J Neurotrauma. 34:1249–1259. doi:10.1089/neu.2016.4757.
- Matsye P, Zheng L, Si Y, Kim S, Luo W, Crossman DK, Bratcher PE, King PH. 2017. HuR promotes the molecular signature and phenotype of activated microglia: implications for amyotrophic lateral sclerosis and other neurodegenerative diseases. Glia. 65:945–963. doi:10.1002/glia.23137.
- Filippova N, Yang X, Ananthan S, Sorochinsky A, Hackney JR, Gentry Z, Bae S, King P, Nabors LB. 2017. Hu antigen R (HuR) multimerization contributes to glioma disease progression. J Biol Chem. 292:16999–17010. doi:10.1074/jbc.M117.797878.
- Filippova N, Yang X, King P, Nabors LB. 2012. Phosphoregulation of the RNA-binding protein Hu antigen R (HuR) by Cdk5 affects centrosome function. J Biol Chem. 287:32277–32287. doi:10.1074/jbc.M112.353912.
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. 2006. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 444:756–760. doi:10.1038/nature05236.
- Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, Lu L, Irvin D, Black KL, Yu JS. 2006. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 5:67. doi:10.1186/1476-4598-5-67.
- Chen J, Li Y, Yu T-S, McKay RM, Burns DK, Kernie SG, Parada LF. 2012. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 488:522. doi:10.1038/nature11287.
- Herrmann A, Cherryholmes G, Schroeder A, Phallen J, Alizadeh D, Xin H, Wang T, Lee H, Lahtz C, Swiderski P, et al. 2014. TLR9 is critical for glioma stem cell maintenance and targeting. Cancer Res. 74:5218–5228. doi:10.1158/0008-5472.can-14-1151.
- Grivennikov S, Karin M. 2008. Autocrine IL-6 signaling: a key event in tumorigenesis?. Cancer Cell. 13:7–9. doi:10.1016/j.ccr.2007.12.020.
- Wang H, Lathia JD, Wu Q, Wang J, Li Z, Heddleston JM, Eyler CE, Elderbroom J, Gallagher J, Schuschu J, et al. 2009. Targeting interleukin 6 signaling suppresses glioma stem cell survival and tumor growth. Stem Cells. 27:2393–2404. doi:10.1002/stem.188.
- Gurgis FM, Yeung YT, Tang MX, Heng B, Buckland M, Ammit AJ, Haapasalo J, Haapasalo H, Guillemin GJ, Grewal T, et al. 2015. The p38-MK2-HuR pathway potentiates EGFRvIII-IL-1beta-driven IL-6 secretion in glioblastoma cells. Oncogene. 34:2934–2942. doi:10.1038/onc.2014.225.
- Tran AN, Boyd NH, Walker K, Hjelmeland AB. 2017. NOS expression and NO function in glioma and implications for patient therapies. Antioxid Redox Signal. 26:986–999. doi:10.1089/ars.2016.6820.
- Han J, Alvarez-Breckenridge CA, Wang Q-E YJ. TGF-β signaling and its targeting for glioma treatment. Am J Cancer Res. 2015;5:945–955.
- Heddleston JM, Hitomi M, Venere M, Flavahan WA, Yang K, Kim Y, Minhas S, Rich JN, Hjelmeland AB. Glioma stem cell maintenance: the role of the microenvironment. Curr Pharm Des. 2011;17:2386–2401. doi:10.2174/138161211797249260.
- Li W, Graeber MB. 2012. The molecular profile of microglia under the influence of glioma. Neuro-Oncology. 14:958–978. doi:10.1093/neuonc/nos116.
- Tummers B, Green DR. 2017. Caspase-8: regulating life and death. Immunol Rev. 277:76–89. doi:10.1111/imr.12541.
- Arakawa Y, Tachibana O, Hasegawa M, Miyamori T, Yamashita J, Hayashi Y. 2005. Frequent gene amplification and overexpression of decoy receptor 3 in glioblastoma. Acta Neuropathol. 109:294–298. doi:10.1007/s00401-004-0956-6.
- Koff JL, Ramachandiran S, Bernal-Mizrachi L. 2015. A time to kill: targeting apoptosis in cancer. Int J Mol Sci. 16:2942–2955. doi:10.3390/ijms16022942.
- Zhao X, Laver T, Hong SW, Twitty GB Jr., Devos A, Devos M, Benveniste EN, Nozell SE, An N. 2011. F-kappaB p65-cIAP2 link is necessary for mediating resistance to TNF-alpha induced cell death in gliomas. J Neurooncol. 102:367–381. doi:10.1007/s11060-010-0346-y.
- LaCasse EC, Mahoney DJ, Cheung HH, Plenchette S, Baird S, Korneluk RG. IAP-targeted therapies for cancer. Oncogene. 2008;27:6252–6275. doi:10.1038/onc.2008.302.
- Tan AC, Heimberger AB, Khasraw M. 2017. Immune checkpoint inhibitors in gliomas. Curr Oncol Rep. 19:23. doi:10.1007/s11912-017-0586-5.
- Coelho MA, de Carne Trecesson S, Rana S, Zecchin D, Moore C, Molina-Arcas M, East P, Spencer-Dene B, Nye E, Barnouin K, et al. 2017. Oncogenic RAS signaling promotes tumor immunoresistance by stabilizing PD-L1 mRNA. Immunity. 47:1083–99 e6. doi:10.1016/j.immuni.2017.11.016.
- Marín-Ramos NI, Thein TZ, Cho H-Y, Swenson SD, Wang W, Schönthal AH, Chen TC, Hofman FM. 2018. NEO212 inhibits migration and invasion of glioma stem cells. Mol Cancer Ther. 17:625–637. doi:10.1158/1535-7163.mct-17-0591.
- D’Agostino VG, Lal P, Mantelli B, Tiedje C, Zucal C, Thongon N, Gaestel M, Latorre E, Marinelli L, Seneci P, et al. 2015. Dihydrotanshinone-I interferes with the RNA-binding activity of HuR affecting its post-transcriptional function. Sci Rep. 5:16478. doi:10.1038/srep16478.
- Moradi F, Berglund P, Linnskog R, Leandersson K, Andersson T, Prasad CP. 2016. Dual mechanisms of action of the RNA-binding protein human antigen R explains its regulatory effect on melanoma cell migration. Transl Res. 172:45–60. doi:10.1016/j.trsl.2016.02.007.
- Muralidharan R, Panneerselvam J, Chen A, Zhao YD, Munshi A, Ramesh R Hu R-targeted nanotherapy in combination with AMD3100 suppresses CXCR4 expression, cell growth, migration and invasion in lung cancer. Cancer Gene Ther 2015. doi:10.1038/cgt.2015.55.
- Zhu VF, Yang J, LeBrun DG, Li M. Understanding the role of cytokines in Glioblastoma multiforme pathogenesis. Cancer Lett. 2012;316:139–150. doi:10.1016/j.canlet.2011.11.001.
- Choi C, Kutsch O, Park J, Zhou T, Seol DW, Benveniste EN. Tumor necrosis factor-related apoptosis-inducing ligand induces caspase-dependent interleukin-8 expression and apoptosis in human astroglioma cells. Mol Cell Biol. 2002;22:724–736.
- Zhang W, Vreeland AC, Noy N. 2016. RNA-binding protein HuR regulates nuclear import of protein. J Cell Sci. 129:4025–4033. doi:10.1242/jcs.192096.
- Fang Y, Fullwood MJ. 2016. Roles, functions, and mechanisms of long non-coding RNAs in cancer. Genomics Proteomics Bioinformatics. 14:42–54. doi:10.1016/j.gpb.2015.09.006.
- Qazi MA, Vora P, Venugopal C, Sidhu SS, Moffat J, Swanton C, Singh SK. 2017. Intratumoral heterogeneity: pathways to treatment resistance and relapse in human glioblastoma. Ann Oncol. 28:1448–1456. doi:10.1093/annonc/mdx169.
- Lopez de Silanes I, Lal A, Gorospe M. HuR: post-transcriptional paths to malignancy. RNA Biol. 2005;2:11–13. doi:10.4161/rna.2.1.1552.
- Friedman GK, Langford CP, Coleman JM, Cassady KA, Parker JN, Markert JM, Yancey Gillespie G. 2009. Engineered herpes simplex viruses efficiently infect and kill CD133+ human glioma xenograft cells that express CD111. J Neurooncol. 95:199–209. doi:10.1007/s11060-009-9926-0.
- Qin H, Niyongere SA, Lee SJ, Baker BJ, Benveniste EN. Expression and functional significance of SOCS-1 and SOCS-3 in astrocytes. J Immunol. 2008;181:3167–3176.