
ABSTRACT
Background: Tumor microenvironment (TME) is a crucial part of tumor hallmarks. Mesenchymal stem cells (MSCs), important components of TME, are the main source of Carcinoma-associated fibroblasts (CAFs), but the mechanism of transformation regulation is still unclear. Transforming growth factor-β1 (TGF-β1), chemokine Stromal cell-derived factor-1 (SDF-1) and its endogenous receptor CXCR4 may play important roles during this process.
Methods: Co-culture technique was used to explore the effects of MSCs on the proliferation, migration and invasion of colorectal carcinoma (CRC) cells and how they induced MSCs to differentiate into CAFs. The expression of α-SMA, Vimentin, S100A4 and FAP were detected as CAFs markers. Inhibitors AMD3100 and cyclophosphamide (Cy) were pre-treated in MSCs to verify the functions of CXCR4/TGF-β1. Finally, the xenograft models in nude mice were generated to further verify this process in vivo.
Results: MSCs promoted the CRCs proliferation, invasion and migration, and induced SDF-1 expression and secretion, which dramatically up-regulated CXCR4 and TGF-β1 expression in MSCs. The levels of CAFs markers elevated in MSCs, indicating CAFs differentiation occurred in MSCs. AMD3100 and Cy treatment significantly blocked this differentiation process of MSCs by suppressing CXCR4 expression and TGF-β1 secretion. In vivo xenograft experiments also demonstrated that MSCs promoted differentiation into CAFs through CXCR4/TGF-β1 signaling in either primary tumor tissues or hepatic metastatic tissues of CRC.
Conclusion: Our studies have revealed the essential role of CXCR4/TGF-β1 axis playing in the transformation of tumor microenvironment by mediating MSCs differentiation into CAFs, promoting CRCs growth and metastasis.
Introduction
Tumor microenvironment (TME), as the name implies, refers to the external environment of tumor cells for growth. TME is mainly composed of immune cells, tumor stromal cells, extracellular matrix (ECM) and other related factors. As the study progressing, medical researchers are beginning to realize that TME may be related to tumorigenesis and development.1 Tumor cells participate in the formation of TME by secreting cytokines, which provide conditions for tumor cells growth and metastasis.2 Colorectal cancer is a common high-risk and metastatic malignant tumor. Recent studies have found that TME plays an important role in promoting colorectal cancer development and metastasis,Citation1 but the underlying specific molecular mechanisms are still unclear.
Tumor stroma cells, a primary constituent of TME, mainly originate from bone marrow, including mesenchymal stem cells (MSCs) and hemangioblasts. Actually, MSCs are multipotent stromal cells that are capable of self-renewing and have the potential to differentiate into a variety of cell types, including osteoblasts, adipocytes and chondrocytes.Citation3 MSCs have multiple therapeutic potentials for regenerative, anti-inflammatory and immunomodulatory purposes due to its stem cell properties and the secreted growth factors. Moreover, MSCs is a promising option to treat tumor-induced osteolysis due to the osteogenic potential and the capability of delivering anti-cancer reagents.Citation4 Unfortunately, MSCs are also a source of Carcinoma-associated fibroblasts (CAFs),Citation5,Citation6 which promote growth and metastasis of cancer cells by secreting pro-tumor factors and inhibiting anti-tumor immunity.Citation7,Citation8 Therefore, to some extent, these characteristics suggest the dual properties of MCSs on either promoting or suppressing tumorgenesis.
Differentiation of MSCs into CAFs can be induced by tumor-conditioned medium (TCM), suggesting the important roles of secreted factors in CAF differentiation.Citation5 Numerous studies demonstrated that transforming growth factor-β (TGF-β) signaling pathway might be essential for CAFs differentiation of MSCs. The key factor TGF-β1 can induce expression of some CAF markers2 such as α-smooth muscle actin (α-SMA), Vimentin, fibroblast surface protein 1 (FSP1, also called S100A4) and fibroblast activated protein (FAP) in MSCs,Citation9 which promote growth and metastasis of cancer cells. Furthermore, CXCR4 is an endogenous receptor for the chemokine CXCL12 (also known as SDF-1),Citation10,Citation11 which increases the secretion of TGF-β1 and thus promotes tumor cell proliferation, migration and invasion.Citation12 Additionally, it has been identified that CXCL12/CXCR4 is an important signaling pathway for the metastasis of CRCs to the liver.Citation13 Moreover, CXCR4 is also expressed on MSCs and modulates MSCs migration and apoptosis.Citation14 Based on these researches, we proposed that CXCR4/TGF-β1 may play an important role in the MSCs by promoting CRCs proliferation and metastasis in TME.
In the present study, we used transwell co-culture technology to investigate the intercellular interaction between MSCs and CRC cells. The effects on CRC cells proliferation, migration and invasion, and the markers of CAFs differentiation from MSCs after co-culture were evaluated. Treatment with AMD3100, a selective CXCR4 antagonist, or cyclophosphamide (Cy), a chemotherapy drug suppressing TGF-β1, further verified the essential role of CXCR4/TGF-β1 axis in the transformation of tumor microenvironment by mediating MSCs differentiation into CAFs in vivo and in vitro.
Materials and methods
Cell culture and drug treatment
The human bone marrow MSCs and human colorectal cancer cell lines HCT116 and HT29 were obtained from the American Type Culture Collection (ATCC, USA). HCT116 and HT29 were cultured in RPMI 1640 medium (Gibco, USA) supplemented with 10% fetal bovine serum (FBS) and 100 U/ml penicillin and streptomycin (Gibco, USA) at 37°C in a 5% CO2 humidified atmosphere, but the MSCs were cultured with α-minimum essential medium (α-MEM, Gibco, USA) and 10% FBS.
To study the intercellular interaction between MSCs and CRC cells, a Transwell co-culture method was performed. Briefly, HCT116/HT29 cells were seeded in the upper chamber (0.45 μm pores, Corning, USA) with MSCs lawns in the lower chambers (24-well or 6-well plates), and then these cells were cultured for 24, 48 and 72 h. The control group was added medium in the lower chambers only.
Inhibitors AMD3100 and Cy were purchased from Sigma (USA) and dissolved in 1% dimethylsulfoxide (DMSO) to made up 10 mM stock solutions.
Colony formation assay
Colony formation assay were performed as reported previously.Citation14 HCT116/HT29 cells (1 × 10Citation3) were seeded in a 60-mm dish and cultured with medium collected from the co-cultured system of MSCs with HCT116/HT29 for 14 days to allow colony formation. The surviving colonies were fixed with paraformaldehyde (4.0% v/v) and then stained with crystal violet (0.4% w/v) to be visible and counted.
Migration and invasion assay
The migration and invasion assay was performed in Transwell 24-well plates with 8.0 μm pore-size transwell inserts (Corning, USA). The upper side of the membrane was coated with Matrigel (20 μg/well, Sigma, USA) for invasion assay and without materiel coating for migration assay. The CRC cells HCT116/HT29 (1 × 10Citation4) were placed in the upper chamber and MSCs (1 × 10Citation5) were seeded in the lower chamber. After incubation at 37°C for 48 hours, the cells remaining on the upper surface of the membrane were removed gently with a cotton swab. The membranes were fixed with 4% (w/v) paraformaldehyde and stained with 0.1% crystal violet. Then migrated or invased cells were counted under an optical microscope (Olympus, Japan).
Immunofluorescence
After culture and treatment, cells were fixed with 4% (w/v) paraformaldehyde at room temperature for 20 min, washed with Phosphate-Buffered Saline (PBS) and then permeabilized with 0.1% (v/v) Triton X-100 for 10 min. After blocked with 10% FBS at room temperature for 30 min, the cells were incubated overnight at 4°C in the presence of primary antibodies against α-SMA (1:250, Abcam, England) and Vimentin (1:200, Abcam, England). After washing with PBS, cells were incubated for 1 hour at room temperature with secondary antibodies conjugated with Alexa Fluor 488 (green) or Alexa Fluor 647 (red). Following three washes with PBS, DAPI (ThermoFisher Scientific, USA) was used for nuclear counterstaining. In the end, images of stained cells were captured by a fluorescence microscope (Olympus, Japan).
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the cell or tissue samples using TRIzol reagent (Invitrogen, USA) following conventional processes. Subsequently, cDNA was synthesized using a TaqMan Reverse Transcription Reagents Kit (ThermoFisher Scientific, USA). The relative expression levels of mRNA were determined using a SYBR Green PCR Master Mix kit (Takara, Japan) on a thermocycler (7500, Applied Biosystems, USA), and the data were analyzaed by a 2−ΔΔCt method normalized to GAPDH.
Western Blot and ELISA
The total proteins were extracted by a RIPA Lysis buffer (ThermoFisher Scientific, USA) and then quantified using a BCA assay kit (ThermoFisher Scientific, USA). Equal amounts of the total protein (30 μg) were separated by SDS polyacrylamide gel electrophoresis (SDS-PAGE). Afterward, the separated protein bands were transferred to a PVDF membrane (Millipore, USA), which were then blocked by 5% skim milk for one hour at room temperature. Primary antibodies diluted in 5% skim milk (1:1000) were used to incubate the membranes at 4°C overnight, followed by horseradish peroxidase (HRP)-conjugated secondary antibody incubation for 1 hour at room temperature. The protein bands were visualized by ECL reagent (ThermoFisher Scientific, USA) under a chemiluminescence system (Bio-Rad, USA). The primary antibodies for α-SMA, Vimentin, FAP, S100A4, TGF-β1 and CXCR4 were purchased from Abcam (England) but the anti-E-cadherin antibody, anti-N-cadherin antibody and the β-actin antibody were purchased from Cell Signaling Technology (USA).
The SDF-1 and TGF-β1 concentrations in the cell supernatants were quantitatively measured using commercial kits (Abcam, England) following the manufacturer’s instruction.
Xenograft model
Animal experiments were performed under a protocol approved by the Laboratory Animal Welfare and Ethics Committee of Chinese Association for Laboratory Animal Sciences (CALAS) and the Medical Ethics Committee of Hunan Provincial People’s Hospital. Five- to six-week-old BALB/C mice (Jackson Labs) were housed under pathogen-free conditions and were given autoclaved food and water ad libitum. 1 × 10Citation6 HCT116 cells were subcutaneously injected in the right flank to generate CRC xenograft models. When these cells developed palpable tumors, the CRC model mice were randomly divided into four groups (n = 8 for each group): Alone, MSCs, MSCs+AMD3100, and MSCs+Cy. The mice of latter three groups were intratumor injected with MSCs which were pretreated with DMSO, AMD3100 (20 μM) and Cy (5 μM) for 6 hrs, respectively. Thereafter, DMSO/AMD3100/Cy was sequentially intratumor injected every other day. The volume of tumors was measured every five days. After 30 days administration with MSCs and inhibitors, the mice were sacrificed and tumor samples were collected for further analysis.
To establish the liver metastasis models, HCT116 was injected via tail vein without or with MSCs, which were pretreated DMSO, AMD3100 (50 μM) and Cy (5 μM) for 6 hrs, respectively. Thereafter, these mice of MSCs-injected groups were sequentially administrated with DMSO/AMD3100/Cy by intraperitoneal injected every other day for 30 days. Then the liver samples were harvested for metastatic pathological observation and analysis.
Immunohistochemistry staining
Tissue samples fixed in formalin (at least 24 h) were embedded in paraffin and then cut into 4 μm sections and placed onto poly-prep slides (Sigma, USA). After deparaffinized and hydrated in 100% and 95% ethanol successively, the slides were treated with 10 mM sodium citrate (pH 6.0) for 10 min at 95°C for antigen retrieval. When cooled to room temperature, the slices were blocked with 5% normal horse serum, followed by incubation with primary antibodies specific for α-SMA and Vimentin (1: 500, Abcam, USA) at 4°C for overnight. Positive reactions were visualized as brown by a 3, 3-diamino-benzidine (DAB) reagent (Sigma, USA) and the cell nuclears were counterstained with hemotoxylin.
Statistical analysis
All data presented here were carried out at least three times with consistent results and expressed as mean ± standard deviation (SD). Data were analyzed by GraphPad Prism 6.0 (GraphPad Software Inc., San Diego, CA). Statistical differences were performed using one-way analysis of variance (ANOVA) followed by Turkey post hoc test for multiple comparison. A p < .05 was considered statistically significant.
Results
Mscs promoted CRC cells proliferation, migration and invasion and increased SDF-1 secretion
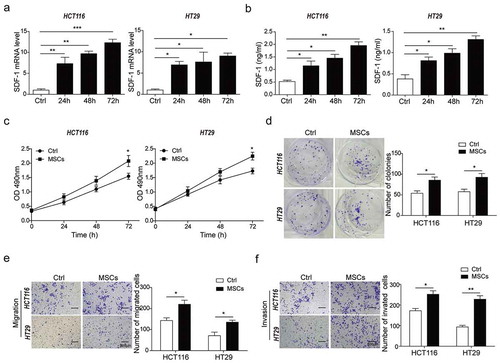
To explore the intercellular interaction between MSCs and CRC cells, the MSCs were co-cultured with HCT116 and HT29 cells respectively for 24, 48 and 72 h. In the co-culture system, the expression and secretion of stromal cell-derived factor-1 (SDF-1) significantly increased with the extension of co-culture time ()), indicating that MSCs promoted syntheses and secretion of SDF-1 in CRC cells. Simultaneously, faster growth rate (,)), higher migration capacity ()) and higher invasion capacity ()) were observed in MSCs-cocultured CRC cells (HCT116 and HT29). These results demonstrated that MSCs induced expression and secretion of SDF-1 and promoted proliferation, migration and invasion in CRC cells.
Figure 1. MSCs promoted CRC cells proliferation, migration and invasion and increased SDF-1 secretion.
(a) qPCR analysis for mRNA level of SDF-1 in response to co-culture time (24h, 48h and 72h) in HCT116 and HT29 cells with or without MSCs. (b) ELISA analysis for protein level of SDF-1 secretion after 24h, 48h, or 72h co-culture. (c) CCK-8 assay for cell proliferation of HCT116 and HT29 cells after 24h, 48h, or 72h co-culture. (d) Colony formation assay for cell proliferation of HCT116 and HT29 cells co-cultured with or without MSCs for 48h. (e) Matrigel-free transwell assay for cell migration of HCT116 and HT29 cells as described in D. (f) Matrigel transwell assay for cell invasion of HCT116 and HT29 cells co-cultured with or without MSCs for 48h. * p < .05, ** p < .01, *** p < .001.
CRC cells induced CAFs differentiation in MSCs
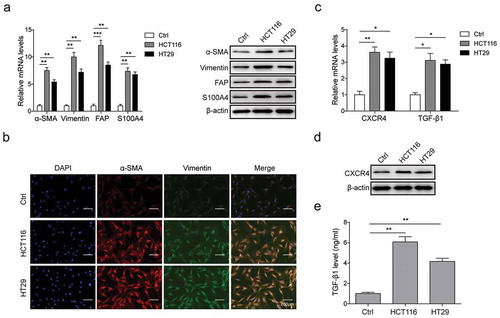
To identify CAFs differentiation induced by tumor cells in CRC, the expression of CAFs biomarkers was investigated in MSCs after co-cultured with HCT116/HT29 cells for 48 h. As indicated by qRT-PCR and Western Blot, the mRNA and protein levels of CAFs biomarkers including α-SMA, Vimentin, S100A4 and FAP presented a significant increase in MSCs co-cultured with HCT116 (HCT116-MSCs) and HT29 cells (HT9-MSCs) compared to the no-co-cultured group (Ctrl) ()). Simultaneously, higher expression levels of α-SMA and Vimentin were observed by immunofluorescence staining ()) in HCT116-MSCs and HT9-MSCs, indicating CAFs differentiation was occurred in MSCs. In addition, it was noted that the expression levels of TGF-β1 and CXCR4 were significantly increased in HCT116-MSCs and HT29-MSCs compared to the Ctrl group (,)), as well as the secretion of TGF-β1 ()). These data suggested that co-cultured with CRC cells, MSCs may be induced to initiate differentiation to CAFs, and the up-regulated CXCR4 expression and TGF-β1 secretion may be closely related to the differentiation process.
Figure 2. CRC cells induced CAFs differentiation in MSCs.
(a) qPCR and western blot analysis for expression of CAFs markers α-SMA, Vimentin, S100A4 and FAP in mRNA and protein levels in MSCs co-cultured with HCT116/HT29 cells or not. (b) Immunofluorescence staining for α-SMA and Vimentin in MSCs with or without HCT116/HT29 cells co-culture. (c) qPCR and western blot analysis for mRNA levels of CXCR4 and TGF-β1 in MSCs co-cultured with HCT116/HT29 cells or not. (d) Western blot analysis for protein expression level of CXCR4 in MSCs in the presence or absence of co-culture. (E) ELISA analysis for TGF-β1 secretion from MSCs after HCT116/HT29 cells co-culture or not. * p < .05, ** p < .01, *** p < .001.
Inhibition of CXCR4 expression and TGF-β1 secretion prevented CRC-induced MSCs self-differentiation to CAFs
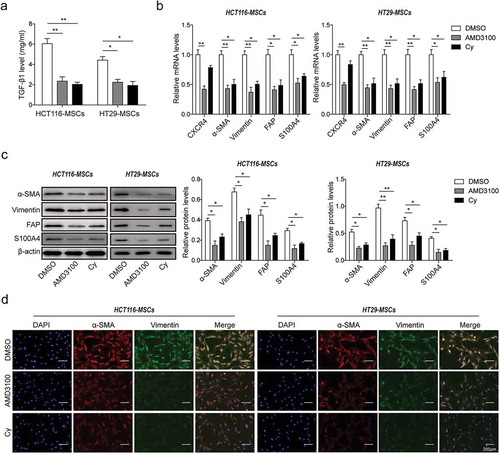
To further confirm the importance of CXCR4 and TGF-β1 in CRCs-MSCs interaction, inhibitors AMD3100 (20 μM, for CXCR4 inhibition) and Cy (5 μM, for TGF-β1 inhibition) were treated in HCT116-MSCs and HT29-MSCs, respectively. DMSO treatment was performed as a control. The concentration of TGF-β1 in supernatants significantly decreased after AMD3100 and Cy treatment compared to DMSO treatment ()), suggesting that TGF-β1 secretion was suppressed by AMD3100 and Cy. As indicated by qRT-PCR and Western Blot assays, the mRNA and protein expression levels of CXCR4 significantly reduced by AMD3100 treatment while no significant difference after Cy treatment compared to DMSO treatment ()), so it is inferred that TGF-β1 secretion may be modulated by CXCR4 signaling activation. Furthermore, the mRNA and protein expression of CAFs biomarkers α-SMA, Vimentin, S100A4 and FAP were significantly declined in HCT116-MSCs and HT29-MSCs after AMD3100 or Cy treatment ()), indicating the CAFs differentiation in HCT116-MSCs and HT29-MSCs was blocked by AMD3100 and Cy. Consistent results were obtained by immunofluorescence staining analysis ()). According to these results, we confirmed that the CXCR4/TGF-β1 signaling axis was involved in the process of CAFs differentiation from HCT116/HT29-induced MSCs.
Figure 3. Inhibition of CXCR4 expression and TGF-β1 secretion prevented CRC-induced MSCs self-differentiation to CAFs.
(a) ELISA analysis for TGF-β1 secretion in HCT116-MSCs and HT29-MSCs (short for MSCs co-cultured with HCT116/HT29 cells) pretreated with DMSO, AMD3100 or Cy. (b) qPCR analysis in mRNA level, and (c) western blot analysis in protein level for the expression of CXCR4 and CAFs markers α-SMA, Vimentin, S100A4 and FAP in HCT116-MSCs and HT29-MSCs cells treated with DMSO, AMD3100 or Cy. The quantitative analysis of protein bands grey intensity were displayed in the hisograms. (d) Immunofluorescence staining for α-SMA and Vimentin in HCT116-MSCs and HT29-MSCs cells treated with DMSO, AMD3100 or Cy. * p < .05, ** p < .01
Blocking CXCR4/TGF-β1 signaling inhibited CRC cells migration and invasion in vitro
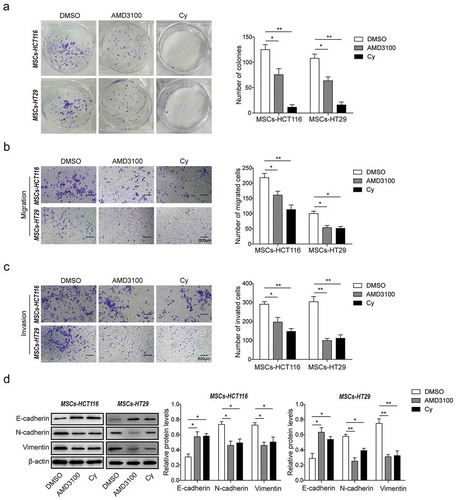
To further confirm the effects of CXCR4/TGF-β1 signaling on CRC cells proliferation, migration and invasion, colony formation assay and Transwell assay were performed, and the epithelial-mesenchymal transition (EMT) in CRC cells was evaluated. The number of colonies was significantly reduced in MSCs-co-cultured HCT116 cells (MSCs-HCT116) and HT29 cells (MSCs-HT29) after AMD3100 and Cy treatment compared to DMSO treatment ()). Consistently, the migration and invasion capacities of MSCs-HCT116 and MSCs-HT29 cells were also observably inhibited by AMD3100 and Cy (,)). Moreover, compared to the control group (DMSO), the expression level of epithelial marker E-cadherin was significantly increased and the expression levels of mesenchymal markers N-cadherin and Vimentin were remarkably decreased in MSCs-HCT116 and MSCs-HT29 cells treated with AMD3100 and Cy ()). All these data indicated that blocking CXCR4/TGF-β1 signaling by inhibitors significantly suppressed the increase of CRC cells proliferation, migration and invasion promoted by MSCs, and it was probably due to the attenuated CAFs differentiation in MSCs by AMD3100 and Cy.
Figure 4. Blocking CXCR4/TGF-β1 signaling inhibits CRC cells migration and invasion in vitro.
(a) Colony formation assay for cell proliferation in MSCs-HCT116 and MSCs-HT29 cells (short for HCT116/HT29 cells co-cultured with MSCs) pretreated with DMSO, AMD3100 or Cy. (b) Transwell assays without matrigel for cell migration, and (c) with matrigel for cell invasion analysis were measured in MSCs-HCT116 and MSCs-HT29 cells pretreated with DMSO, AMD3100 or Cy. (d) Western blot analysis for protein levels of EMT markers E-cadherin, N-cadherin and Vimentin were detected in MSCs-HCT116 and MSCs-HT29 cells pretreated with DMSO, AMD3100 or Cy. The quantitative analyses of protein bands grey intensity were displayed in the hisograms. * p < .05, ** p < .01
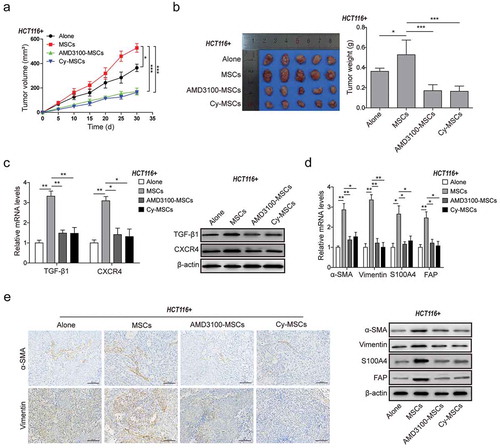
MSCs accelerated CRC xenograft tumor growth and CAFs differentiation in vivo via CXCR4/TGF-β1 axis
To further verify the role of CXCR4/TGF-β1 axis in MSC-induced CRC development in vivo, CRC subcutaneous xenograft models in nude mice were established (n = 20, five in each group). Compared to HCT116 cells injected alone (Alone group), tumors grew fastest after injection with MSCs pretreated with DMSO (MSCs group). However, the rate of tumor growth induced by MSCs injection was significantly inhibited by AMD3100 and Cy treatment (AMD3100-MSCs and Cy-MSCs) ()). The size and weight of tumor after 30-days treatment were significantly higher in MSCs group than the Alone group, but they were remarkably decreased after AMD3100 and Cy treatment ()). In addition, the mRNA and protein expression of CXCR4, TGF-β1 and CAFs markers (α-SMA, Vimentin, S100A4 and FAP) were significantly up-regulated in MSCs group, which were remarkably down-regulated by AMD3100 and Cy treatment (,)). Consistent results were obtained by immunofluorescence staining ()). These experiments demonstrated that MSCs accelerated CRC xenograft tumor growth and CAFs differentiation in vivo, which may be blocked through suppressing CXCR4/TGF-β1 signaling.
Figure 5. MSCs accelerated CRC xenograft tumor growth and CAFs differentiation in vivo via CXCR4/TGF-β1 axis.
(a) Tumor volume growth delay curves in athymic nude mice (n = 20, five in each group) measured every five days for HCT116 cell xenografts treated with or without MSCs in the presence of DMSO, AMD3100 or Cy administration for 30 days. (b) Observation of the xenograft tumors from nude mice of each group (n = 5) after 30 days growth and drug treatment. The weights of these xenograft tumors were presented in the histogram. (c) qPCR and western blot analysis for expression of TGF-β1 and CXCR4 in the xenograft tumor tissues from the HCT116 cell xenografts mice treated with or without MSCs in the presence of DMSO, AMD3100 or Cy administration for 30 days. (d) qPCR and western blot analysis for expression of CAFs markers α-SMA, Vimentin, S100A4 and FAP, and (e) Immunohistochemistry analysis for the levels of α-SMA and Vimentin in the xenograft tumor tissues after MSCs treatment with or without AMD3100/Cy treatment. * p < .05, ** p < .01, *** p < .001
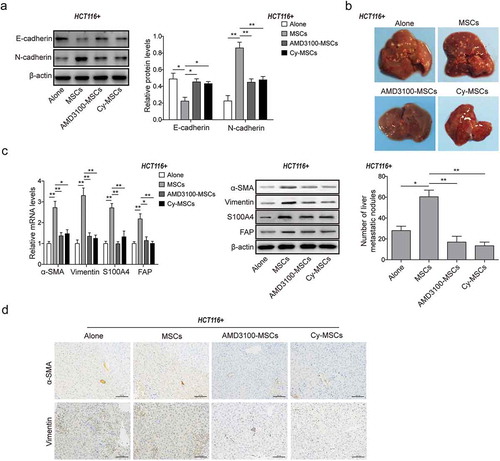
MSCs promoted CRC liver metastasis by self-differentiation to CAFs via CXCR4/TGF-β1 axis
The effects of MSCs on CRC liver metastasis were investigated in vivo. In the primary CRC xenograft tumors, the EMT were enhanced by decreased E-cadherin and increased N-cadherin in MSCs group compared to the Alone group, which were however attenuated by AMD3100 and Cy treatment by rescuing the expression of E-cadherin and N-cadherin ()). Furthermore, more liver metastatic nodules were observed in MSCs group than the Alone group. However, the number of nodules significantly decreased in groups for AMD3100 and Cy treatment ()), indicating that inhibitors AMD3100 and Cy prevented CRC liver metastasis induced by MSCs. The mRNA and protein expression of CAFs markers (α-SMA, Vimentin, S100A4 and FAP) were significantly up-regulated in the metastatic liver tissues of MSCs group, which were dramatically reduced in the metastatic tissues of groups with AMD3100 and Cy treatment (,)). These data suggested that MSCs promoted the liver metastasis of CRC by self-differentiation to CAFs via CXCR4/TGF-β1 axis.
Figure 6. MSCs promoted CRC liver metastasis by self-differentiation to CAFs via CXCR4/TGF-β1 axis.
(a) Western blot analysis for protein levels of E-cadherin and N-cadherin in the primary xenograft tumor tissues treated with or without MSCs and DMSO, AMD3100 or Cy administration. The quantitative analyses of protein bands grey intensity were displayed in the hisograms. (b) Observation and counting of metastatic nodules on the liver surface from HCT116 nude mice of each group treated with or without MSCs and DMSO, AMD3100 or Cy administration for 30 days. (c) qPCR and western blot analysis for expression of CAFs markers α-SMA, Vimentin, S100A4 and FAP, and (d) Immunohistochemistry analysis for the levels of α-SMA and Vimentin in the metastatic liver tissues from HCT116 nude mice of each group treated with or without MSCs and DMSO, AMD3100 or Cy administration. * p < .05, ** p < .01.
Discussion
CAFs are a heterogeneous population of fibroblast-like cells with a tumor promoting function, which may be due to the variation of cell origins and the molecular constitution of tumor stroma. CAFs have been found to originate from bone marrow MSCs and TGF-β may be involved in the transition as MSCs transduced with a lentivirus vector which inhibited TGF-β/smad signaling, expressed a decrease in CAF markers when conditioned for 10 days in tumor cell conditioned medium in comparison to naïve MSCs .Citation15 In addition, treatment of MSCs with the endoplasmic reticulum chaperone, GRP78, activated TGF-β/smad signaling and induced the differentiation to a CAF like phenotype .Citation16 These are consistent with what we found in the present study. When HCT116 and HT29 co-cultured with MSCs, the mRNA and protein expression level of multiple CAF markers were significantly enhanced. Simultaneously, higher expression level of α-SMA and Vimentin was observed in co-culture HCT116-MSCs and HT29-MSCs, indicating the increased differentiation of CAFs in co-culture cells. Considering that many processes between gene transcription and protein translation and especially the half-life of different proteins could vary dramatically from minutes to days while mRNA degradation rate is usually within hours .Citation17 Thus, the correlation of fold difference between mRNA level and protein level could vary a lot. Therefore, it is clear that TGF-β plays a major role in the differentiation from MSC to CAF, promoting the formation of tumor microenvironment, however it is unclear to what degree it affects the secretory profile of the cells and their functional characteristics .Citation18
In this study, both CRCs and MSCs cells express CXCR4 and secret TGF-β1. Transforming growth factor β (TGF-β) is a widely-acknowledged chondrogenic induction factor for MSCsCitation19-Citation21 and TGF-β-stimulated signaling pathway might be essential for CAF differentiation of MSCs. TGF-β1-enhanced CXCR4 expression was reported in bone marrow-derived stem cells (BMSCs) .Citation22 It is possible for TGF-β1 as a multi-potent factor to assist in the SDF-1, which is important in promoting both tumor growth and angiogenesis and driving migration by targeting CXCR4 .Citation23 Furthermore, it was first elucidated that the TGF-β1 augmented CXCR4 expression is mediated by the phosphorylation of ERK pathway .Citation24 In addition to the induction of CAF differentiation, SDF-1 released by CAFs directly mediates pro-tumor effects of CAFs by promoting tumor growth and angiogenesis .Citation25 Furthermore, inhibition of CXCR4 expression and TGF-β1 secretion significantly blocked their differentiation from HCT116-MSCs or HT29-MSCs to CAFs in tumor microenvironments both in vitro and in vivo, which would be consistent with the findings of Lei et al .Citation15 These findings verified the essential role of CXCR4/TGF-β1 signaling in mediating the differentiation of HCT116/HT29-induced MSCs to CAFs and pro-tumor effects.
Furthermore, the present study showed that when HCT116 and HT29 co-cultured with MSCs the mRNA and protein expression level of multiple CAF markers including α-SMA, Vimentin, FSP and FAP were significantly enhanced compare to the control group, indicating that MSCs may promote tumor growth and metastasis, which is consistent with several coinjection experiments .Citation5,Citation26 To determine the effect of MSCs on metastasis in vivo, we used an experimental lung metastasis model, and we found that MSCs enhanced the metastatic potential of CRCs. Thus, it is reasonable to suppose that MSC promoted EMT in HCT116 cells to enhance their metastatic potentialCitation27 and AMD3100 and Cyclophosphamide treatment inhibited this process. The metastatic mechanisms of EMT have been widely recognizedCitation28 and recent observations have revealed that chemokine networks in cancer cells are utilized to modulate the host microenvironment in favor of cancer progression .Citation29 Interactions between CXCR4 and CXCL12 have been considered to play a role in the progression of colon cancerCitation30,Citation31 and also EMT is mediated by the CXCR4/CXCL12 system .Citation32,Citation33
To sum up, the present study has revealed an essential role of CXCR4/TGF-β1 axis playing in the transformation of tumor microenvironment by mediating MSCs self-differentiation to CAFs, promoting CRC growth and metastasis. Inhibition on CXCR4/TGF-β1 axis may be a potential approach to improve the safety of MSCs therapies in CRC even other cancers. A better understanding of interplay between MSCs and CRCs within the tumor microenvironment will be important in developing strategies for improving tumor therapy which considering the effects of the tumor microenvironment on tumor survival and growth.
Conflict
The authors declare no conflict of interest.
Acknowledgments
This work was supported by Natural Science Foundation of China (No. 81460379).
References
- Friedl P, Alexander S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell. 2011;147(5):992–1009. doi:10.1016/j.cell.2011.11.016.
- Lyden D, Hattori K, Dias S, Costa C, Blaikie P, Butros L, Chadburn A, Heissig B, Marks W, Witte L, et al. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat Med. 2001;7(11):1194–1201. doi:10.1038/nm1101-1194.
- Phinney DG, Prockop DJ. Concise review: mesenchymal stem/multipotent stromal cells: the state of transdifferentiation and modes of tissue repair–current views. Stem Cells. 2007;25(11):2896–2902. doi:10.1634/stemcells.2007-0637.
- Fritz V, Noël D, Bouquet C, Opolon P, Voide R, Apparailly F, Louis-Plence P, Bouffi C, Drissi H, Xie C, et al. Antitumoral activity and osteogenic potential of mesenchymal stem cells expressing the urokinase-type plasminogen antagonist amino-terminal fragment in a murine model of osteolytic tumor. Stem Cells. 2008;26(11):2981–2990. doi:10.1634/stemcells.2008-0139.
- Mishra PJ, Mishra PJ, Humeniuk R, Medina DJ, Alexe G, Mesirov JP, Ganesan S, Glod JW, Banerjee D. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008;68(11):4331–4339. doi:10.1158/0008-5472.CAN-08-0943.
- Spaeth EL, Dembinski JL, Sasser AK, Watson K, Klopp A, Hall B, Andreeff M, Marini F. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS One. 2009;4(4):e4992. doi:10.1371/journal.pone.0004992.
- Ostman A, Augsten M. Cancer-associated fibroblasts and tumor growth–bystanders turning into key players. Curr Opin Genet Dev. 2009;19(1):67–73. doi:10.1016/j.gde.2009.01.003.
- Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, Gopinathan A, Tuveson DA, Fearon DT. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science. 2010;330(6005):827–830. doi:10.1126/science.1195300.
- Wang D, Park JS, Chu JSF, Krakowski A, Luo K, Chen DJ, Li S. Proteomic profiling of bone marrow mesenchymal stem cells upon transforming growth factor beta1 stimulation. J Biol Chem. 2004;279(42):43725–43734. doi:10.1074/jbc.M407368200.
- Yoshitake N, Fukui H, Yamagishi H, Sekikawa A, Fujii S, Tomita S, Ichikawa K, Imura J, Hiraishi H, Fujimori T, et al. Expression of SDF-1 alpha and nuclear CXCR4 predicts lymph node metastasis in colorectal cancer. Br J Cancer. 2008;98(10):1682–1689. doi:10.1038/sj.bjc.6604363.
- Kollmar O, Rupertus K, Scheuer C, Junker B, Tilton B, Schilling MK, Menger MD. Stromal cell-derived factor-1 promotes cell migration and tumor growth of colorectal metastasis. Neoplasia. 2007;9(10):862–870. doi:10.1593/neo.07559.
- Zhang L, Yeger H, Das B, Irwin MS, Baruchel S. Tissue microenvironment modulates CXCR4 expression and tumor metastasis in neuroblastoma. Neoplasia. 2007;9(1):36–46. doi:10.1593/neo.06670.
- Minagawa M, Makuuchi M, Torzilli G, Takayama T, Kawasaki S, Kosuge T, Yamamoto J, Imamura H. Extension of the frontiers of surgical indications in the treatment of liver metastases from colorectal cancer: long-term results. Ann Surg. 2000;231(4):487–499. doi:10.1097/00000658-200004000-00006.
- Lee RH, Seo MJ, Pulin AA, Gregory CA, Ylostalo J, Prockop DJ. The CD34-like protein PODXL and alpha6-integrin (CD49f) identify early progenitor MSCs with increased clonogenicity and migration to infarcted heart in mice. Blood. 2009;113(4):816–826. doi:10.1182/blood-2007-12-128702.
- Shangguan L, Ti X, Krause U, Hai B, Zhao Y, Yang Z, Liu F. Inhibition of TGF-β/Smad signaling by BAMBI blocks differentiation of human mesenchymal stem cells to carcinoma-associated fibroblasts and abolishes their protumor effects. Stem Cells. 2012;30(12):2810–2819. doi:10.1002/stem.v30.12.
- Peng Y, Li Z, Li Z. GRP78 secreted by tumor cells stimulates differentiation of bone marrow mesenchymal stem cells to cancer-associated fibroblasts. Biochem Biophys Res Commun. 2013;440(4):558–563. doi:10.1016/j.bbrc.2013.09.108.
- Vogel C, Marcotte EM. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet. 2012;13(4):227–232. doi:10.1038/nrg3185.
- Ridge SM, Sullivan FJ, Glynn SA. Mesenchymal stem cells: key players in cancer progression. Mol Cancer. 2017;16(1):31. doi:10.1186/s12943-017-0597-8.
- Dong M, Blobe GC. Role of transforming growth factor-beta in hematologic malignancies. Blood. 2006;107(12):4589–4596. doi:10.1182/blood-2005-10-4169.
- Zhang Y, Li J, Davis ME, Pei M. Delineation of in vitro chondrogenesis of human synovial stem cells following preconditioning using decellularized matrix. Acta Biomater. 2015;20:39–50. doi:10.1016/j.actbio.2015.04.001.
- Zhang Y, Pizzute T, Li J, He F, Pei M. sb203580 preconditioning recharges matrix-expanded human adult stem cells for chondrogenesis in an inflammatory environment - A feasible approach for autologous stem cell based osteoarthritic cartilage repair. Biomaterials. 2015;64:88–97. doi:10.1016/j.biomaterials.2015.06.038.
- Zhang S-J, Song X-Y, He M, Yu S-B. 2016. Effect of TGF-β1/SDF-1/CXCR4 signal on BM-MSCs homing in rat heart of ischemia/perfusion injury. Eur Rev Med Pharmacol Sci. 20(5):899–905.
- Wang Y, Chen J, Fan W, Zhang J, Hua B, Sun B, Zhu L, Niu X, Yan Z, Guo C, et al. 2017. Stromal cell-derived factor-1α and transforming growth factor-β. Am J Transl Res. 9(5):2656–2667.
- Liu X, Duan B, Cheng Z, Jia X, Mao L, Fu H, Che Y, Ou L, Liu L, Kong D, et al. SDF-1/CXCR4 axis modulates bone marrow mesenchymal stem cell apoptosis, migration and cytokine secretion. Protein Cell. 2011;2(10):845–854. doi:10.1007/s13238-011-1097-z.
- Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121(3):335–348. doi:10.1016/j.cell.2005.02.034.
- Mishra PJ, Glod JW, Banerjee D. Mesenchymal stem cells: flip side of the coin. Cancer Res. 2009;69(4):1255–1258. doi:10.1158/0008-5472.CAN-08-3562.
- Xue Z, Wu X, Chen X, Liu Y, Wang X, Wu K, Nie Y, Fan D. Mesenchymal stem cells promote epithelial to mesenchymal transition and metastasis in gastric cancer though paracrine cues and close physical contact. J Cell Biochem. 2015;116(4):618–627. doi:10.1002/jcb.25013.
- Tse JC, Kalluri R. Mechanisms of metastasis: epithelial-to-mesenchymal transition and contribution of tumor microenvironment. J Cell Biochem. 2007;101(4):816–829. doi:10.1002/(ISSN)1097-4644.
- Sakai N, Yoshidome H, Shida T, Kimura F, Shimizu H, Ohtsuka M, Takeuchi D, Sakakibara M, Miyazaki M. CXCR4/CXCL12 expression profile is associated with tumor microenvironment and clinical outcome of liver metastases of colorectal cancer. Clin Exp Metastasis. 2012;29(2):101–110. doi:10.1007/s10585-011-9433-5.
- Schimanski CC, Schwald, S., Simiantonaki, N., Jayasinghe, C., Gönner, U., Wilsberg, V., Junginger, T., Berger, M.R., Galle, P.R., Moehler, M, et al. Effect of chemokine receptors CXCR4 and CCR7 on the metastatic behavior of human colorectal cancer. Clin Cancer Res. 2005;11(5):1743–1750. doi:10.1158/1078-0432.CCR-04-1195.
- Zeelenberg IS, Ruuls-Van Stalle L, Roos E. The chemokine receptor CXCR4 is required for outgrowth of colon carcinoma micrometastases. Cancer Res. 2003;63(13):3833–3839.
- Onoue T, Uchida D, Begum NM, Tomizuka Y, Yoshida H, Sato M. 2006. Epithelial-mesenchymal transition induced by the stromal cell-derived factor-1/CXCR4 system in oral squamous cell carcinoma cells. Int J Oncol. 29(5):1133–1138.
- Taki M, Higashikawa K, Yoneda S, Ono S, Shigeishi H, Nagayama M, Kamata N. 2008. Up-regulation of stromal cell-derived factor-1alpha and its receptor CXCR4 expression accompanied with epithelial-mesenchymal transition in human oral squamous cell carcinoma. Oncol Rep. 19(4):993–998.