
ABSTRACT
Introduction: Transporters comprising the blood-brain barrier complicate delivery of many therapeutics to the central nervous system. The present study ascertained whether the natural product botryllamide G is viable for in vivo inhibition of ABCG2 using lapatinib as a probe for ABCB1 and ABCG2-mediated efflux from the brain. Methods: Wild-type and Mdr1a/Mdr1b (-/-) mice were treated with botryllamide G and lapatinib (“doublet therapy”), and while a separate cohort of wild-type mice was treated with botryllamide, tariquidar and lapatinib (“triplet therapy”). Results: Botryllamide G demonstrates biphasic elimination with a rapid distribution, decreasing below the in vitro IC50 of 6.9 µM within minutes, yet with a relatively slower terminal half-life (4.6 h). In Mdr1a/Mdr1b (-/-) mice, doublet therapy resulted in a significant increase in brain lapatinib AUC at 8 h (2058 h*ng/mL vs 4007 h*ng/mL; P = .031), but not plasma exposure (P = .15). No significant differences were observed after 24 h. Lapatinib brain exposure was greater through 1 h when wild-type mice were administered triplet therapy (298 h*pg/mg vs 120 h*pg/mg; P < .001), but the triplet decreased brain AUC through 24 h vs. mice administered lapatinib alone (2878 h*pg/mg vs 4461hr*ng/mL; P < .001) and did not alter the brain:plasma ratio. Conclusions: In summary, the ABCG2 inhibitor, botryllamide G, increases brain exposure to lapatinib in mice lacking Abcb1, although the combination of botryllamide G and tariquidar increases brain exposure in wild-type mice only briefly (1 h). Additional research is needed to find analogs of this compound that have better pharmacokinetics and pharmacodynamic effects on ABCG2 inhibition.
Introduction
Cancers of the brain and central nervous system are notoriously difficult to treat with chemotherapy due in part to the presence of drug efflux transporters that constitute a portion of the blood-brain barrier (BBB). From 1999 to 2015, overall cancer death rates have decreased by 1.6% per year on average, yet death rates from brain cancer specifically have risen by 0.5%.Citation1 This increase is despite a 0.7% lower incidence rate of brain cancer in both men and women. Clearly, drug delivery and retention in the brain are of importance. Two transporters within the BBB are abundantly expressed (the 7th and 8th most abundant of 304 total transporters found) and therefore responsible for a majority of drug efflux: ABCG2 (aka breast cancer resistance protein, BCRP) and ABCB1 (aka P-glycoprotein; P-gp; MDR1).Citation2 A number of anti-cancer therapies have been shown to have limited brain penetration due to ABCB1- and ABCG2-mediated efflux.Citation3–Citation21 Indeed, these two transporters serve a compensatory function – with deletion of either transporter unable to increase concentrations of substrate drugs in the brain, while deletion or inhibition of both transporters is able to markedly increase brain penetration.Citation22 Accordingly, much research has recently focused on the inhibition of one or both of these transporters to prolong the mean residence time of anti-cancer therapies in the brain.Citation23–Citation32 However, there has been no sustainable clinical improvement in this field; thus, the search continues for a new class of ABCB1 and/or ABCG2 inhibitors. Furthermore, many targeted small molecule- or chemo-therapeutics are substrates for more than one transporter.Citation22 Thus, in order to study the in vivo efficacy of botryllamide G, a probe drug was chosen that mimics real-world brain efflux, i.e. from more than one transporter.
Lung and breast cancers have a high frequency of brain metastases (approximately 19.9% and 5.1% respectively),Citation33 and many of these tumors demonstrate HER2 positivity (2% of lung cancers and 15-30% of breast cancers).Citation34–Citation37 Lapatinib is approved for the treatment of HER2-positive breast cancer,Citation38 and targeting HER2 mutations may be useful in certain subpopulations of patients with HER2+ lung cancer.Citation39 Lapatinib penetration into and retention within the brain is significantly limited by the blood-brain barrier (BBB), specifically ABCB1 and ABCG2.Citation40,Citation41 A transgenic animal study demonstrated that the lapatinib brain-to-plasma ratio is increased 40-fold in mice lacking both murine-type ABCB1 and ABCG2.Citation42 Thus, inhibiting drug efflux through ATP-binding cassette (ABC) transporters presents an attractive method for improving brain exposure to lapatinib.
We therefore hypothesized that dual inhibition of ABCB1 and ABCG2 could improve brain retention of lapatinib, a known substrate for both transporters. However, clinically viable ABCG2 inhibitors have not yet been identified. The natural product, botryllamide G (NSC-794459)Citation43 was identified in a large screen of 89,229 potential ABCG2 inhibitorsCitation44 that was further characterized as a selective inhibitor of ABCG2 (IC50 = 6.9 µM), but not ABCB1 (IC50 > 50 µM).Citation45,Citation46 We thus theorized that combined inhibition of ABCB1 with tariquidar and ABCG2 with botryllamide G could improve brain uptake of lapatinib. To that end, we undertook preclinical characterization of lapatinib brain uptake in animals treated with both agents. Concurrently, we aimed to characterize the pharmacokinetics of botryllamide G and the degree to which botryllamide G limits murine-type ABCG2 in Mdr1a/Mdr1b (-/-) mice.
Materials and methods
Chemical reagents and animals
Both wild-type FVB (FVB/NTac) and double knockout FVB (FVB.129P2-Abcb1atm1BorAbcb1btm1BorN12) mice were purchased from Taconic Biosciences (Hudson, NY). Botryllamide G was provided by the NCI Molecular Targets Program (Frederick, MD). Lapatinib was purchased from US Biological (Salem, MA). 13[C],2[H]7-Lapatinib for assay internal standard was purchased from Alsachim (Illkirch Graffenstaden, France). Tariquidar was purchased from Selleck Chemicals (Houston, TX). Optima grade methanol and acetonitrile were purchased from Fisher Scientific (Pittsburgh, PA). All water used was deionized and ultra-filtered (0.2 um) using a MilliPore Milli-Q Gradient purification system (EMD Millipore, Billerica, MA). All animal experiments were granted approval by NCI Animal Care and Use Committee (ACUC) and were conducted under NCI ACUC guidelines.
Dosage, administration, and sample processing
Studies were conducted using male FVB wild-type and FVB (Mdr1a/Mdr1b knockout mice). Mice received either botryllamide G or vehicle i.v. at 13.4 mg/kg in the solution ([80/10/10, v/v/v], saline/EtOH/TWEEN80). After ~2mins, mice were orally gavaged with 90 mg/kg lapatinib formulated in DMSO (200 mg/mL) then diluted with Labrasol before administration. Animals treated with the addition of tariquidar were treated at 4 mg/kg i.v. in ([30/5/65, v/v/v], Propylene Glycol/TWEEN80/D5W). Botryllamide G and lapatinib treatments were the same for this group. Tariquidar treatment occurred immediately following botryllamide G injection. Mice were euthanized at 0.25, 0.5, 1, 4, 8, 18, and 24 h post lapatinib dose for all cohorts. Blood was collected into heparinized tubes and centrifuged to separate out plasma. Plasma was stored at −80°C until analysis. Brains were resected, snap-frozen, and stored at −80°C.
LC-MS/MS conditions
Botryllamide G plasma concentrations were measured using a validated LC-MS/MS assay with a calibration range of 20–50,000 ng/mL. Briefly, plasma (50 µL) was added to an Ostro® phospholipid removal plate (Waters Corp, Milford, MA) before the addition of 3x volume methanol to precipitate proteins. The contents of each well were mixed before pushing the supernatent through the wells with compressed nitrogen gas using a Waters Positive Pressure Manifold. Samples were injected onto an ACQUITY UPLC® BEH C18 column (2.1x50 mm, 1.7 µm). Botryllamide G was chromatographically separated using an isocratic elution of (30/70, v/v) 0.1% formic acid (aq) and 0.1% formic acid in methanol at a flow rate of 0.3 mL/min (run time 3 min). The resulting plasma concentrations were plotted vs time post-IV bolus administration, with biphasic elimination rates and AUCLAST, using Bailer’s approach to calculating AUC with destructive samplingCitation47 as calculable PK parameters.
Lapatinib plasma concentrations were measured using a validated LC-MS/MS assay with a calibration range of 10–10,000 ng/mL, with minimal sample preparation involving 100 µL of sample being diluted with 4x volume of acetonitrile containing 200 ng/mL of stable isotope-labeled lapatinib (13[C],2[H]7-lapatinib). Samples were mixed to precipitate plasma proteins before being centrifuged, and further diluted with 200 µL of (80/20, v/v) H2O/ACN before analysis by LC-MS/MS.
Lapatinib brain tissue concentration was measured using a separately validated LC-MS/MS assay with a calibration range of 5–50,000 pg/mg. Approximately 50–100 mg of tissue per sample was needed. For each mg of tissue, 10 µL of water were added in order to produce a 100 mg/mL homogenate. A 3-fold dilution of 50 µL of this homogenate was performed with acetonitrile containing 200 ng/mL of 13[C],2[H]7-lapatinib, vortexed, centrifuged, and ~135 µL of supernatent dried and reconstituted in 100 µL of (80/20, v/v) water/ACN for analysis by LC-MS/MS.
Both lapatinib plasma and tissue samples utilized the same LC-MS/MS assay, albeit with different concentration ranges and units. Ten microlitres of sample were injected onto a Waters XSelect® HSS PFP column (2.1x50 mm, 1.7 um) and chromatographically separated using a gradient elution of 5 mM ammonium formate, pH 3.0 (A) and 0.5% formic acid in acetonitrile (B) at a flow rate of 0.8 mL/min (run time 8 min).
Statistical analysis
Bailer’s Method was used to calculate lapatinib plasma and brain AUC to the last time point (24 h measurement period). The elimination rate (kEL) was determined from natural log-transformed concentrations during the terminal phase. Half-life (t1/2) was calculated as natural log of 2 divided by the kEL. Only measured concentrations above the LLOQ were used in the calculation of PK parameters. A one-tailed Z-test was performed to assess statistical differences (alpha = 0.05) between lapatinib control (alone) and lapatinib + botryllamide G.
Results
Botryllamide G pharmacokinetics
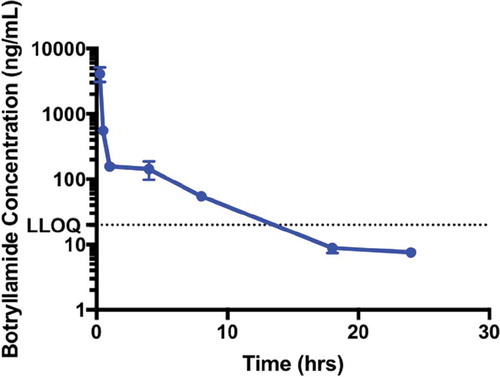
Botryllamide G was administered intravenously at the maximum soluble dose (13.4 mg/kg in 80/10/10 (v/v/v), saline/EtOH/TWEEN80). In wild-type mice, botryllamide G plasma AUC was 2088 h*ng/mL (), with an average (n = 3) measured Cmax of 4106 ng/mL sampled just after tail vein injection. Botryllamide G demonstrates biphasic elimination with a rapid distribution, decreasing below the in vitro IC50 of 6.9 µM (3236 ng/mL) within minutes, and a terminal half-life of 4.6 h that is mostly cleared from plasma by 24 h.
Figure 1. The Pharmacokinetics of Botryllamide G in Mice. The maximum soluble dose (13 mg/kg) of botryllamide G was administered to mice (n = 3 for each timepoint) via IV tail vein injection. Botryllamide G demonstrate biphasic elimination, which involves rapid distribution into tissues and a plasma exposure that quickly dropped below the in vitro IC50 of 6.9 µM. Error bars represent mean standard deviation.
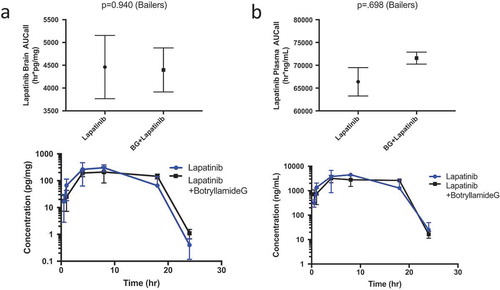
Botryllamide G ineffective at increasing brain exposure of lapatinib in wild-type mice
Lapatinib brain exposures (AUC0-24hr) were similar between the two treatment groups (4462 h*pg/mg for lapatinib alone, 4398 h*pg/mg for the lapatinib/botryllamide G combination; P = .940; )). Lapatinib plasma AUC was also comparable when botryllamide G was administered with lapatinib (66,386 h*ng/mL for lapatinib alone vs 71,576 h*pg/mg for the lapatinib/botryllamide G combination; P = .698; )). The lapatinib brain:plasma exposure (AUC) ratios were equivalent between lapatinib only (0.067) and lapatinib+botryllamide groups (0.061). This is consistent with prior work showing that Abcg2 inhibition alone is not sufficient to block blood-brain barrier efflux, since.Citation42
Figure 2. Lapatinib AUC in the brain of wild-type mice (n = 3 at each timepoint) when treated with lapatinib alone or in combination with botryllamide G (a) and exposure curve over 24 h. Lapatinib AUC in the plasma of wild-type mice (n = 3 at each timepoint) when treated with lapatinib alone or in combination with botryllamide G (b) and exposure curve over 24 h. Error bars represent mean standard deviation.
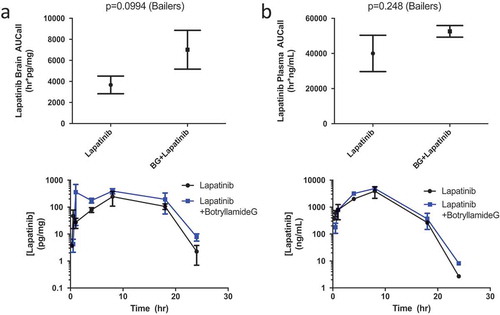
Botryllamide G increases brain and plasma exposure of lapatinib in Mdr1a/Mdr1b knockout mice
To eliminate the brain exclusion of lapatinib through murine-type Abcb1 (aka Mdr1), brain and plasma concentrations of lapatinib were ascertained after treatment with botryllamide G in Mdr1a/Mdr1b (-/-) animals. Consistent with the rapid early distribution of botryllamide G, a significantly greater lapatinib brain AUC was observed for 8 h following botryllamide G administration when compared to mice treated with lapatinib alone (2058 h*pg/mg vs 4007 h*pg/mg; P = .031). There was also slightly greater plasma exposure to lapatinib in mice given botryllamide G through 8 h, likely due to inhibited Abcg2-mediated gut efflux, however this was not statistically significant (P = .15). The trend remained; however, the difference in lapatinib exposure between cohorts became non-significant for both brain (P = .099; )) and plasma (P = .25; )) at the 24-h exposure timepoint. This Abcb1a/b knockout with Abcg2 inhibition with botryllamide G model demonstrated results consistent with Polli et al.,Citation42 however not with the same magnitude effect.
Figure 3. Lapatinib AUC in the brain of Mdr1a/Mdr1b knockout mice (n = 3 for each timepoint) when treated with lapatinib alone or in combination with botryllamide G (a) and exposure curve over 24 h. Lapatinib AUC in the plasma of wild-type mice (n = 3 for each timepoint) when treated with lapatinib alone or in combination with botryllamide G (b) and exposure curve over 24 h. Error bars represent mean standard deviation.
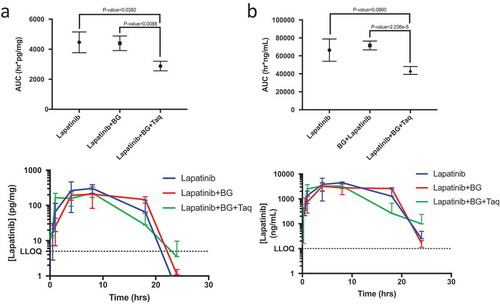
Combination treatment of botryllamide G and tariquidar modulates brain exposure to lapatinib
We next ascertained whether a specific and potent ABCB1 inhibitor, tariquidar,Citation48 could recapitulate the Mdr1a/1b knockout phenotype when co-administered with botryllamide G in wild-type mice. Due to the rapid systemic distribution of botryllamide G and its short half-life, differences in early exposure (AUC0-1 h) were compared between the treatment groups. The triplet cohort (lapatinib + botryllamide G + tariquidar) had significantly greater lapatinib brain exposure through 1 h (298 ± 44.6 h*pg/mg) than either singlet (120 ± 39.9 h*pg/mg; p = .003) or doublet therapy (48.3 ± 14.9 h*pg/mg; p < .0001). This increase in brain exposure in the first hour was greater in magnitude than the increased plasma exposure within the same duration of time in triplet vs singlet (p = .063) or doublet cohorts (P = .004).
Unexpectedly, the triplet combination decreased 24 h brain exposure (2878 h*pg/mg) when compared to lapatinib alone (4461 h*pg/mg; P = .038) or lapatinib with botryllamide G (4398 h*pg/mg; P = .009) in wild-type mice (). Triplet therapy also significantly decreased plasma exposure through 24 h when compared to the singlet or doublet therapy, however, there was no difference (P = .12) up through 8 h.
Figure 4. Lapatinib AUC in the brain of wild-type mice (n = 3 for each timepoint) when treated with lapatinib alone, in combination with botryllamide G alone, or botryllamide G and tariquidar (a) and exposure curve over 24 h. Lapatinib AUC in the plasma of wild-type mice (n = 3 for each timepoint) when treated with lapatinib alone, in combination with botryllamide G alone, or botryllamide G and tariquidar (b) and exposure curve over 24 h. Error bars represent mean standard deviation.
Discussion
There have been several specific and nonspecific ABCG2 inhibitors in various stages of development, yet very few have shown promise in preclinical studies.Citation49 Elacridar, a very promising dual ABCG2/ABCB1 inhibitor, has shown modest increases in brain penetration of several targeted therapies, such as gefitinib,Citation4 dasatinib,Citation5 vemurafenib,Citation7 sunitinib,Citation8 crizotinib,Citation24 and erlotinib.Citation50 Ko143 is considered a potent ABCG2 inhibitor,Citation51,Citation52 but is highly unstable due to a labile ester group that is rapidly metabolized into an inactive metabolite (T1/2 = 0.2 h in mice).Citation27,Citation31,Citation53 High doses of Ko143 (15 mg/kg) increased brain AUC of aCitation11C-labeled ABCG2 substrate in Mdr1a/1b (-/-) mice; however, Ko143 did not increase brain concentration of such a substrate in wild-type mice.Citation54
We sought an ABCG2 inhibitor more clinically suitable than Ko143 and identified the natural product botryllamide G, which is more metabolically stable and specific for Abcg2 than Ko143.Citation45,Citation46,Citation53 Similar to results with Ko143, botryllamide G also increased brain exposure of an Abcg2 substrate, lapatinib, but only in Mdr1a/1b (-/-) mice. Further, in addition to significantly increasing brain exposure to lapatinib, the plasma exposure was also higher (albeit non-significant) in the presence of botryllamide G, likely due to Abcg2 inhibition in the gut. This latter observation suggests that murine-type Abcb1 and Abcg2 regulate the brain permeability to lapatinib to a greater extent than they regulate hepatobiliary elimination, or that there are other mechanisms regulating plasma levels. This result is also consistent with multiple individual reports of substrate drugs in ABCB1 and/or ABCG2 knockout animals, showing plasma concentrations are modulated much less, or not at all, compared to brain concentrations.Citation22 Comparing brain:plasma ratios (AUC0-24, brain/AUC0-24hr, plasma) between lapatinib alone given to wild-type (0.067) vs Mdr1 knockout mice (0.092) show an improvement in lapatinib brain exposure, and when botryllamide G was present, the ratio increased to 0.133. This suggests a clear drug effect, albeit short lived. Improving on botryllamide G administration (e.g. prolonged infusion at the EC50) or increasing the dose from an improved formulation might prolong Abcg2 inhibition in the BBB. While this result is encouraging, it occurred in a transgenic knockout mouse model.
To provide a more clinically relevant animal model, wild-type mice were also dosed with botryllamide G and lapatinib and not surprisingly, the presence of uninhibited Abcb1 proved sufficient to effectively efflux lapatinib despite Abcg2 inhibition. Tariquidar was added to the regimen to inhibit P-glycoprotein, and these results suggest that while tariquidar did not improve brain retention of lapatinib over a 24-hr period, the co-inhibition of Abcg2 and Mdr1 was effective early (at 1 h) when botryllamide G plasma concentration was at (or near) its known IC50, 6.9uM. Notably, the brain:plasma AUC ratios over 24 h for lapatinib (0.067), lapatinib + botryllamide G (0.061), and triplet therapy (0.066) were very similar in wild-type mice, suggesting no sustained drug effect whatsoever. Perhaps a more potent and selective Abcb1/Mdr1a/b inhibitor is needed to replicate the results of the Mdr1a/b knockout mice.
Interestingly, the presence of tariquidar and botryllamide G actually lowered lapatinib plasma exposure through 24 h, which in turn lowered the overall 24-hr brain exposure. Although inhibition of these efflux transporters (in the gut, liver) would be expected to increase plasma exposure, this result of Abcb1 inhibition by tariquidar has been observed previously.
Abcb1 inhibition with tariquidar causes an increase in the peripheral tissue concentration of several target substrates: docetaxel,Citation55 verapamil,Citation30 bortezomib,Citation25 and others.Citation26,Citation56,Citation57 The addition of tariquidar to botryllamide G significantly increased the brain exposure of lapatinib through 1 h due to rapid early botryllamide G distribution. Yet, over a 24 h period, tariquidar (in the presence of low botryllamide G exposure) counter-intuitively decreased lapatinib plasma and brain AUC. It is possible that through tariquidar-mediated inhibition of Abcb1 gut efflux, lapatinib is able to more widely distribute into peripheral tissues that are not normally permeable to lapatinib, thereby lowering the exposure of plasma as well as any given tissue. However, we did not observe this decreased lapatinib brain exposure in the Mdr1a/1b (-/-) mice, which had similar brain AUC to wild-type mice. Another explanation could be that tariquidar is a substrate of ABCG2 at low concentrations and an inhibitor at higher concentrations.Citation58 This point is consistent with the increased brain lapatinib exposure through 1 h when tariquidar was administered with botryllamide G. Thus, once botryllamide G blocks hepatic tariquidar efflux through ABCG2,Citation58 ABCB1 efflux of lapatinib from the liver would be more strongly inhibited. Such inhibition would be expected to promote more-rapid hepatic metabolism of lapatinib, resulting in lower systemic exposure further post-dose, and hence a lack of greater brain concentrations. We suggest that the latter possibility is the most likely, and optimal dual inhibition of both transporters would be achieved by combining botryllamide G with an ABCB1 inhibitor that is not itself an ABCG2 substrate.
This pilot study provides proof-of-principle that botryllamide G can inhibit ABCG2 in vivo, but further optimization of dosing and schedule, as well as the need to give a concomitant ABCB1 inhibitor, are required before this compound can be considered for clinical use. The dose in the present study (13.4 mg/kg) was limited by the poor aqueous solubility of botryllamide G in an intravenous formulation. Since plasma concentrations rapidly fell below the IC50 of ABCG2 inhibition in cell culture (6.9 μM), further increasing the dose would likely improve ABCG2 the inhibitory capacity of botryllamide G. Improving the formulation of botryllamide G is therefore required for the future development of this agent. Additionally, more-frequent dosing will likely extend the time that botryllamide plasma concentration exceeds 6.9 μM. Safety studies are also underway to test whether botryllamide G administration can be improved. Since Polli et al. observed that Abcg2 knockout animals had an approximately 40-fold increase in lapatinib brain concentrations, such improvements to botryllamide G dosing and schedule are expected to further increase the brain concentration of lapatinib.Citation42
Taken together, the present study demonstrates botryllamide G may be a viable option to improve drug delivery into the brain, granted an improved formulation can allow for increased dose and frequency, as well as optimizing chemical ABCB1 inhibition. While the present study applies to lapatinib administration, numerous anticancer therapeutics are also dual ABCB1 and ABCG2 substrates and should be amenable to animal studies: topotecan, dasatinib, gefitinib, sorafenib, erlotinib, imatinib, tandutinib, flavopiridol, mitoxantrone.Citation23 In sum, attempts to block both ABCB1 and ABCG2 should be continued so that brain tissue becomes accessible to anticancer therapy. Characterizing the efficacy of botryllamide G analogs, hopefully with an improved solubility and PK profile, may also be of importance.
Disclaimer
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Acknowledgments
This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Additional information
Funding
References
- Cronin KA, Lake AJ, Scott S, Sherman RL, Noone A-M, Howlader N, Henley SJ, Anderson RN, Firth AU, Ma J, et al. Annual report to the nation on the status of cancer, part I: national cancer statistics. Cancer. 2018;124(13):2785–2800. doi:10.1002/cncr.31551.
- Suhy AM, Webb A, Papp AC, Geier EG, Sadee W. Expression and splicing of ABC and SLC transporters in the human blood-brain barrier measured with RNAseq. Eur J Pharm Sci. 2017;103:47–51. doi:10.1016/j.ejps.2017.02.010.
- Marchetti S, de Vries NA, Buckle T, Bolijn MJ, van Eijndhoven MAJ, Beijnen JH, Mazzanti R, van Tellingen O, Schellens JHM. Effect of the ATP-binding cassette drug transporters ABCB1, ABCG2, and ABCC2 on erlotinib hydrochloride (Tarceva) disposition in in vitro and in vivo pharmacokinetic studies employing Bcrp1-/-/Mdr1a/1b-/- (triple-knockout) and wild-type mice. Mol Cancer Ther. 2008;7(8):2280–2287. doi:10.1158/1535-7163.MCT-07-2250.
- Kawamura K, Yamasaki T, Yui J, Hatori A, Konno F, Kumata K, Irie T, Fukumura T, Suzuki K, Kanno I, et al. In vivo evaluation of P-glycoprotein and breast cancer resistance protein modulation in the brain using [(11)C]gefitinib. Nucl Med Biol. 2009;36(3):239–246. doi:10.1016/j.nucmedbio.2008.12.006.
- Lagas JS, van Waterschoot RA, van Tilburg VA, Hillebrand MJ, Lankheet N, Rosing H, Beijnen JH, Schinkel AH. Brain accumulation of dasatinib is restricted by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) and can be enhanced by elacridar treatment. Clin Cancer Res. 2009;15(7):2344–2351. doi:10.1158/1078-0432.CCR-08-2253.
- Poller B, Iusuf D, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH. Differential impact of P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) on axitinib brain accumulation and oral plasma pharmacokinetics. Drug Metab Dispos. 2011;39(5):729–735. doi:10.1124/dmd.110.037317.
- Durmus S, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH. Oral availability and brain penetration of the B-RAFV600E inhibitor vemurafenib can be enhanced by the P-GLYCOprotein (ABCB1) and breast cancer resistance protein (ABCG2) inhibitor elacridar. Mol Pharm. 2012;9(11):3236–3245. doi:10.1021/mp3003144.
- Tang SC, Lagas JS, Lankheet NA, Poller B, Hillebrand MJ, Rosing H, Beijnen JH, Schinkel AH.Brain accumulation of sunitinib is restricted by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) and can be enhanced by oral elacridar and sunitinib coadministration. Int J Cancer. 2012;130(1):223–233. doi:10.1002/ijc.26000.
- Tang SC, Lankheet NA, Poller B, Wagenaar E, Beijnen JH, Schinkel AH. P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) restrict brain accumulation of the active sunitinib metabolite N-desethyl sunitinib. J Pharmacol Exp Ther. 2012;341(1):164–173. doi:10.1124/jpet.111.186908.
- Durmus S, Xu N, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH. P-glycoprotein (MDR1/ABCB1) and breast cancer resistance protein (BCRP/ABCG2) restrict brain accumulation of the JAK1/2 inhibitor, CYT387. Pharmacol Res. 2013;76:9–16. doi:10.1016/j.phrs.2013.06.009.
- Lin F, Hoogendijk L, Buil L, Beijnen JH, van Tellingen O. Sildenafil is not a useful modulator of ABCB1 and ABCG2 mediated drug resistance in vivo. Eur J Cancer. 2013;49(8):2059–2064. doi:10.1016/j.ejca.2012.12.028.
- Lin F, Marchetti S, Pluim D, Iusuf D, Mazzanti R, Schellens JHM, Beijnen JH, van Tellingen O. Abcc4 together with abcb1 and abcg2 form a robust cooperative drug efflux system that restricts the brain entry of camptothecin analogues. Clin Cancer Res. 2013;19(8):2084–2095. doi:10.1158/1078-0432.CCR-12-3105.
- Marchetti S, Pluim D, van Eijndhoven M, van Tellingen O, Mazzanti R, Beijnen JH, Schellens JHM. Effect of the drug transporters ABCG2, Abcg2, ABCB1 and ABCC2 on the disposition, brain accumulation and myelotoxicity of the aurora kinase B inhibitor barasertib and its more active form barasertib-hydroxy-QPA. Invest New Drugs. 2013;31(5):1125–1135. doi:10.1007/s10637-013-9923-1.
- Tang SC, de Vries N, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH. Impact of P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) gene dosage on plasma pharmacokinetics and brain accumulation of dasatinib, sorafenib, and sunitinib. J Pharmacol Exp Ther. 2013;346(3):486–494. doi:10.1124/jpet.113.205583.
- Marchetti S, Pluim D, Beijnen JH, Mazzanti R, van Tellingen O, Schellens JH. “Effect of the drug transporters ABCB1, ABCC2, and ABCG2 on the disposition and brain accumulation of the taxane analog BMS-275,183”. Invest New Drugs. 2014;32(6):1083–1095. doi:10.1007/s10637-014-0143-0.
- Durmus S, Sparidans RW, van Esch A, Wagenaar E, Beijnen JH, Schinkel AH. Breast cancer resistance protein (BCRP/ABCG2) and P-glycoprotein (P-GP/ABCB1) restrict oral availability and brain accumulation of the PARP inhibitor rucaparib (AG-014699). Pharm Res. 2015;32(1):37–46. doi:10.1007/s11095-014-1442-z.
- Kort A, Durmus S, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH. Brain and testis accumulation of regorafenib is restricted by breast cancer resistance protein (BCRP/ABCG2) and P-glycoprotein (P-GP/ABCB1). Pharm Res. 2015;32(7):2205–2216. doi:10.1007/s11095-014-1609-7.
- Kort A, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH. Brain accumulation of the EML4-ALK inhibitor ceritinib is restricted by P-glycoprotein (P-GP/ABCB1) and breast cancer resistance protein (BCRP/ABCG2). Pharmacol Res. 2015;102:200–207. doi:10.1016/j.phrs.2015.09.003.
- Kort A, van Hoppe S, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH. Brain accumulation of ponatinib and its active metabolite, N-desmethyl ponatinib, is limited by P-Glycoprotein (P-GP/ABCB1) and breast cancer resistance protein (BCRP/ABCG2). Mol Pharm. 2017;14(10):3258–3268. doi:10.1021/acs.molpharmaceut.7b00257.
- van Hoppe S, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH. Breast cancer resistance protein (BCRP/ABCG2) and P-glycoprotein (P-gp/ABCB1) transport afatinib and restrict its oral availability and brain accumulation. Pharmacol Res. 2017;120:43–50. doi:10.1016/j.phrs.2017.01.035.
- Wang J, Gan C, Sparidans RW, Wagenaar E, van Hoppe S, Beijnen JH, Schinkel AH. P-glycoprotein (MDR1/ABCB1) and breast cancer resistance protein (BCRP/ABCG2) affect brain accumulation and intestinal disposition of encorafenib in mice. Pharmacol Res. 2018;129:414–423. doi:10.1016/j.phrs.2017.11.006.
- Robey RW, Pluchino KM, Hall MD, Fojo AT, Bates SE, Gottesman MM. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat Rev Cancer. 2018;18(7):452–464. doi:10.1038/s41568-018-0005-8.
- Agarwal S, Hartz AM, Elmquist WF, Bauer B. Breast cancer resistance protein and P-glycoprotein in brain cancer: two gatekeepers team up. Curr Pharm Des. 2011;17(26):2793–2802. doi:10.2174/138161211797440186.
- Tang SC, Nguyen LN, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH. Increased oral availability and brain accumulation of the ALK inhibitor crizotinib by coadministration of the P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) inhibitor elacridar. Int J Cancer. 2014;134(6):1484–1494. doi:10.1002/ijc.v134.6.
- Foran E, Kwon DY, Nofziger JH, Arnold ES, Hall MD, Fischbeck KH, Burnett BG. CNS uptake of bortezomib is enhanced by P-glycoprotein inhibition: implications for spinal muscular atrophy. Neurobiol Dis. 2016;88:118–124. doi:10.1016/j.nbd.2016.01.008.
- Joosen MJA, Vester SM, Hamelink J, Klaassen SD, van Den Berg RM. Increasing nerve agent treatment efficacy by P-glycoprotein inhibition. Chem Biol Interact. 2016;259(Pt B):115–121. doi:10.1016/j.cbi.2016.06.012.
- Li Y, Woo J, Chmielecki J, Xia CQ, Liao M, Chuang B-C, Yang JJ, Guan MY, Plesescu M, Prakash SR, et al. Synthesis of a new inhibitor of breast cancer resistance protein with significantly improved pharmacokinetic profiles. Bioorg Med Chem Lett. 2016;26(2):551–555. doi:10.1016/j.bmcl.2015.11.077.
- Saunders NR, Habgood MD, Mollgard K, Dziegielewska KM. 2016. The biological significance of brain barrier mechanisms: help or hindrance in drug delivery to the central nervous system? F1000 Res. 5. DOI:10.12688/f1000research.
- Xu P, Ling ZL, Zhang J, Li Y, Shu N, Zhong Z-Y, Chen Y, Di X-Y, Wang Z-J, Liu L, et al. Unconjugated bilirubin elevation impairs the function and expression of breast cancer resistance protein (BCRP) at the blood-brain barrier in bile duct-ligated rats. Acta Pharmacol Sin. 2016;37(8):1129–1140. doi:10.1038/aps.2016.25.
- Bauer M, Wulkersdorfer B, Karch R, Philippe C, Jäger W, Stanek J, Wadsak W, Hacker M, Zeitlinger M, Langer O, et al. Effect of P-glycoprotein inhibition at the blood-brain barrier on brain distribution of (R)-[(11) C]verapamil in elderly vs. young subjects. Br J Clin Pharmacol. 2017;83(9):1991–1999. doi:10.1111/bcp.v83.9.
- Liu K, Zhu J, Huang Y, Li C, Lu J, Sachar M, Li S, Ma X. Metabolism of KO143, an ABCG2 inhibitor. Drug Metab Pharmacokinet. 2017;32(4):193–200. doi:10.1016/j.dmpk.2017.02.003.
- de Gooijer MC, de Vries NA, Buckle T, Buil LCM, Beijnen JH, Boogerd W, van Tellingen O. Improved brain penetration and antitumor efficacy of temozolomide by inhibition of ABCB1 and ABCG2. Neoplasia. 2018;20(7):710–720. doi:10.1016/j.neo.2018.05.001.
- Barnholtz-Sloan JS, Sloan AE, Davis FG, Vigneau FD, Lai P, Sawaya RE. Incidence proportions of brain metastases in patients diagnosed (1973 to 2001) in the Metropolitan Detroit Cancer Surveillance System. J Clin Oncol. 2004;22(14):2865–2872. doi:10.1200/JCO.2004.12.149.
- Mitri Z, Constantine T, The OR. HER2 receptor in breast cancer: pathophysiology, clinical use, and new advances in therapy. Chemother Res Pract. 2012;2012:743193.
- Burstein HJ. The distinctive nature of HER2-positive breast cancers. N Engl J Med. 2005;353(16):1652–1654. doi:10.1056/NEJMp058197.
- Witzel I, Loibl S, von Minckwitz G, Eidtmann H, Fehm T, Khandan F, Schmatloch S, Hauschild M, Bischoff J, Fasching PA, et al. Predictive value of HER2 serum levels in patients treated with lapatinib or trastuzumab – a translational project in the neoadjuvant GeparQuinto trial. Br J Cancer. 2012;107(6):956–960. doi:10.1038/bjc.2012.353.
- World Cancer Report 2014. World Health Organization;2014.
- Novartis. Tykerb tablet package insert; 2017. In:.
- Peters S, Zimmermann S. Targeted therapy in NSCLC driven by HER2 insertions. Transl Lung Cancer Res. 2014;3(2):84–88. doi:10.3978/j.issn.2218-6751.2014.02.06.
- Polli JW, Humphreys JE, Harmon KA, Castellino S, O’Mara MJ, Olson KL, John-Williams LS, Koch KM, Serabjit-Singh CJ. The role of efflux and uptake transporters in [N-{3-chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine (GW572016, lapatinib) disposition and drug interactions. Drug Metab Dispos. 2008;36(4):695–701. doi:10.1124/dmd.107.018374.
- Taskar KS, Rudraraju V, Mittapalli RK, Samala R, Thorsheim HR, Lockman J, Gril B, Hua E, Palmieri D, Polli JW, et al. Lapatinib distribution in HER2 overexpressing experimental brain metastases of breast cancer. Pharm Res. 2012;29(3):770–781. doi:10.1007/s11095-011-0601-8.
- Polli JW, Olson KL, Chism JP, John-Williams LS, Yeager RL, Woodard SM, Otto V, Castellino S, Demby VE. An unexpected synergist role of P-glycoprotein and breast cancer resistance protein on the central nervous system penetration of the tyrosine kinase inhibitor lapatinib (N-{3-chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine; GW572016. Drug Metab Dispos. 2009;37(2):439–442. doi:10.1124/dmd.108.024646.
- Rao MR, Faulkner DJ. Botryllamides E-H, four new tyrosine derivatives from the ascidian Botrylloides tyreum. J Nat Prod. 2004;67(6):1064–1066. doi:10.1021/np0499618.
- Henrich CJ, Bokesch HR, Dean M, Bates SE, Robey RW, Goncharova EI, Wilson JA, McMahon JB. A high-throughput cell-based assay for inhibitors of ABCG2 activity. J Biomol Screen. 2006;11(2):176–183. doi:10.1177/1087057105284576.
- Henrich CJ, Robey RW, Takada K, Bokesch HR, Bates SE, Shukla S, Ambudkar SV, McMahon JB, Gustafson KR. Botryllamides: natural product inhibitors of ABCG2. ACS Chem Biol. 2009;4(8):637–647. doi:10.1021/cb900134c.
- Takada K, Imamura N, Gustafson KR, Henrich CJ. Synthesis and structure-activity relationship of botryllamides that block the ABCG2 multidrug transporter. Bioorg Med Chem Lett. 2010;20(4):1330–1333. doi:10.1016/j.bmcl.2010.01.016.
- Bailer AJ. Testing for the equality of area under the curves when using destructive measurement techniques. J Pharmacokinet Biopharm. 1988;16(3):303–309. doi:10.1007/BF01062139.
- Palmeira A, Sousa E, Vasconcelos MH, Pinto MM. Three decades of P-gp inhibitors: skimming through several generations and scaffolds. Curr Med Chem. 2012;19(13):1946–2025. doi:10.2174/092986712800167392.
- Pena-Solorzano D, Stark SA, Konig B, Sierra CA, Ochoa-Puentes C. ABCG2/BCRP: specific and nonspecific modulators. Med Res Rev. 2017;37(5):987–1050. doi:10.1002/med.2017.37.issue-5.
- Verheijen RB, Yaqub M, Sawicki E, van Tellingen O, Lammertsma AA, Nuijen B, Schellens JHM, Beijnen JH, Huitema ADR, Hendrikse NH, et al. Molecular imaging of ABCB1 and ABCG2 inhibition at the human blood-brain barrier using elacridar and (11)C-Erlotinib PET. J Nucl Med. 2018;59(6):973–979. doi:10.2967/jnumed.117.195800.
- Matsson P, Pedersen JM, Norinder U, Bergstrom CA, Artursson P. Identification of novel specific and general inhibitors of the three major human ATP-binding cassette transporters P-gp, BCRP and MRP2 among registered drugs. Pharm Res. 2009;26(8):1816–1831. doi:10.1007/s11095-009-9896-0.
- Allen JD, van Loevezijn A, Lakhai JM, van der Valk M, van Tellingen O, Reid G, Schellens JHM, Koomen G-J, Schinkel AH. 2002. Potent and specific inhibition of the breast cancer resistance protein multidrug transporter in vitro and in mouse intestine by a novel analogue of fumitremorgin C. Mol Cancer Ther. 1(6):417–425.
- Weidner LD, Zoghbi SS, Lu S, Shukla S, Ambudkar SV, Pike VW, Mulder J, Gottesman MM, Innis RB, Hall MD, et al. The inhibitor Ko143 is not specific for ABCG2. J Pharmacol Exp Ther. 2015;354(3):384–393. doi:10.1124/jpet.115.225482.
- Thomas Wanek CK, Bankstahl JP, Bankstahl M, Stanek J, Sauberer M, Muller M, Loscher W, Langer O. Inhibition of breast cancer resistance protein at the murine blood-brain barrier by Ko143 studied with [11C]tariquidar and PET. BMC Pharmacol. 2011; 11:A48.
- Kelly RJ, Draper D, Chen CC, Polli JW, Olson KL, Chism JP, John-Williams LS, Yeager RL, Woodard SM, Otto V, et al. A pharmacodynamic study of docetaxel in combination with the P-glycoprotein antagonist tariquidar (XR9576) in patients with lung, ovarian, and cervical cancer. Clin Cancer Res. 2011;17(3):569–580. doi:10.1158/1078-0432.CCR-10-1725.
- Brandt C, Bethmann K, Gastens AM, Loscher W. The multidrug transporter hypothesis of drug resistance in epilepsy: proof-of-principle in a rat model of temporal lobe epilepsy. Neurobiol Dis. 2006;24(1):202–211. doi:10.1016/j.nbd.2006.06.014.
- Zoghbi SS, Liow JS, Yasuno F, Hong J, Tuan E, Lazarova N, Gladding RL, Pike VW, Innis RB. 11C-loperamide and its N-desmethyl radiometabolite are avid substrates for brain permeability-glycoprotein efflux. J Nucl Med. 2008;49(4):649–656. doi:10.2967/jnumed.107.047308.
- Kannan P, Telu S, Shukla S, Ambudkar, S.V., Pike, V.W., Halldin, C., Gottesman, M.M., Innis, R.B, Hall, M.D. The “specific” P-glycoprotein inhibitor Tariquidar is also a substrate and an inhibitor for breast cancer resistance protein (BCRP/ABCG2). ACS Chem Neurosci. 2011;2(2):82–89. doi:10.1021/cn100078a.