
ABSTRACT
c-Met is a well-characterized oncogene that is associated with poor prognosis in many solid tumor types. While responses to c-Met inhibitors have been observed in clinical trials, activity appears to be limited to those with MET gene amplifications or mutations. We developed a c-Met targeted antibody-drug conjugate (ADC) with preclinical activity in the absence of MET gene amplification or mutation, and activity even in the context of moderate protein expression. The ADC utilized a high-affinity c-Met antibody (P3D12), that induced c-Met degradation with minimal activation of c-Met signaling, or mitogenic effect. P3D12 was conjugated to the tubulin inhibitor toxin MMAF via a cleavable linker (vc-MMAF). P3D12-vc-MMAF demonstrated potent in vitro activity in c-Met protein-expressing cell lines regardless of MET gene amplification or mutation status, and retained activity in cell lines with medium-low c-Met protein expression. In contrast, the c-Met tyrosine kinase inhibitor (TKI) PHA-665752 slowed tumor cell growth in vitro only in the context of MET gene amplification or very high protein expression. This differential activity was even more marked in vivo. P3D12-vc-MMAF demonstrated robust inhibition of tumor growth in the MET gene amplified MKN-45 xenograft model, and similar results in H1975, which expresses moderate levels of wild type c-Met without genomic amplification. By comparison, the c-Met TKI, PHA-665752, demonstrated modest tumor growth inhibition in MKN-45, and no inhibition at all in H1975. Taken together, these data suggest that P3D12-vc-MMAF may have a superior clinical profile in treating c-Met positive malignancies in contrast to c-Met pathway inhibitors.
Introduction
Many tumors depend on the continued expression and/or amplification of a single oncogene for the initiation and maintenance of a malignant state, and its product can be targeted with small-molecule drugs or biotherapeutics. c-Met, the receptor for hepatocyte growth factor (HGF), is a receptor tyrosine kinase overexpressed and activated in 75–90% of gastric cancers and 41–72% of lung cancers,Citation1 and a constitutively active signaling pathway of c-Met has been linked to malignant cell growth.Citation2 C-Met protein expression is also associated with poor prognosis in many solid tumor types.Citation3,Citation4 So far, several clinically available TKIs (tyrosine kinase inhibitors) have shown efficacy in a subset of patients with tumors exhibiting MET gene amplification (e.g., 6% of gastric cancers and 1% of lung cancers)Citation5 or exon 14-skipping mutations.Citation6 However, these tumors eventually acquire resistance, and long-term efficacy of the treatment is ineffective.Citation7,Citation8
Other therapeutic approaches using anti-c-Met antibodies or anti-HGF antibodies to target c-Met initially looked promising. Several anti-c-Met antibodies have been generated,Citation9,Citation10 however, some of them mimicked unwanted HGF agonism by inducing receptor dimerization. Others, like onartuzumab, which does not show agonistic activity of c-Met signaling, failed to demonstrate significant efficacy in a phase III clinical trial of NSCLC.Citation11
We developed an antibody-based therapeutic that targets amplified and non-amplified c-Met-overexpressing tumors without activating c-Met signaling. We identified a high affinity, specific c-Met antibody (P3D12) that induces c-Met degradation with minimal activation of ERK, and no measurable mitogenic activity. By conjugating P3D12 to the cytotoxic drug vc-MMAF using maleimide-based conjugation, we achieved the specificity of a targeted therapy and the potency and efficacy of a chemotherapeutic agent in a single agent. The ADC, P3D12-vc-MMAF, showed a much higher potency than the c-Met TKI, PHA-665752, in in vitro cytotoxicity assays against a panel of gastric and lung cancer cell lines and in vivo xenograft models.
Results
Generation of anti-c-Met antibodies and identification of candidate P3D12 which induces c-Met degradation with minimal agonistic activity
Generation of anti-c-Met antibodies to inhibit cancers with constitutive c-Met signaling has been challenging.Citation12 Some antibodies had strong agonistic activity, including the bivalent 5D5 (Genentech, Inc) and DO-24 antibodies,Citation13 which could lead to cancer cell proliferation. We developed a three-step screening method for generating anti-c-Met antibodies with high c-Met affinity, high c-Met internalizing and degrading properties while maintaining low to no agonistic activity.
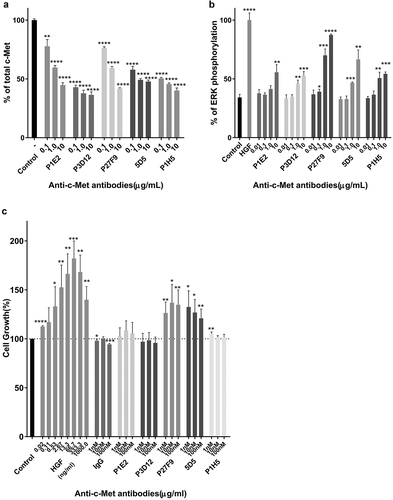
Mice were immunized with human c-Met-Fc recombinant protein. Twenty thousand hybridomas were produced and high-affinity antibodies were selected that bound specifically to human c-Met by ELISA and were rapidly internalized detected by FACS (data not shown). In the second step, about 500 selected antibodies were assayed for induction of c-Met degradation as a surrogate marker for delivery of the mAbs to the lysosomal compartment, as opposed to receptor recycling (). The tested antibodies showed different degrees of c-Met degradation, and one candidate, P3D12, showed the highest activity. In addition, selected hit antibodies were assessed in a c-Met agonist assay using ERK phosphorylation status as a surrogate marker for c-Met activation and downstream signaling (). Three c-Met antibody candidates (P1E2, P3D12, P1H5) demonstrated strongly reduced agonistic activity compared to c-Met’s natural ligand, hepatocyte growth factor (HGF) and the bivalent 5D5 antibody.
Figure 1. Identification of the lead P3D12 anti-c-Met antibody. cMet antibody candidates were tested for efficient c-Met internalization/degradation, minimal activation (non-agonistic) of the ERK pathway (low phosphorylation) and low cell growth induction (slow cell proliferation).
(a) c-Met degradation in SNU-16 cells was measured with the SECTOR Imager 2400. Cells were treated with anti-c-Met antibodies and incubated for 24 h. Values are plotted as mean ± S.D. (n = 3). % total c-Met; * p < .05, ** p < .01, *** p < .001 and **** p < .0001 compared to control. Significance was determined with student t-test. (b) ERK phosphorylation in MKN-45 cells was measured with the SECTOR Imager 2400. MKN-45 cells were incubated with c-Met antibodies for 15 min. Values are plotted as mean ± S.D. (n = 3). % P-ERK; * p < .05, ** p < .01, *** p < .001 and **** p < .0001 compared to control. Significance was determined with student t-test. (c) c-Met-induced cell proliferation of lead P3D12 cMet antibody. The proliferation of 4MBr-5 cells was measured by CTG assay after treatment with IgG, bivalent 5D5, and anti-c-Met antibodies for 5 days. All groups were normalized to cell growth of the control group (no antibody). Values are generated from three independent experiments with duplicate samples each. Values are plotted as the mean ± S.D. % Cell growth; * p < .05, ** p < .01, *** p < .001 and **** p < .0001 compared to control. Significance was determined with student t-test.
P3D12 did not induce cMet-dependent cell proliferation
To further assess the potential for agonistic activity of the selected anti-c-Met antibodies, they were assayed in an HGF-dependent monkey lung epithelial cell line 4MBr-5 proliferation assay (). HGF induced 4MBr-5 cell proliferation in a dose-dependent manner, as did bivalent 5D5 and one of our candidates P27F9. Although bivalent 5D5 has high affinity to c-Met and leads to strong c-Met degradation, it also triggers agonistic proliferative activity which could be counter-productive. Altogether, the data indicate that P3D12 induces binding and internalization of the c-Met receptor, without triggering c-Met signaling in the 4MBr-5 cell line.
Determination of affinity and cross-reactivity of anti-c-Met antibodies
The affinity of the selected c-Met antibodies was tested using recombinant c-Met from various species. All our candidates showed good reactivity with human and cynomolgus monkey c-Met, and EC50 were almost same between human and cynomolgus (1.2〜1.4 nM), but no binding to mouse or canine c-Met in an ELISA assay (). Interestingly, P3D12 was the only candidate that reacted with rat c-Met in ELISA (4.8 nM). The affinity of P3D12 (0.75 nM) and P1E2 (0.89 nM) to human c-Met was essentially identical to the bivalent c-Met antibody 5D5 (0.80 nM), as determined by surface plasmon resonance (SPR) (Supplementary Table1). P1E2 and P27F9 demonstrated lower affinity for c-Met than bivalent 5D5.
Figure 2. Species cross-reactivity of lead P3D12 cMet antibody. c-Met antibodies and isotype control IgG were serially diluted and tested for cross-reactivity to various c-Met species (human, NHP: non-human primate, canine, rat, mouse) by ELISA.
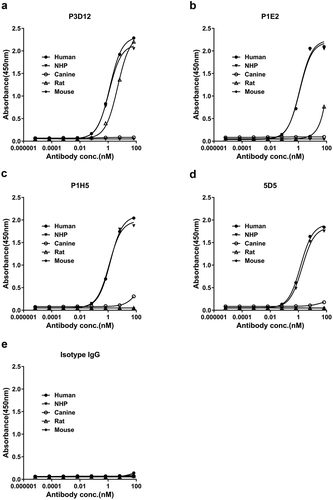
Taken together, P3D12 was the best candidate, based on its biological properties (non-agonist and robust internalization upon binding) and biophysical properties (protein stability, high affinity to human c-Met and cross-reactivity to cynomolgus monkey and rat c-Met).
An anti-cMet-vc-MMAF ADC has superior potency over c-Met TKIs in c-Met non-amplified and amplified cancer cell lines in vitro
To investigate the effect of TKIs and anti-c-Met antibody-vc-MMAF in in vitro cytotoxicity assays, cell viability was measured by CTG (Cell Titer Glo) reagent 5 days after inhibitor or antibody-drug conjugate treatment. In the high c-Met expressing cell lines, MKN-45 (c-Met amplified) and SNU-620 (c-Met amplified) (, ), P3D12-vc-MMAF and P1E2-vc-MMAF showed higher potency than the c-Met-selective kinase inhibitor (PHA-665752). Under these conditions, unconjugated P3D12 and P1E2 had no effect on cytotoxicity at concentrations up to 50 nM. In the medium-low c-Met expressing cell lines without c-Met gene amplification, H1975, SNU-16, H441 (), and PHA-665752 did not induce cell death as expected, while P3D12-vc-MMAF and P1E2-vc-MMAF led to cytotoxicity. However, the efficacy of these two ADCs was around 50% in H441, and 80% in H1975 and 90% in SNU-16.
Table 1. Potency of lead cMet-ADC P3D12-vcMMAF in several cMet-positive cancer cell lines. Potency of P3D12-vc-MMAF, P1E2-vcMMAF, cMet kinase inhibitor PHA-665752 and Erlotinib was assessed in 8 cMet-positive cell lines and a negative control cell line. c-Met receptor number, c-Met copy number and exon 14 status is indicated for all 9 cancer cell lines.
Figure 3. In vitro cytotoxicity and specificity of lead P3D12 c-Met ADC. Several cMet ADCs, non-targeting ADC and a c-Met- selective Tyrosine Kinase Inhibitor (TKI), PHA-665752, were tested on five cMet-positive and one cMet-negative (SNU-1) cancer cell lines. cMet receptor number (receptors/cell) for each cell line is mentioned in the figure.
(a) High c-Met expressing cell line MKN-45, (b) High c-Met expressing cell line SNU-620, (c) Low-Medium c-Met expressing cell line H1975, (d) Low-Medium c-Met expressing cell line SNU-16, (e) Low-Medium c-Met expressing cell line H441, (f)c-Met negative cell line SNU-1, G: c-Met-mutant cell line Hs-746 T, H: c-Met-mutant cell line H596.
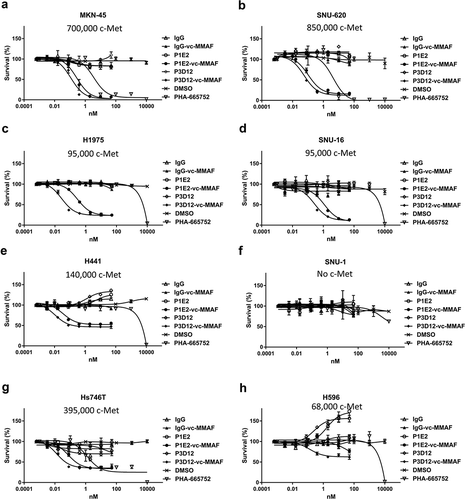
We also tested, a lung adenosquamous carcinoma cell line H596 (c-Met non-amplified) and a gastric adenocarcinoma cell line Hs746 T (c-Met amplified), both of which have a unique type of c-Met alteration leading to exon 14 skipping (). Exon 14 of c-Met includes the coding region for the Cbl binding site which is associated with c-Met internalization.Citation14 In H596 cells, PHA-665752 did not show any effect while P3D12-vc-MMAF had a pronounced effect on viability but with reduced potency (3 x less) and efficacy (50% killing) (). In the Hs746 T cell line, the two ADCs and the TKI were equally potent as in the high c-Met cell lines (IC50 = 23pM: P3D12-vc-MMAF, 72pM: P1E2-vc-MMAF,1.3 nM: PHA-665752). However, efficacy was highly reduced in comparison to high c-Met cell lines (75% killing). c-Met internalization is hampered in H596 and Hs746 T cell lines (Supplementary Figure 1), which leads to a suboptimal intracellular accumulation of c-Met ADC. In the c-Met-negative cell line SNU-1 (), both c-MET inhibitor and c-Met-ADCs (P3D12-vc-MMAF, P1E2-vc-MMAF) had no effect on viability, which emphasizes the specificity and stability of the ADCs and TKI.
In vivo pharmacokinetics and antitumor efficacy of P3D12 and P3D12-vc-MMAF in non-tumor-bearing mice
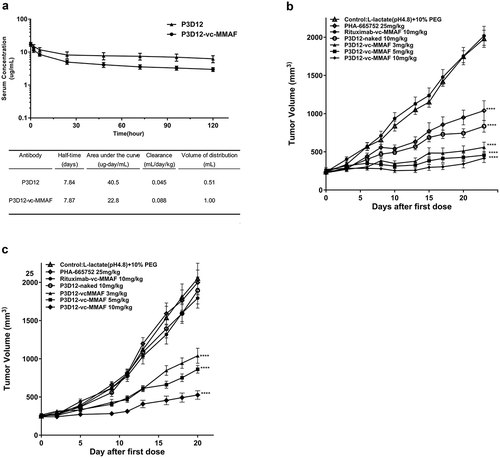
The stability of unconjugated P3D12 and P3D12-vc-MMAF in serum was determined in a mouse PK study () with animals given a single i.v. bolus injection of 1 mg/kg unconjugated or MMAF-conjugated P3D12 antibody. (a) illustrates the pharmacokinetic parameters calculated by non-compartmental analysis. The circulating half-lives in mouse serum were 7.84 and 7.87 days, respectively. The drug-linker conjugation to the c-Met antibody had no effect on stability in mouse serum. However, the exposure of antibody-drug conjugates, as measured by the area under the curve, was decreased in comparison to unmodified antibody, ranging from 40.5 µg-day/mL for unconjugated P3D12 to 22.8 µg-day/mL for P3D12-vc-MMAF. Clearance values increased from 0.045 mL/day/kg for unmodified P3D12 to 0.088 mL/day/kg for the ADC. Similarly, the volume of distribution was found to correlate with drug concentration in the blood of the animals. These data indicated that P3D12-vc-MMAF did not show accelerated clearance by linker de-conjugation through maleimide elimination reaction.
Figure 4. In vivo assessment of lead P3D12 cMet ADC.
(a) Time concentration curves of P3D12 and P3D12-vc-MMAF and their pharmacokinetic parameters in mice. C57BL/6 mice were injected via the vein with 1 mg/kg P3D12 and P3D12-vcMMAF. Plasma samples were analyzed by ELISA to determine the antibody or antibody-drug conjugate concentration. Pharmacokinetic parameters of P3D12 and P3D12-vc-MMAF antibody-drug conjugate were calculated with WinNonlin software. (b) Antitumor activity of P3D12 and P3D12-vc-MMAF in the high c-Met gastric cancer xenograft model MKN-45. P3D12 unconjugated antibody was administered intravenously at 10.0 mg/kg and P3D12-vcMMAF at 3.0, 5.0, and 10.0 mg/kg and compared to the control ADC Rituximab-vc-MMAF (Rituxan®: Rituximab, 10.0 mg/kg). Group size n = 9, Tumor volume was plotted as mean ± S.E.M. Statistical significance was determined with a two-way ANOVA followed by Bonferroni post hoc test (**** p < .0001 compared to control). (c) Antitumor activity of P3D12 and P3D12-vc-MMAF in low-medium c-Met lung cancer xenograft model H1975 with L858 R/T790 M-EGFR mutations. P3D12 unconjugated antibody was administered intravenously at 10.0 mg/kg and P3D12-vc-MMAF was at 3.0, 5.0, and 10.0 mg/kg and compared with the control ADC Rituximab-vc-MMAF (Rituxan®: Rituximab, 10.0 mg/kg). Group size n = 9, Tumor volume was plotted as mean ± S.E.M. 9 mice per group were used for the H1975 studies. Statistical significance was determined with a two-way ANOVA followed by Bonferroni post hoc test (**** p < .0001 compared to control).
The efficacy of unconjugated P3D12, P3D12-vc-MMAF and c-Met TKI (PHA-665752) was evaluated in xenograft models of gastric cancer (MKN-45) and lung cancer (H1975) in immuno-compromised nude mice. Tumor-bearing mice were given intravenous injections of the c-Met inhibitor PHA-665752 at a dosage of 25 mg/kg/day for 5 days, or at the same dosing frequency as the corresponding vehicle control (50 mM L-lactate [pH 4.8] and 10% polyethylene glycol). The other treatment groups were given single intravenous injections of unconjugated P3D12 (10 mg/kg), P3D12-vc-MMAF (3, 5, 10 mg/kg), or non-targeting antibody Rituximab-vc-MMAF as control.
In the MKN-45 model, compared to the c-Met inhibitor (PHA-665752: 25 mg/kg, 5 doses), all dose concentrations (3, 5, 10 mg/kg) of P3D12-vc-MMAF showed strong tumor growth inhibition (). In contrast, unconjugated P3D12 and c-Met specific TKI (PHA-665752) showed significantly less activity than P3D12-vc-MMAF in this model. Interestingly, unconjugated P3D12 demonstrated some tumor growth inhibition, even though it did not exhibit cytotoxicity in vitro at all. Administration of the control ADC Rituximab-vc-MMAF at 10 mg/kg had no impact on tumor growth, suggesting that the observed therapeutic responses were specific for the c-Met-targeting ADC. The antitumor effect of P3D12-vc-MMAF was also evaluated in a non-amplified medium-low c-Met-expressing and EGFR-TKI-resistant (L858 R/T790 M-EGFR) lung cancer cell line H1975 (). In this model, unconjugated P3D12 and c-Met inhibitor (PHA-665752) did not show any antitumor effect at all. On the other hand, P3D12-vc-MMAF induced tumor growth inhibition in all treatment groups in a dose-dependent manner. These data are consistent with the in vitro cytotoxicity data ().
All administered doses of cMet-ADC were well tolerated in mice and no bodyweight change of more than 12% was seen at all administered doses (Supplementary Figure 2). The most bodyweight loss was seen in vehicle control groups and non-targeting ADC with study progression due to high tumor-burden in the mice.
Discussion
Amplification and over-expression of c-Met are observed in several cancer indications (ovarian cancer, gastric cancer, NSCLC and HCC),Citation15 and its expression is associated with poor prognosis and resistance to EGFR- and VEGF-targeted therapies.Citation16-Citation18 Therefore, there has been a major effort in the last few years to develop c-Met pathway inhibitorsCitation19 and inhibitory antibodies.Citation20 A number of antibody-based targeted therapies against c-Met are, or were, in clinical development including onartuzumab: Genentech (discontinued), emibetuzumab: Eli Lilly, LY-3164530, Eli Lilly (discontinued) ficlatuzumab, AVEO; ARGX-111 arGEN-X NV; ABT-700, Abbvie, Sym-015, SymphogenA/S; SAIT-301, Samsung. In addition, there are also a number of small molecule c-Met antagonists including crizotinib, cabozantinib, capmatinib, tepotinib, and glesatinib in clinical development. However, identification of patients who can derive clinical benefit from c-Met pathway inhibition has proven challenging. For example, the c-Met monovalent antibody onartuzumab and the small molecule inhibitor tivantinib failed to show efficacy in Phase III trials in late-stage NSCLC.Citation11,Citation21,Citation22 Recent clinical data suggest that tepotinib may have activity in NSCLC with MET exon 14 skipping mutations, but demonstration of clinical activity in other contexts such as overexpression and gene amplification has been complicated.Citation6 Therefore, significant effort is needed to expand the antitumor activity of c-Met-targeting therapies.
Although c-Met antibodies can have superior specificity in comparison to c-Met tyrosine kinase inhibitors, therapeutic c-Met antibody generation can be difficult because mAbs can trigger c-Met downstream signaling and potentially promote malignant cell proliferation.
We produced a c-Met monoclonal antibody which induced c-Met degradation, had very low agonistic activity and was cross-reactive with cynomolgus monkey and rat c-Met. The rat cross-reactivity of our c-Met antibody was an important feature because it allowed us to assess the on-target toxicity of the c-Met ADC during lead optimization. To reduce potential immunogenicity in patients our c-Met antibody P3D12 will be humanized in the future.
To enhance the breadth of efficacy of c-Met antibody-based therapy, we used our c-Met antibody to generate an antibody-drug conjugate (ADC), c-Met-vc-MMAF.Citation23,Citation24 We chose the non-membrane permeable tubulin-inhibitor monomethyl auristatin F (MMAF) which is about 100 times less cytotoxic in vitro than MMAE.Citation25 It is thought that MMAF has low cytotoxicity because its charged carboxy group hinders diffusion into cells, but once delivered into the cell by an ADC is very potent. As expected, the c-Met-ADCs (P3D12-vc-MMAF, P1E2-vc-MMAF) demonstrated potent in vitro cytotoxicity in c-Met expressing cell lines, including in lower-expressing cells lines where the c-Met TKI did not show any effect. In contrast to small-molecule inhibitor, the c-Met ADCs worked on all c-Met overexpressing cell lines independent of gene amplification status (). This dramatically increases the potential treatable patient population considering that only a minor fraction of patients have c-Met amplification (e.g., 4–10% in gastric cancer and NSCLC).Citation1
Some critical parameter that could limit the efficacy of a c-Met ADC are receptor number, internalization rate, intracellular trafficking and release of the drug. Our data suggest a relationship between c-Met receptor number and in vitro EC50 ().
Antibody-induced c-Met degradation was relatively low in H596 and Hs746T cells (Supplementary Figure 1), reflective of inefficient internalization mediated by the impaired Cbl degradation pathway in these cell lines. The efficacy of the ADCs was also as a result reduced in these cell lines. Moreover, cell growth was observed in H441 and H596, when cells were treated with unmodified c-Met antibody. Choosing a more potent drug-linker or increasing the amount of internalized ADC could mitigate this issue.
In general, a higher drug-to-antibody ratio (DAR) can give higher potency in in vitro cytotoxicity assays, but it induces faster plasma clearance by destabilization of the IgG structure.Citation26 The DAR during the conjugation reaction was controlled to be in the range of 2–3, in order to avoid stability issues of the ADC. In the PK study, beta phase clearance and half-life of ADC resembled that of unmodified-P3D12, which suggested that there was little influence of the conjugation process on stability. As P3D12 is non-cross-reactive with mouse c-Met (), it was not affected by target-mediated clearance in the mouse PK study.
In this study, we demonstrated that the potency differences that were seen between P3D12-vc-MMAF and c-Met TKI (PHA-665752) in vitro translated to in vivo xenograft models. In the MKN-45 model, a single dose of P3D12-vc-MMAF via intravenous injection at 3, 5,10 mg/kg resulted in a stronger tumor inhibition than 5 doses of 25 mg/kg c-Met TKI (PHA-665752). Interestingly, at a dose of 10 mg/kg unconjugated P3D12 also showed significant antitumor effect in the MKN45 model, similar to the c-Met TKI (PHA-665752). This effect may be based on antibody-dependent cell-mediated cytotoxicity (ADCC) of the antibody, or possibly antibody-mediated pathway inhibition in this MET gene-amplified model (Supplementary Figure 3). There are currently two different opinions regarding ADCC activity: It may be beneficial for the anti-c-Met antibody to have ADCC activity to enhance efficacyCitation27 and conversely, the lack of ADCC activity may avoid potential adverse effects.Citation28 We do not think that the ADCC activity of our c-Met antibody is of any significant concern, as a recent report has shown that the anti-c-Met antibody ARGX-111, which does have ADCC activity, showed no major adverse events at doses up to 30 mg/kg administered weekly in cynomolgus monkeys. In future studies, the removal of the effector cell function could be further investigated, although we believe our c-Met ADC would be efficacious enough without additional ADCC activity. We also plan to assess potential on-target toxicities in normal tissues that express c-Met (e.g., colon, liver, and lung) with our rat and cynomolgus monkey cross-reactive c-Met ADC in rodent and NHP pharmacology studies. The localization of c-Met in the organs expressing the receptor will be critical for the accessibility of the ADC to its target and the degree of potential toxicity.
It is known that c-Met plays an important role in tumor progression in NSCLC, and c-Met and EGFR pathways have been also found to be associated with receptor cross-activation in NSCLC.Citation29,Citation30 H1975 is a human NSCLC cell line that expresses wild type c-Met without genomic amplification and has the EGFR L858R/T790M double mutation. It is thought that these mutations convey EGFR-TKIs resistance, such as to erlotinib and gefitinib.Citation31 Therefore, this cell line is widely used as a model to develop new targeting drugs to overcome TKI resistance.Citation32,Citation33 In the H1975 xenograft model, P3D12-vc-MMAF demonstrated dose-dependent tumor growth inhibition, while the c-Met inhibitor (PHA-665752) did not show any effect at all. These data support the hypothesis that c-Met could be targeted by an ADC rather than a pathway inhibitor to overcome mutant EGFR-mediated resistance to EGFR-specific TKIs in NSCLC.Citation34 Despite initially promising clinical results using many TKIs, including c-Met- and EGFR-TKIs, the rise of acquired resistance is still an unsolved problem.Citation35,Citation36 Current opinion suggests that the causes of acquired resistance to TKIs are genomic alterations and pathway addiction by excessive downstream signaling associated with continuous exposure of patients to TKIs. Monoclonal antibodies against c-Met and EGFR are used to overcome the shortcomings of TKI, but these agents themselves are also prone to resistance mechanisms.Citation37,Citation38 c-Met ADCs may provide a new treatment option for patients that failed prior TKIs or therapeutic antibody treatment. The advantage of an antibody-drug conjugate is that it will target c-Met positive cancer cells independent of amplification or mutational status of the targeted RTKs. The receptor itself functions as a delivery vehicle into the cell but its biological function is not directly targeted.
In summary, we have developed a novel c-Met-targeting antibody, P3D12, that induces c-Met degradation with minimal ERK phosphorylation. Additionally, when P3D12 was linked to the antimitotic agent monomethyl auristatin F (MMAF) it produced a more potent and highly c-Met selective anti-tumor activity than c-Met TKI (PHA-665752), in vitro and in vivo. Finally, P3D12-vc-MMAF showed strong tumor growth inhibition in xenograft models, in c-Met amplified and non-amplified cell lines. Taken together, these data show that P3D12-vc-MMAF is a highly potent and selective agent and that patients with c-Met positive malignancies may have a significant clinical benefit from this new drug candidate.
Materials and methods
Antibody generation
Mice were immunized with recombinant human c-Met ECD (extra-cellular domain) fused to human Fc, and spleen B cells were harvested. B cells were fused with SP2/0 cells to generate hybridomas, and were seeded in semi-solid agar plates. Hybridoma colonies were transferred to 96-well plates and supernatants were screened for binding to c-Met protein (ELISA) and c-Met protein internalization and degradation (FACS).
ELISA for hybridoma screening
Microtiter plates (Nunc) were coated overnight at 4°C with 2 μg/mL human c-Met/Fc protein. After blocking with SuperBlock (Thermo Fisher Scientific), hybridoma supernatants were added, and plates were incubated for 1 h at room temperature. After washing, goat anti-mouse Fc-HRP (horseradish peroxidase) secondary antibody (Jackson Immunology # 209-005-098) was added for 20 min at room temperature. c-Met-bound antibodies were detected by addition of TMB (3, 3ʹ, 5, 5ʹ -tetramethyl-benzidine) substrate (KPL). The reaction was stopped with 2 N H2SO4, and the absorbance was measured at 450 nm on a SPECTROstar Nano microplate reader (BMG LABTECH).
FACS analysis of antibody-induced c-Met internalization and degradation
The hybridoma supernatants were added to c-Met-positive cell lines and incubated for 18 h at 37°C. After washing, cells were suspended in phosphate-buffered saline (PBS) with 2% bovine serum albumin, 0.05% sodium azide, and 5 mM EDTA. Goat anti-hMET antibody (R&D #: AF276) was added, and cells were then incubated on ice for 1 h. After washing, PE-conjugated (phycoerythrin) anti-goat secondary antibody (R&D #: F0107) was added. After further washing, the samples were analyzed on the BD Accuri instrument (BD Bioscience).
Quantitative flow cytometry
The Quantum Simply Cellular Microbeads kit (Bangs Laboratories, IN, USA) was used to quantify the expression of c-Met on the cell surface of various cancer cell lines. In the analysis of c-Met receptor number, anti-c-Met conjugated with Alexa Fluor 488 was used. Samples were analyzed on the BD Accuri flow cytometer (BD Biosciences, San Jose, CA, USA). At least 5,000 cell live events were evaluated in each sample. Dead cells were excluded by 7-Amino-actinomycin (7-AAD, Sigma, St. Louis, MO, USA) staining. The median values of fluorescence intensity were converted to antibody-binding capacity (ABC) units using Quick Cal v2.3 software (Bangs Laboratories, IN, USA).
C-Met degradation and ERK phosphorylation assay
Total c-Met degradation in SNU-16 and Erk phosphorylation in MKN-45 cells were measured with the SECTOR Imager 2400 (MSD, Gaithersburg, MD). SNU-16 cells were treated with anti-c-Met antibodies and incubated for 24 h. MKN-45 cells were incubated with c-Met antibodies for 15 min. Samples were analyzed using the following MSD reagents: Phospho/Total Met whole cell lysate kit (total c-Met), Phospho/Total ERK1/2 whole cell lysate kit (p-ERK 1/2 and total ERK 1/2). All assays were carried out according to the manufacturer’s specifications.
Cell lines
The gastric cancer cell line MKN-45 was purchased from DSMZ (Deutsche Sammlung von Mikroorganismen und Zellkulturen, Germany). The gastric cancer cell line SNU-620 was purchased from KCLB (Korean Cell Line Bank, S. Korea). Other gastric and lung cancer cell lines SNU-1, 16, H441, H596, H1975, N87 and Hs746 T were purchased from the American Tissue Type Collection (ATCC). All cell lines were maintained according to the cell bank’s recommendations or in normal growth medium RPMI-1640 with 2 mM glutamine and 10% FBS at 37°C and 5% CO2.
Cell proliferation assay
Cell proliferation in response to c-Met antibody treatment was assessed in vitro using an HGF bioassay and the rhesus monkey lung epithelial cell line 4MBr-5 (CCL-208; ATCC, Manassas, VA). HGF or c-Met stimulation is required for 4MBr-5 cells proliferation. 4MBr-5 cells were plated at a density of 2 × 103 cells per well in F-12 K medium (ATCC, Manassas, VA) with 5% FBS into a 96-well plate (Corning, Inc., Corning, NY, USA.). After 5-h incubation, antibodies and control human recombinant HGF (R&D) diluted in the above-described growth medium were added. After 5 days of incubation, cell proliferation was measured by detecting ATP with the Celltiter-Glo reagent (Promega, Madison, WI) on an EnSpire multimode plate reader (Perkin Elmer, Waltham, MA).
ELISA for cross-reactivity
Microtiter plates (Nunc) were coated overnight at 4°C with 5 μg/mL of either recombinant human-Fc fused to human, rat, mouse, canine or cynomolgus c-Met extracellular domain (Sino Biological Inc). After blocking with SuperBlock solution (Thermo Fisher Scientific), c-Met antibody candidates were added, and plates were incubated for 1 h at room temperature. After washing, goat anti-mouse IgG-HRP conjugate (Thermo Fisher Scientific) was added for 20 min at room temperature. Bound c-Met antibodies were detected by adding TMB substrate (KPL). The reaction was stopped with 2 N H2SO4, and the absorbance at 450 nm was measured on an EnSpire multimode plate reader (Perkin Elmer, Waltham, MA).
Antibody drug-linker conjugation
Drug-linker conjugation for c-Met antibody, P3D12 and P1E2, and Rituximab were performed described as follow. Antibodies in PBS containing 50 mM sodium borate, pH 8.0, were treated with Bond-Breaker (Thermo Fisher Scientific) at 37°C for 2 h. After buffer exchange (Zeba Spin Desalting Columns, Thermo Fisher Scientific), the reduced antibodies were added to the linker-toxin (MC-Val-Cit-PAB-MMAF, CONCORTIS, San Diego, CA) on ice. After 30 min, the reactions were quenched with excess cysteine. After buffer exchange, the conjugates were concentrated by centrifugal ultrafiltration as needed. Protein and drug concentrations were determined by spectral analysis. The conjugates P3D12-vc-MMAF, P1E2-vc-MMAF, and Rituximab -vc-MMAF contained an average of 1.8, 2.0, 2.0 toxin molecules per antibody, respectively.
Cytotoxicity assay
Cells diluted in 10% FBS containing growth media were plated in 96-well flat-bottom plates (Corning, Inc., Corning, NY, USA.) and incubated with the indicated ADCs, controls, or c-Met inhibitor PHA-665752 (Sigma Aldrich) for 5 days. Cell viability was measured by detecting ATP with Celltiter-Glo reagent (Promega, Madison, WI) on a Perkin Elmer EnSpire multimode plate reader (Perkin Elmer, Waltham, MA). Half-maximal inhibitory concentrations (IC50) were calculated by non-linear regression analysis using a sigmoidal curve fitting with Prism 7 software (GraphPad, La Jolla, CA).
Pharmacokinetic study in mice
The pharmacokinetics of P3D12 and P3D12-vc-MMAF were evaluated in C57/BL6 mice. C57/BL6 mice (n = 5) were given 1 mg/kg of test material by a single bolus i.v. injection. Blood samples were collected from each mouse via the tail vein at 5 min, 2 h, 6 h, 1 day, 2 days, 3 days, 4 days, 5 days after injection, into heparin-coated tubes followed by centrifugation (14,000 x g, 5 min) to isolate plasma. Plasma concentrations of P3D12 and P3D12-vc-MMAF were measured by ELISA. Plates were coated with human c-Met recombinant protein. After blocking with SuperBlock solution (Thermo Fisher Scientific), plasma samples were added, and plates were incubated for 1 h at room temperature. After washing, goat anti-mouse IgG-HRP conjugate (Thermo Fisher Scientific) was added for 20 min at room temperature. Bound P3D12 and P3D12-vc-MMAF were detected by adding TMB substrate (KPL, Milford MA). The reaction was stopped with 2N H2SO4, and the absorbance at 450 nm was measured on an EnSpire multimode plate reader (Perkin Elmer, Waltham, MA). Non-compartmental pharmacokinetic parameters were calculated using WinNonlin software (Pharsight, Mountain View, CA).
Tumor xenograft study
All in vivo mouse experimental protocols were approved by Tanabe Research Laboratories, U.S.A., Inc. Company Institutional Animal Care guidelines. MKN-45 or H1975 cells (5 x 106 cells) were implanted subcutaneously into the flank of nu/nu female mice (Charles River Laboratories). When the average tumor volume reached 200–300 mm3, animals were randomized to treatment groups according to tumor volume and body weight. Mice were given intravenous injections of the c-Met inhibitor PHA-665752 at a dosage of 25 mg/kg/day for 5 days, and other control mice were given vehicle (50 mM L-lactate [pH 4.8] and 10% polyethylene glycol). Mice were also given single intravenous injections of P3D12 (10 mg/kg), P3D12-vc-MMAF (3, 5,10 mg/kg), and control mice were given the non-targeting antibody Rituximab-vc-MMAF (10 mg/kg). Tumor volumes and body weights were measured 2–3 times a week for a total study period. The tumor volume (V) was calculated as follows: V (mm3) = {0.5236 x long axis length (mm) x (short axis length (mm))2}. Tumor volumes ± SEM were plotted in Prism 7 (GraphPad, San Diego, CA). Statistical significance was determined with a two-way ANOVA followed by Bonferroni post hoc test in Prism 7.
Antibody affinity measurement
Affinity determinations were performed by surface plasmon resonance using a ProteOn XPR36 (BIO-RAD). Running buffer was 10 mM HEPES pH 7.4, 150 mM NaCl, 0.005% tween-20. Data were collected at 25°C. After each injection, the chip was regenerated using 3 M MgCl2 and 10 mM glycine pH1.5. Binding response was corrected by subtracting the RU from a blank flow cell.
Authors’ Contributions
Conception and design: RF, VB, MG, JC,
Development of methodology: RF, VL, VB, MG
Acquisition of data: RF, EW, CS, VL, RR, AB, JV
Analysis and interpretation of data: RF, EW, MG
Writing, review, and/or revision of the manuscript: RF, MG, JC, RN, VB
Administrative, technical, or material support: VB, AT
Study supervision: VB, JC, RN
Supplemental Material
Download MS Word (453.3 KB)Supplementary material
Supplemental data for this article can be accessed on publisher’s website.
References
- You W-K, Lee DH, Sung E-S, Ahn J-H, An S, Huh J. Development of antibody-based c-Met inhibitors for targeted cancer therapy. Immunol Targets Ther. 2015:35. doi:10.2147/ITT.
- Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. 2012;12(2):89–103. doi:10.1038/nrc3205.
- Yu S, Yu Y, Zhao N, Cui J, Li W, Liu T. c-Met as a prognostic marker in gastric cancer: a systematic review and meta-analysis. PLoS One. 2013;8(11):e79137. doi:10.1371/journal.pone.0079137.
- Liu X, Yao W, Newton RC, Scherle PA. Targeting the c-MET signaling pathway for cancer therapy. Expert Opin Investig Drugs. 2008;17(7):997–1011. doi:10.1517/13543784.17.7.997.
- Jardim DL, Tang C, Gagliato Dde M, Falchook GS, Hess K, Janku F, Fu S, Wheler JJ, Zinner RG, Naing A, et al. Analysis of 1,115 patients tested for MET amplification and therapy response in the MD Anderson phase I clinic. Clin Cancer Res. 2014;20(24):6336–6345. doi:10.1158/1078-0432.CCR-14-1293.
- Paik P, Cortot A, Felip E, Sakai H, Mazieres J, Horn L, Griesinger F, Bruns R, Scheele J, Straub J, Veillon R. Tepotinib in patients with advanced non-small cell lung cancer (NSCLC) harboring MET exon 14-skipping mutations: phase II trial. ASCO. 2018. PMID: 32131323. DOI: 10.1093/annonc/mdz063.080
- Kosaka T, Yatabe Y, Endoh H, Yoshida K, Hida T, Tsuboi M, Tada H, Kuwano H, Mitsudomi T. Analysis of epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer and acquired resistance to gefitinib. Clin Cancer Res. 2006;12(19):5764–5769. doi:10.1158/1078-0432.CCR-06-0714.
- Engelman JA, Settleman J. Acquired resistance to tyrosine kinase inhibitors during cancer therapy. Curr Opin Genet Dev. 2008;18(1):73–79. doi:10.1016/j.gde.2008.01.004.
- Jin H, Yang R, Zheng Z, Romero M, Ross J, Bou-Reslan H, Carano RA, Kasman I, Mai E, Young J, et al. MetMAb, the one-armed 5D5 anti-c-Met antibody, inhibits orthotopic pancreatic tumor growth and improves survival. Cancer Res. 2008;68(11):4360–4368. doi:10.1158/0008-5472.CAN-07-5960.
- Oh YM, Song YJ, Lee SB, Jeong Y, Kim B, Kim GW, Kim KE, Lee JM, Cho MY, Choi J, et al. A new anti-c-Met antibody selected by a mechanism-based dual-screening method: therapeutic potential in cancer. Mol Cells. 2012;34(6):523–529. doi:10.1007/s10059-012-0194-z.
- Rolfo C, Van Der Steen N, Pauwels P, Cappuzzo F. Onartuzumab in lung cancer: the fall of Icarus? Expert Rev Anticancer Ther. 2015;15(5):487–489. doi:10.1586/14737140.2015.1031219.
- Greenall SA, Gherardi E, Liu Z, Donoghue JF, Vitali AA, Li Q, Murphy R, Iamele L, Scott AM, Johns TG. Non-agonistic bivalent antibodies that promote c-MET degradation and inhibit tumor growth and others specific for tumor related c-MET. PLoS One. 2012;7(4):e34658. doi:10.1371/journal.pone.0034658.
- Prat M, Crepaldi T, Pennacchietti S, Bussolino F, Comoglio PM. Agonistic monoclonal antibodies against the Met receptor dissect the biological responses to HGF. J Cell Sci. 1998;111(Pt 2):237–247.
- Kong-Beltran M, Seshagiri S, Zha J, Zhu W, Bhawe K, Mendoza N, Holcomb T, Pujara K, Stinson J, Fu L, et al. Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res. 2006;66(1):283–289. doi:10.1158/0008-5472.CAN-05-2749.
- Wang K, Lim HY, Shi S, Lee J, Deng S, Xie T, Zhu Z, Wang Y, Pocalyko D, Yang WJ, et al. Genomic landscape of copy number aberrations enables the identification of oncogenic drivers in hepatocellular carcinoma. Hepatology. 2013;58(2):706–717. doi:10.1002/hep.26402.
- Bean J, Brennan C, Shih J-Y, Riely G, Viale A, Wang L, Chitale D, Motoi N, Szoke J, Broderick S, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A. 2007;104(52):20932–20937. doi:10.1073/pnas.0710370104.
- Zhang Y-W, Su Y, Volpert OV, Vande Woude GF. Hepatocyte growth factor/scatter factor mediates angiogenesis through positive VEGF and negative thrombospondin 1 regulation. Proc Natl Acad Sci USA. 2003;100(22):12718–12723. doi:10.1073/pnas.2135113100.
- Spigel DR, Ervin TJ, Ramlau RA, Daniel DB, Goldschmidt JH, Blumenschein GR, Krzakowski MJ, Robinet G, Godbert B, Barlesi F, et al. Randomized phase II trial of Onartuzumab in combination with erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2013;31(32):4105–4114. doi:10.1200/JCO.2012.47.4189.
- Lath DL, Buckle CH, Evans HR, Fisher M, Down JM, Lawson MA, Chantry AD, Amodio N. ARQ-197, a small-molecule inhibitor of c-Met, reduces tumour burden and prevents myeloma-induced bone disease in vivo. PLoS One. 2018;13(6):e0199517. doi:10.1371/journal.pone.0199517.
- Wang J, Goetsch L, Tucker L, Zhang Q, Gonzalez A, Vaidya KS, Oleksijew A, Boghaert E, Song M, Sokolova I, et al. Anti-c-Met monoclonal antibody ABT-700 breaks oncogene addiction in tumors with MET amplification. BMC Cancer. 2016;16(1):105. doi:10.1186/s12885-016-2138-z.
- Charakidis M, Boyer M. Targeting MET and EGFR in NSCLC-what can we learn from the recently reported phase III trial of onartuzumab in combination with erlotinib in advanced non-small cell lung cancer? Transl Lung Cancer Res. 2014;3(6):395–396. doi:10.3978/j..2218-6751.2014.09.03.
- Rimassa L, Assenat E, Peck-Radosavljevic M, Pracht M, Zagonel V, Mathurin P, Rota Caremoli E, Porta C, Daniele B, Bolondi L, et al. Tivantinib for second-line treatment of MET-high, advanced hepatocellular carcinoma (METIV-HCC): a final analysis of a phase 3, randomised, placebo-controlled study. Lancet Oncol. 2018;19(5):682–693. doi:10.1016/S1470-2045(18)30146-3.
- Dubowchik GM, Firestone RA. Cathepsin b-sensitive dipept1de prodrugs. 1. A model study of structural requirements for efficient release of doxorubicin. Bioorg Med Chem Lett. 1998;8(23):3341–3346. doi:10.1016/S0960-894X(98)00609-X.
- McCombs JR, Owen SC. Antibody drug conjugates: design and selection of linker, payload and conjugation chemistry. AAPS J. 2015;17(2):339–351. doi:10.1208/s12248-014-9710-8.
- Chari RV, Miller ML, Widdison WC. Antibody-drug conjugates: an emerging concept in cancer therapy. Angew Chem Int Ed Engl. 2014;53(15):3796–3827. doi:10.1002/anie.201307628.
- Hamblett KJ, Senter PD, Chace DF, Sun MM, Lenox J, Cerveny CG, Kissler KM, Bernhardt SX, Kopcha AK, Zabinski RF, et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res. 2004;10(20):7063–7070. doi:10.1158/1078-0432.CCR-04-0789.
- Hultberg A, Morello V, Huyghe L, De Jonge N, Blanchetot C, Hanssens V, De Boeck G, Silence K, Festjens E, Heukers R, et al. Depleting MET-expressing tumor cells by ADCC provides a therapeutic advantage over inhibiting HGF/MET signaling. Cancer Res. 2015;75(16):3373–3383. doi:10.1158/0008-5472.CAN-15-0356.
- Merchant M, Ma X, Maun HR, Zheng Z, Peng J, Romero M, Huang A, Yang N-Y, Nishimura M, Greve J, et al. Monovalent antibody design and mechanism of action of onartuzumab, a MET antagonist with anti-tumor activity as a therapeutic agent. Proc Natl Acad Sci USA. 2013;110(32):E2987–96. doi:10.1073/pnas.1302725110.
- Agarwal S, Zerillo C, Kolmakova J, Christensen JG, Harris LN, Rimm DL, Digiovanna MP, Stern DF. Association of constitutively activated hepatocyte growth factor receptor (Met) with resistance to a dual EGFR/Her2 inhibitor in non-small-cell lung cancer cells. Br J Cancer. 2009;100(6):941–949. doi:10.1038/sj.bjc.6604937.
- Jo M, Stolz DB, Esplen JE, Dorko K, Michalopoulos GK, Strom SC. Cross-talk between epidermal growth factor receptor and c-Met signal pathways in transformed cells. J Biol Chem. 2000;275(12):8806–8811. doi:10.1074/jbc.275.12.8806.
- Godin-Heymann N, Ulkus L, Brannigan BW, McDermott U, Lamb J, Maheswaran S, Settleman J, Haber DA. The T790M “gatekeeper” mutation in EGFR mediates resistance to low concentrations of an irreversible EGFR inhibitor. Mol Cancer Ther. 2008;7(4):874–879. doi:10.1158/1535-7163.MCT-07-2387.
- Yu Z, Boggon TJ, Kobayashi S, Jin C, Ma PC, Dowlati A, Kern JA, Tenen DG, Halmos B. Resistance to an irreversible epidermal growth factor receptor (EGFR) inhibitor in EGFR-mutant lung cancer reveals novel treatment strategies. Cancer Res. 2007;67(21):10417–10427. doi:10.1158/0008-5472.CAN-07-1248.
- Kobayashi S, Ji H, Yuza Y, Meyerson M, Wong KK, Tenen DG, Halmos B. An alternative inhibitor overcomes resistance caused by a mutation of the epidermal growth factor receptor. Cancer Res. 2005;65(16):7096–7101. doi:10.1158/0008-5472.CAN-05-1346.
- Tang Z, Du R, Jiang S, Wu C, Barkauskas DS, Richey J, Molter J, Lam M, Flask C, Gerson S, et al. Dual MET-EGFR combinatorial inhibition against T790M-EGFR-mediated erlotinib-resistant lung cancer. Br J Cancer. 2008;99(6):911–922. doi:10.1038/sj.bjc.6604559.
- Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362(25):2380–2388. doi:10.1056/NEJMoa0909530.
- Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, Campos D, Maoleekoonpiroj S, Smylie M, Martins R, et al. Erlotinib in previously treated non–small-cell lung cancer. N Engl J Med. 2005;353(2):123–132. doi:10.1056/NEJMoa050753.
- Van Emburgh BO, Sartore-Bianchi A, Di Nicolantonio F, Siena S, Bardelli A. Acquired resistance to EGFR-targeted therapies in colorectal cancer. Mol Oncol. 2014;8(6):1084–1094. doi:10.1016/j.molonc.2014.05.003.
- Sekhar SC, Kasai T, Satoh A, Shigehiro T, Mizutani A, Murakami H, El-Aarag BY, Salomon DS, Massaguer A, de Llorens R, et al. Identification of caveolin-1 as a potential causative factor in the generation of trastuzumab resistance in breast cancer cells. J Cancer. 2013;4:391–401. doi:10.7150/jca.6470.