
ABSTRACT
Angiomyolipoma (AML) is classified as a perivascular epithelioid cell neoplasm, mostly occurring in the kidney. Twenty percent of patients with renal AML have tuberous sclerosis complex (TSC) caused by germline variation in the TSC1 or TSC2 gene. In this paper, we report the first case of renal AML harboring somatic missense mutations of the TSC2 gene and concomitant copy-neutral loss of heterozygosity (CN-LOH). The patient presented with solitary renal AML and pulmonary lymphangiomyomatosis and without other findings suggestive of TSC. Exome sequencing analysis of the renal AML, however, identified a pathogenic somatic missense mutation in the TSC2 gene (NM_000548:c.5228G>A:p. R1743Q), although no other somatic mutation was detected. Furthermore, no germline mutation in TSC1 or TSC2 was detected. Interestingly, the mutant allele ratio was too high for a somatic heterozygous mutation without loss of heterozygosity (LOH). Furthermore, no copy number variation was detected around the TSC2 locus (16p13.3). To clarify the allelic status, we analyzed heterozygous single-nucleotide polymorphisms (SNPs) in chromosome 16. In these SNPs, an unbalanced allele ratio was accumulated inside the 16p13.3 region. These results suggested copy-neutral LOH (CN-LOH). Consequently, we concluded that the missense mutation of the TSC2 gene and CN-LOH of the TSC2 locus caused renal AML.
Introduction
Angiomyolipoma (AML) is classified as a kind of perivascular epithelioid cell neoplasm (PEComa).Citation1 Most AML occurs in the kidney, and renal AML is the most common PEComa.Citation1,Citation2 Although renal AML is a histologically benign tumor, it often causes clinically critical events such as hemorrhage and renal failure.Citation3 Twenty percent of patients with renal AML have tuberous sclerosis complex (TSC), an autosomal dominant genetic disease due to functional loss of the TSC1 or TSC2 gene.Citation2 The recent development of sequencing technology makes it possible to elucidate the etiology and molecular pathology of various diseases and syndromes, especially in genetic disorders and tumors. In this paper, we performed exome sequencing analysis of renal AML and reported the first case of renal AML harboring a missense mutation of the TSC2 gene with a copy-neutral loss of heterozygosity (CN-LOH).
Case report
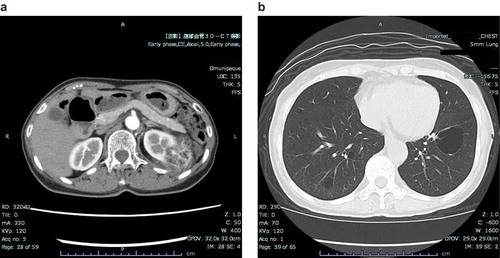
A woman in her seventies had a renal tumor and multiple lung cysts, which were incidentally found by computed tomography (CT). Her medical history included a laryngeal cyst and a uterine cyst. She had no history of pneumothorax, seizure or mental retardation. Among her five siblings, one of her younger sisters had a history of a solitary renal AML. On physical examination, no skin lesions related to TSC, such as facial papules, plaques, periungual papules or hypopigmented macules, were observed. Her complete blood count, biochemistry, urinalysis and pulmonary function were normal. Contrast-enhanced CT of the abdomen revealed a solitary tumor involving the cortex of the left kidney, 86 × 42 × 58 mm in size (). The tumor consisted of a mixture of areas with fat density and areas with high CT values. CT images of the chest revealed approximately 40 cysts of various size with thin walls in the bilateral lungs, which suggested the radiological diagnosis of lymphangiomyomatosis (LAM) (). In this case, it was essential to perform differential diagnosis distinguishing Birt-Hogg-Dubé syndrome, which is disease involving multiple pulmonary cysts, tumors of the kidneys, and fibrofolliculoma and/or trichodiscoma of the skin, associated with folliculin gene (FLCN) abnormality. However, the characteristic skin lesions of Birt-Hogg-Dubé syndrome were not found.
Figure 1. Computed tomography (CT).
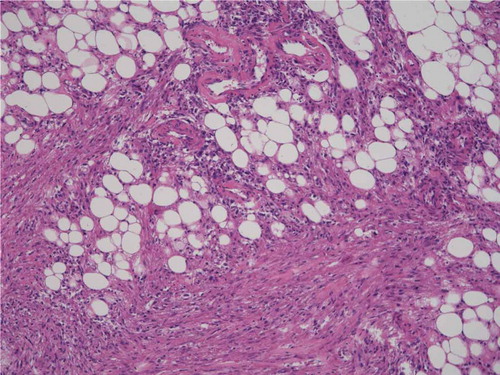
Complete resection of the renal tumor was performed. Histopathological examination revealed a tumor that consisted of fat lobules intermingled with nodular areas with spindle and epithelioid cells (). Blood vessels with a thick wall were conspicuous in the tumor. Immunohistochemically, the spindle cells were positive for α-SMA and vimentin and partially positive for MART-1 and HMB-45. A histopathological diagnosis of the renal tumor was AML. Biopsy of the lung cysts was not performed.
Figure 2. Histopathology of the left renal tumor.
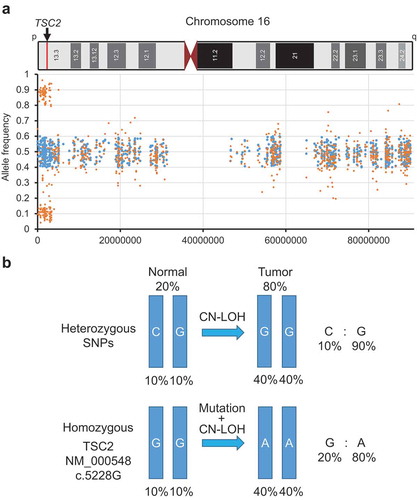
To elucidate the molecular pathogenesis, we performed exome sequencing. The comparison between the sequence of renal AML and the peripheral blood sample identified a somatic missense mutation in exon 41 of the TSC2 gene (NM_000548:c.5228G>A:p. R1743Q), although no other somatic mutation was detected. Therefore, we considered this TSC2 mutation to be responsible for the renal AML in this case. However, germline mutations of the TSC1, TSC2, and FLCN genes were not detected. Interestingly, the mutant allele ratio in the AML sample was 76.1%, and the rest of the reads indicated only a normal reference sequence. Furthermore, no copy number variation (CNV) around the TSC2 locus (16p13.3) was detected by exome sequencing, which means that there is no typical loss of heterozygosity (LOH) of the TSC2 gene. This mutant ratio was too high for a somatic heterozygous mutation without LOH. It was unlikely that both alleles had identical mutations by chance. To clarify the allele status, we analyzed heterozygous single-nucleotide polymorphisms (SNPs) in chromosome 16 (). In these SNPs, an unbalanced allele ratio was accumulated near 0.1 and 0.9 throughout approximately 3.4 Mb inside the 16p13.3 region, although the ratio should be approximately 0.5 if these alleles preserved heterozygosity. Generally, an unbalanced allele ratio indicates LOH, but exome sequencing did not indicate CNV. To explain this inconsistency, we assumed copy-neutral loss of heterozygosity (CN-LOH), also known as uniparental disomy. CN-LOH is an unconventional type of LOH induced by the duplication of a maternal (unimaternal) or paternal (unipaternal) chromosome or chromosomal region and concurrent loss of the other allele.Citation4 Considering that any tumor samples include genomic DNA derived from normal tissues to a certain extent, the minor allele ratio (0.1) was considered approximately equal the proportion of one heterozygous allele derived from normal tissues. Therefore, it was estimated that the major allele ratio (0.9) consisted of 0.8 homozygous alleles from the tumor tissues and 0.1 another allele from the normal tissues (). The proportion of tumor cells calculated on these assumptions was fairly close to the actual mutant allele ratio (0.76). From the above results, we concluded that missense mutation of the TSC2 gene and CN-LOH of the TSC2 locus caused the renal AML.
Figure 3. Copy-neutral loss of heterozygosity (CN-LOH) in chromosome 16.
Discussion
According to the clinical diagnostic criteria for TSC,Citation5 the patient had one major feature (LAM), one partial major feature (single renal AML) and no minor features. Although a definite diagnosis of TSC is established when two major features are observed, it is provided as an exception that the combination of LAM and AML without other features does not meet the clinical diagnostic criteria for a definite diagnosis. The majority of TSC is caused by a germline heterogeneous inactivating mutation of TSC1 (~26%) or TSC2 (~69%).Citation6 Therefore, the possibility of TSC was denied. Neither germline mutations of TSC1 nor TSC2 supported the diagnosis.
A few studies reported the genetic background of AML without TSC. First, LOH of the TSC2 region (16p13) was reported.Citation7–Citation9 Then, point mutations of the TSC2 gene were identified.Citation10,Citation11 These findings suggest that AML without TSC is caused by biallelic (i.e., two-hit) inactivation of the TSC2 gene, as in the case of AML with TSC. A recent exome sequencing analysis revealed that AML without TSC requires two-hit inactivation of the TSC2 gene for its development.Citation12 In our case, the AML had one missense mutation of TSC2 (p.R1743Q). On the other hand, according to three previous studies of 30 TSC2 mutations found in renal AMLs without TSC, 29 were truncating mutations such as nonsense or frameshift mutations, and one was a missense mutation (p.A460T).Citation11 Furthermore, TSC2 p.R1743Q has already been reported in TSC patients, and several studies indicate that p.R1743Q induces functional impairment of TSC2 protein (Tuberin).Citation13,Citation14 The same somatic mutation was also detected in subependymal giant cell astrocytoma (SEGA) without TSC.Citation15 Therefore, it is speculated that this missense mutation strongly abrogates TSC2 function, resulting in pathogenic events.
Interestingly, we detected CN-LOH of the TSC2 gene in this case (). In brief, this means that the normal allele of the TSC2 gene was deleted and the mutant allele was duplicated. Although recent exome sequencing analysis also identified CN-LOH of the TSC2 gene in 62% of AMLs with or without TSC, all cases with CN-LOH contained truncating mutations such as frameshift or nonsense mutations, not missense mutations.Citation12 This result also supports that the missense mutation p.R1743Q has as severe a deleterious effect on TSC2 as a truncating mutation or deletion of the TSC2 gene, resulting in the development of AML.
Initially, we assumed that the patient had a germline mutation responsible for AML development because one of her sisters also had renal AML. However, we could not detect any germline mutations by exome sequencing analysis. Unfortunately, no samples from her sister were available. Therefore, we could not conclude whether the AMLs of the sisters developed by a mechanism based on the same genetic background or sporadically by chance. The accumulation of additional cases would be expected.
Materials and methods
DNA preparation
Genomic DNA was extracted from fresh frozen tissue and peripheral blood samples using the QIAamp DNA Mini Kit (QIAGEN) following the manufacturer’s instructions. The TaqMan RNase P Detection Reagents Kit (Thermo) was used to quantify purified DNA.
Next-generation sequencing
One hundred nanograms of DNA was used for multiplex PCR amplification with an Ion Ampliseq Exome RDY Kit (Thermo), enabling the targeted coverage of all exons. Library preparation and sequencing with the Ion Proton sequencer were performed according to the manufacturer’s protocol. The templates were sequenced after emulsion PCR using Ion PI chip and the Ion PI HI-Q Chef kit (Thermo).
Data analysis
The obtained sequencing data formatted in FASTQ files were aligned to a human genome assembly (hg38) using Hisat2 software,Citation16 and the obtained SAM files were sorted and converted to BAM files as previously described.Citation17–Citation19 Next, mutations, deletions, insertions, and copy number variations were called using Varscan2 software.Citation20 Amino acid substitutions were annotated using ANNOVAR software.Citation21
Disclosure of interest
The authors report no conflict of interest.
References
- Folpe AL, Kwiatkowski DJ. Perivascular epithelioid cell neoplasms: pathology and pathogenesis. Hum Pathol. 2010;41:1–15. doi:10.1016/j.humpath.2009.05.011.
- Eble JN. Angiomyolipoma of kidney. Semin Diagn Pathol. 1998;15:21–40.
- Bissler JJ, Kingswood JC. Renal angiomyolipomata. Kidney Int. 2004;66:924–934. doi:10.1111/j.1523-1755.2004.00838.x.
- O’Keefe C, McDevitt MA, Maciejewski JP. Copy neutral loss of heterozygosity: a novel chromosomal lesion in myeloid malignancies. Blood. 2010;115:2731–2739. doi:10.1182/blood-2009-10-201848.
- Krueger DA, Northrup H. International Tuberous Sclerosis Complex Consensus G. Tuberous sclerosis complex surveillance and management: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:255–265. doi:10.1016/j.pediatrneurol.2013.08.002.
- Northrup H, Koenig MK, Pearson DA, Au KS. Tuberous sclerosis complex. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. Seattle (WA): GeneReviews((R)); 1993. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1220/
- Henske EP, Neumann HP, Scheithauer BW, Herbst EW, Short MP, Kwiatkowski DJ. Loss of heterozygosity in the tuberous sclerosis (TSC2) region of chromosome band 16p13 occurs in sporadic as well as TSC-associated renal angiomyolipomas. Genes Chromosomes Cancer. 1995;13:295–298. doi:10.1002/gcc.2870130411.
- Smolarek TA, Wessner LL, McCormack FX, Mylet JC, Menon AG, Henske EP. Evidence that lymphangiomyomatosis is caused by TSC2 mutations: chromosome 16p13 loss of heterozygosity in angiomyolipomas and lymph nodes from women with lymphangiomyomatosis. Am J Hum Genet. 1998;62:810–815. doi:10.1086/301804.
- Pan CC, Chung MY, Ng KF, Liu CY, Wang JS, Chai CY, Huang S-H, Chen P-H, Ho D. Constant allelic alteration on chromosome 16p (TSC2 gene) in perivascular epithelioid cell tumour (PEComa): genetic evidence for the relationship of PEComa with angiomyolipoma. J Pathol. 2008;214:387–393. doi:10.1002/path.2289.
- Qin W, Bajaj V, Malinowska I, Lu X, MacConaill L, Wu CL, Kwiatkowski DJ. Angiomyolipoma have common mutations in TSC2 but no other common genetic events. PLoS One. 2011;6:e24919. doi:10.1371/journal.pone.0024919.
- Yang HM, Choi HJ, Hong DP, Joo SY, Lee NE, Song JY, Choi Y-L, Lee J, Choi D, Kim B, et al. The analysis of mutations and exon deletions at TSC2 gene in angiomyolipomas associated with tuberous sclerosis complex. Exp Mol Pathol. 2014;97:440–444. doi:10.1016/j.yexmp.2014.09.013.
- Giannikou K, Malinowska IA, Pugh TJ, Yan R, Tseng YY, Oh C, Kim J, Tyburczy ME, Chekaluk Y, Liu Y, et al. Whole exome sequencing identifies TSC1/TSC2 Biallelic loss as the primary and sufficient driver event for renal angiomyolipoma development. PLoS Genet. 2016;12:e1006242. doi:10.1371/journal.pgen.1006242.
- Pal R, Xiong Y, Sardiello M. Abnormal glycogen storage in tuberous sclerosis complex caused by impairment of mTORC1-dependent and -independent signaling pathways. Proc Natl Acad Sci U S A. 2019;116:2977–2986. doi:10.1073/pnas.1812943116.
- Zhang J, Kim J, Alexander A, Cai S, Tripathi DN, Dere R, Tee AR, Tait-Mulder J, Di Nardo A, Han JM, et al. A tuberous sclerosis complex signalling node at the peroxisome regulates mTORC1 and autophagy in response to ROS. Nat Cell Biol. 2013;15:1186–1196. doi:10.1038/ncb2822.
- Ichikawa T, Wakisaka A, Daido S, Takao S, Tamiya T, Date I, Koizumi S, Niida Y. A case of solitary subependymal giant cell astrocytoma: two somatic hits of TSC2 in the tumor, without evidence of somatic mosaicism. J Mol Diagn. 2005;7:544–549. doi:10.1016/S1525-1578(10)60586-7.
- Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–360. doi:10.1038/nmeth.3317.
- Ohashi T, Idogawa M, Sasaki Y, Tokino T. p53 mediates the suppression of cancer cell invasion by inducing LIMA1/EPLIN. Cancer Lett. 2017;390:58–66. doi:10.1016/j.canlet.2016.12.034.
- Idogawa M, Ohashi T, Sasaki Y, Nakase H, Tokino T. Long non-coding RNA NEAT1 is a transcriptional target of p53 and modulates p53-induced transactivation and tumor-suppressor function. Int J Cancer. 2017;140:2785–2791. doi:10.1002/ijc.v140.12.
- Idogawa M, Nakase H, Sasaki Y, Tokino T. Prognostic effect of long noncoding RNA NEAT1 expression depends on p53 mutation status in cancer. J Oncol. 2019;2019:4368068. doi:10.1155/2019/4368068.
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi:10.1101/gr.129684.111.
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi:10.1093/nar/gkq603.