
ABSTRACT
The efficacy of trastuzumab, a treatment for HER2+ breast cancer, can be limited by the development of resistance. Cyclin E (CCNE) overexpression has been implicated in trastuzumab resistance. We sought to uncover a potential mechanism for this trastuzumab resistance and focused on a model of CCNE overexpressing HER2+ breast cancer and noncanonical phosphorylation of the TGF-β signaling protein, SMAD3. Network analysis of transcriptional activity in a HER2+, CCNE overexpressing, trastuzumab-resistant cell line (BT474R2) identified decreased SMAD3 activity was associated with treatment resistance. Immunoblotting showed SMAD3 expression was significantly downregulated in BT474R2 cells (p < .01), and noncanonical phosphorylation of SMAD3 was increased in these CCNE-overexpressing cells. Also, in response to CDK2 inhibition, expression patterns linked to restored canonical SMAD3 signaling, including decreased cMyc and increased cyclin-dependent inhibitor, p15, were identified. The BT474R2 cell line was modified through overexpression of SMAD3 (BT474R2-SMAD3), a mutant construct resistant to CCNE-mediated noncanonical phosphorylation of SMAD3 (BT474R2-5M), and a control (BT474R2-Blank). In vitro studies examining the response to trastuzumab showed increased sensitivity to treatment for BT474R2-5M cells. These findings were then validated in NSG mice inoculated with BT474R2-5M cells or BT474R2 control cells. After treatment with trastuzumab, the NSG mice inoculated with BT474R2-5M cells developed significantly lower tumor volumes (p < .001), when compared to mice inoculated with BT474R2 cells. Taken together, these results indicate that for patients with HER2+ breast cancer, a mechanism of CCNE-mediated trastuzumab resistance, regulated through noncanonical SMAD3 phosphorylation, could be treated with CDK2 inhibition to help enhance the efficacy of trastuzumab therapy.
Introduction
Each year, approximately 250,000 women are diagnosed with breast cancer.Citation1 Of these patients, 20–25% overexpress the human epidermal growth factor receptor 2 (HER2), which has been associated with poor patient outcomes.Citation2 Despite being a marker for more aggressive disease, the HER2 receptor serves as a therapeutic target.Citation3 Trastuzumab, a recombinant IgG antibody directed against HER2, binds with the HER2 extracellular domain to suppress the activation of PI3K signaling.Citation4 Trastuzumab binding also reduces the cleavage of HER2 into active p95HER2 and blocks dimerization with HER3 receptors, thus facilitating cell cycle arrest and apoptosis.Citation5,Citation6
For newly diagnosed HER2+ breast cancer patients, trastuzumab treatment results in improved disease-free survival, and in the metastatic setting, treatment with trastuzumab delays disease progression.Citation5 However, in a significant percentage of patients, the clinical applications of trastuzumab treatment have been limited by either primary or acquired treatment resistance.Citation7,Citation8 Several mechanisms of trastuzumab resistance have been proposed, including alterations in the HER2 complex, deregulation of HER2 downstream signaling, and crosstalk activation of additional signaling pathways including IGFR1 and MET.Citation9–12 Simultaneously, these mechanisms have yet to be thoroughly examined and validated in the clinical setting.Citation6 Therefore, more information is needed to elucidate the clinically relevant and targetable molecular events that lead to trastuzumab resistance, thus allowing for the development of therapies to facilitate sensitivity to this critical anti-HER2 breast cancer treatment.
Overexpression of the cell cycle regulatory protein cyclin E (CCNE), has been linked to aggressive breast cancer subtypes, poor patient outcomes, and resistance to trastuzumab.Citation13–17 During cell cycle regulation, CCNE forms a complex with its catalytic signaling partner, CDK2, to induce cell cycle G1-S transition.Citation18 CCNE/CDK2 action has also been associated with noncanonical phosphorylation of SMAD3, a downstream transcriptional mediator in the TGF-β signaling pathway.Citation16 In the canonical setting, TGF-β/SMAD3 signaling acts to facilitate tumor suppression through upregulation of cell cycle inhibitory protein p15 and downregulation of mitogenic cMyc.Citation19,Citation20 Noncanonical SMAD3 phosphorylation, mediated by CCNE/CDK2 activity, primarily in the linker region of the SMAD3 protein, resulted in blockade of the tumor suppressant actions of TGFβ/SMAD3. This noncanonical CDK2 mediated phosphorylation was thus associated with breast cancer cell cycle progression, tumor cell migration, and larger tumor size in murine models.Citation19,Citation21 Therapeutic blockade of CDK2 has shown promise in preclinical breast cancers modes of CCNE overexpression. This finding may be enacted through the restoration of canonical TGFβ/SMAD3 phosphorylation and associated transcription of proteins mediating cell cycle control.Citation16,Citation17,Citation22,Citation23
This interaction among the HER2 pathway, CCNE/CDK2 and TGFβ through SMAD3 has led to the proposed hypothesis that noncanonical CCNE/CDK2 mediated deregulation of SMAD3 contributes to trastuzumab resistance in HER2+ breast cancers. The goal of this study was to uncover a potential mechanism for trastuzumab resistance, focusing on altered, noncanonical phosphorylation of SMAD3 in CCNE overexpressing HER2+ breast cancer cells, to help identify a new approach to restore the efficacy of this critical anti-HER2 breast cancer therapy.
Materials and methods
Cells and reagents
HER2+ BT474 and SK-BR-3 cells were obtained from the ATCC. Trastuzumab resistant cells, BT474R2, (BT474 cells with acquired trastuzumab resistance and increased CCNE copy number), were generously provided by M. Scaltriti (Memorial Sloan Kettering Cancer Center).Citation22 Transgenic cell lines (BT474R2-5M, BT474R2-SMAD3, BT474-CCNE, SK-BR-3-CCNE) were created to express a mutant SMAD3 construct, resistant to CDK2 phosphorylation or a wild-type SMAD3 construct or CCNE, respectively. The 5M construct consisted of the SMAD3 sequence with inactivating mutations in each of five CDK phosphorylation sites (T8/T179/S204/S208/S213), thus driving upregulation of canonical SMAD3 signaling. These study cell lines were generated through lentiviral infection of the parental cell line followed by puromycin selection for 2 weeks. Transgene expression of the was confirmed through immunoblotting (Figure S1).
Lentivirus
Lentivirus was produced by co-transfecting lentiviral packaging vectors (pMDL-GagPol, pRSV-Rev, pIVS-VSV-G) and lentiviral vectors using JetPrime (Polyplus) into HEK-293T cells, as previously described.Citation24 Viral supernatant was collected after 48 h in culture and concentrated using PEG-it (Systems Biosciences). Virus was re-suspended in phosphate buffered saline (PBS) and stored at −80°C until use.
Reporter arrays
A transcriptional activity cell array (TRACER) was used to identify central transcription factors controlling resistance to trastuzumab in BT474R2 cells.Citation25–27 BT474 and BT474R2 cells were plated into a black 384 well plate with an existing library of transcription factor activity reporters. After 48 h, growth media was exchanged for media containing 630 μM d-luciferin and either 10 μg/mL trastuzumab or vehicle as a control. Transcription factor activity was monitored 2, 4, 6, 8, and 24 h after addition of trastuzumab using an IVIS Spectrum (Perkin Elmer). TRACER data were processed as previously described.Citation27 Briefly, activity measurements were background subtracted, normalized to the empty control reporter and log2 transformed prior to analysis.Citation26 Data were taken from a minimum of three replicates and presented as the mean. Statistical analysis was performed using the limma R package.Citation28 P values were adjusted using the false-discovery rate correction.Citation29
Network analysis
NTRACER (networks for TRACER) was used to analyze connections between dynamic TF activity measurements .Citation27,Citation30 This method employs a combination of inference methods [PLSR,Citation31 similarity index,Citation32 linear ordinary differential equations based on TIGRESS,Citation33 random forest,Citation34 ARACNE,Citation35 CLR,Citation36 MRNETCitation37] to infer high-confidence connections between factors based on their dynamic activity. Prior to this analysis, normalized data was mean centered and variance scaled, and an aggregated data set was constructed from sampling the means of each factor for a total of 500 different potential sets of TF activity. A total of 1000 runs were performed to ensure robust inference of connected factors. Features of the network were found from the top 10% of edges from multiple inference methods. Central nodes were identified through eigenvector centrality. Networks were visualized and analyzed using the R package iGraph.Citation38
Immunoblotting
Immunoblotting was performed following transgene insertion into parental study cells. Cells were collected and lysed using M-PER reagent mixed with the recommended amount of protease/phosphatase inhibitor (Cell Signaling Technologies). Cell lysate was mixed with 2X Laemmli sample buffer containing 5% β-mercaptoethanol. Samples were loaded into a precast 4–15% polyacrylamide gel and run for 1 h. Protein was transferred to a nitrocellulose membrane and blocked with 5% bovine serum albumin solution for 1 h. Membranes were incubated with primary antibody overnight, followed by secondary antibody for 1 h. ECL reagent was used to develop the membrane and blots imaged using a Bio-Rad Chemidoc Touch Imaging System (Bio-Rad). Blot intensity was quantified using ImageJ and normalized to beta-actin expression prior to analysis.
The following antibodies were used from Cell Signaling Technologies: SMAD3 (C67H9), cMyc (D84 C12). Abcam: pSMAD3-T179 (ab74062), pSMAD3-S204 (ab63402), pSMAD3-S213 (ab63403), pSMAD3-S423/425 (ab52903), p15 (ab53034), FLAG-tag (ab1162). All antibodies were grown in rabbit and developed using anti-rabbit IgG (Cell Signaling Technologies).
Proliferation assay
BT474, BT474R2, BT474R2-SMAD3, or BT474R2-5M cells were plated at 500 cells/well in 96 well plates. Two days later, cells were treated with a therapeutic dose (10 μg/mL) of trastuzumab or CDK2/9 inhibitor fadraciclibCitation39 (CYC065, 600 nM, generously provided by Cyclacel Ltd). Media was changed every other day for 7 or 10 days. Cell proliferation was measured by MTS assay (Promega) through addition of 10 μl of MTS reagent to each well. Absorbance at 490 nm was used to quantify proliferation and study cells in treated and untreated conditions were compared through a one-way ANOVA and Kruskal-Wallis test for significance.
Orthotopic tumor models
All animal studies were carried out with the approval of the University of Michigan IACUC under the supervision of the principal investigators. Orthotopic BT474R2 or BT474R2-5M tumors were generated through inoculation of 2 million tumor cells into the fourth right mammary fat pad of NOD/SCID mice. Mice were randomized into two treatment groups: trastuzumab (20 mg/kg) or PBS control (100 μl). These mice were monitored daily for tumor growth until tumors were > 5 mm in any dimension. After tumor formation, mice were treated with the assigned treatment through intraperitoneal injection twice weekly for a total of three weeks. Tumors were snap frozen in isopentane at the end of the study for subsequent immunostaining. Tumor growth was monitored through measurement of the tumor dimensions and volume calculated through the formula V = ½(l x w2). Mice were excluded from analysis if their tumor reached IACUC-defined endpoints (> 20 mm in any dimension) prior to 3 weeks of treatment. Statistical significance among tumor volumes was determined through two-way ANOVA with Tukey correction for multiple comparisons.
Immunohistochemistry
Frozen tumors were embedded in optimal cutting temperature (OCT) compound and cryosectioned into 14 µm thick sections. Prior to staining, sections were fixed in ice-cold acetone, washed with tris-buffered saline, permeabilized with 0.5% triton-x, and blocked with 5% goat-serum. Slides were stained with Ki67 antibody (Novus Biologicals, NB110-89717) at a 1:200 dilution in blocking buffer at 4°C overnight. Slides were washed the next day in tris-buffered saline and stained with an Alexa Fluor 594-conjugated secondary antibody for 1 h (Abcam antibody ab150080). Coverglass was subsequently mounted using DAPI-containing Fluorogel (SouthernBiotech). Slides were imaged using an Axio Observer Z1 (Zeiss) using at 5x/0.16 or 10x/0.75 objective and an ORCA-Flash 4.0 V2 Digital CMOS camera (Hammamatsu Photonics). Red fluorescence was measured at 555 nm. QuPathCitation40 was used to identify and quantify Ki67+ cells from 3 independent sections and presented as percentage of Ki67+ cells ± SEM for each treatment. Differences between conditions were analyzed through two-way ANOVA.
Results
SMAD3 activity is central to response to trastuzumab in HER2±, CCNE-high breast cancer cells
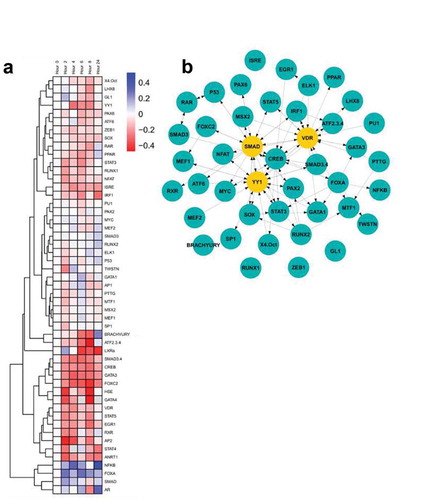
Overexpression of CCNE has been associated with trastuzumab resistance in the HER2- BT474 and SK-BR-3 cell linesCitation22 (Figure S2A and S2B). Cells that spontaneously acquired a copy number increase in CCNE (BT474R2) were resistant to a therapeutic dose of trastuzumab. These cells could be re-sensitized to trastuzumab treatment by blocking CCNE/CDK2 activity through the use of a CDK2 inhibitor (Figure S2A). Overexpression of CCNE in BT474 and SK-BR-3 cells reduced the effect of trastuzumab on cell proliferation by 35% (p < .001) and 5% (p < .05), respectively. We hypothesized that altered transcription factor activity, secondary to CCNE overexpression, contributed to the observed resistance to trastuzumab in these study cells. We therefore used a transcriptional activity cell array, TRACER, to examine differences in transcription factor activity that resulted from trastuzumab treatment in BT474R2 cells ()), along with examination of transcription factors that were identified as regulated during the response to trastuzumab ()). These experiments were conducted over 24 h of treatment time using a trastuzumab dose previously shown to be therapeutic in the parental BT474 cell line in vitro (Figure S3). A total of 50 TFs were measured. For the BT474 cells, a control analysis comparing untreated to trastuzumab treated cells identified 2 factors, MTF1 and MSX2, to be downregulated after trastuzumab treatment at the 24 htime point, with RUNX1 and PU1 as most influential following network analysis (Figure S4). Given the known impact of trastuzumab on BT474 cells, a relative increase in cell death when compared to the BT474R2 cells was noted during this analysis. For the BT474R2 cells treated with trastuzumab during the first 24 h of therapy, 6 TFs were identified with activity that was significantly downregulated: ATF2/3/4, FOXC2, EGR1, ANRT1, BRACHYURY, and GATA3; and 1 TF was identified that had significantly upregulated activity: NFκB, (p < .05) ()). All downregulated factors were significantly changed at the 8-h time point, with 3 factors (ANRT1 at 2 h, GATA3 at 4 h, FOXC2 at 4 and 6 h), significantly downregulated at earlier time points as well. Upregulation of NFκB activity was observed at 4 h as well as 24 h of treatment. Network analysis identified the top 5% of regulatory factors during treatment of BT474R2 cells as SMAD (specifically SMAD3), YY1, and VDR. Of these, SMAD3 activity was most influential in the inferred network, and was thus hypothesized to be associated with trastuzumab resistance in the study cells ()).
Figure 1. (a) TRACER analysis of the response of BT474R2 cells to trastuzumab identified differential dynamics. (b) Central factors controlling response. Yellow nodes were identified as central to the network through eigenvector centrality
SMAD3 phosphorylation is altered in CCNE overexpressing cells
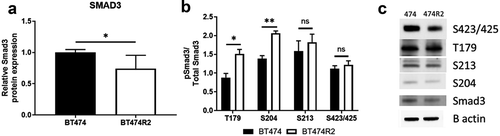
We next performed immunoblotting for the SMAD3 protein and site-specific phosphorylation of the SMAD3 protein. This work was performed to examine a connection between alterations in SMAD3 phosphorylation with the observation that SMAD3 activity was a hub for CCNE-mediated resistance to trastuzumab ()). SMAD3 expression was significantly decreased in BT474R2 cells when compared to parental BT474 cells (p < .01) ()). We next examined canonical phosphorylated SMAD3 (pSMAD3) in BT474R2 cells ()). The pSMAD3/total SMAD3 ratio at the canonical S423 phosphorylation site was not significantly altered in the CCNE-overexpressing HER2+ cell line, consistent with the TF activity data. Noncanonical phosphorylation of SMAD3 can occur upstream of the canonical site, primarily in the linker region of this protein.Citation41,Citation42 The pSMAD3/SMAD3 ratio of expression at the noncanonical S204 Smad3 phosphorylation site was increased by 49%, and expression of the noncanonical T179 Smad3 phosphorylation site was increased by 72%, relative to expression in the parental BT474 cell line control (p < .01 and p < .05, respectively). pSMAD3/SMAD3 at a third noncanonical site in the SMAD3 protein, S213, was not significantly altered between the two cell lines.
Figure 2. Alterations in protein expression for BT474R2 compared with BT474 cell (a) SMAD3, (b) pSMAD3, (c) Blot image for each protein. *p < .05, **p < .01
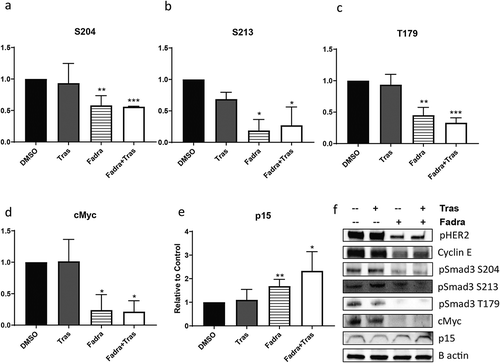
The mitogenic action of CCNE can be blocked through CDK2 inhibition,Citation16 thus we next investigated the effects of CDK2 inhibition on BT474R2 cell survival, sensitivity to trastuzumab, and phosphorylation of SMAD3. Inhibition of CCNE activity with a fadraciclib (CYC065), a potent CDK2 inhibitor, resulted in significantly decreased BT474R2 cell proliferation (Figure S2). Fadraciclib treatment of the BT474R2 cells also led to decreased expression of pSMAD3 at the S204 (p < .01) ()), S213 (p < .05) ()), and T179 (p < .01) ()) phosphorylation sites, in the SMAD3 linker region. Treatment with trastuzumab alone did not significantly alter phosphorylation at any of these sites. Additionally, fadraciclib treatment, alone or in combination with trastuzumab, led to increased expression of p15 (p < .05) and decreased expression of cMyc (p < .05) in comparison with trastuzumab treatment alone, further indicating that CCNE, mediated by CDK2, significantly deceases canonical SMAD3 activity in the study cells (–f)).
Figure 3. (a–c) CDK2 inhibition by fadraciclib significantly decreased phosphorylation at SMAD3 linker region sites in BT474R2 cells: (a) S204, (b) S213, (c) T179. (d, e) CDK2 inhibition decreased cMyc expression (d) and increased p15 expression (e). (f) Immunoblotting images. *p < .05, **p < .01, ***p < .001. Tras = Trastuzumab. Fadra = fadraciclib
SMAD3 linker region phosphorylation significantly alters cell proliferation in vitro
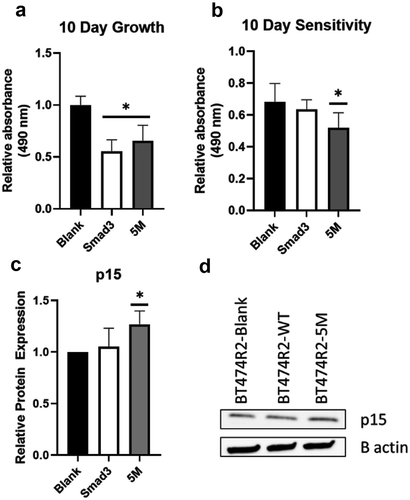
In order to further study the relative influence of canonical SMAD3 and noncanonical SMAD3 linker region phosphorylation on cell proliferation we created several cell lines including BT474R2 cells overexpressing wild-type SMAD3 (BT474R2-SMAD3), study cells expressing a SMAD3 construct with mutations blocking noncanonical linker region CDK2 phosphorylation (BT474R2-5M), and cells expressing a blank control (BT474R2-Blank). These three study cell lines showed significantly different proliferation profiles, with BT474R2-5M and BT474R2-SMAD3 expressing cells proliferating more slowly than the BT474R2-Blank cell line (p < .05), as measured by MTS assay ()). Treatment with trastuzumab resulted in decreased proliferation for all three cell lines, as a percent of untreated cells, by 32, 36, and 48% for BT474R2-Blank, BT474R2-SMAD3, and BT474R2-5M, respectively, with a statistically significant decrease observed for the BT474R2-5M cells (p < .05) ()). Immunoblotting blot analysis for p15 in these cell lines (,d)), also showed significantly increased p15 expression in BT474R2-5M cells treated with trastuzumab, when compared to the BT474R2-Blank cells treated with trastuzumab (26% increase, p < .05). Together, these results suggested that for BT474R2 cells, expression of the mutant SMAD3 construct, resistant to noncanonical CDK2 phosphorylation, contributed to increased sensitivity to trastuzumab.
Figure 4. (a) Overexpressing SMAD3 or noncanonical phosphorylation mutant SMAD3 (5M) resulted in decreased proliferation relative to a blank construct. (b) Expression of the 5M, but not SMAD3, significantly increased response to trastuzumab relative to blank control in BT474R2 cells. (c,d) 72 h treatment with trastuzumab led to upregulation of p15 in the 5M cell line relative to the blank-construct cell line. * p < .05
SMAD3 linker region phosphorylation significantly alters proliferation in vivo
The in vitro results of the MTS assays suggested BT474R2 cells expressing the 5M SMAD3 construct, with mutated noncanonical CDK phosphorylation sites, may be more sensitive to trastuzumab when compared with the parental BT474R2 cell line. We tested this hypothesis using an orthotopic tumor model comparing growth of BT474R2-Blank and BT474R2-5M cells over 21 days of treatment with either trastuzumab or PBS control ()). When comparing tumor growth between the PBS-treated groups (BT474R2-5M and BT474R2-Blank), no statistical difference in tumor growth was identified during the study period (p = .85 at day 14, p = .98 at day 21). Trastuzumab therapy had a moderate effect on BT474R2-Blank tumor growth by day 21, when compared to the PBS-treated BT474R2-Blank tumor growth (p = .999 at day 14, p = .051 at day 21). The effect of trastuzumab treatment on the BT474R2-Blank tumors observed in our study was consistent with a previous report showing that BT474R2 cells were resistant to trastuzumab treatment in vivo.Citation22
Figure 5. (a) Tumor growth was significantly decreased in the 5M cell line with trastuzumab treatment relative to untreated 5M tumors and both treated and untreated R2 tumors. (b) Ki67 staining of tumor sections isolated 21 days post-treatment. (c) Quantification of Ki67+ cells shows decreased proportion of proliferative cells in the 5M trastuzumab group relative to untreated R2-Blank cells (R2-PBS). Tras = trastuzumab treated. Scale bar = 100 µm. C * p < .05, ** p < .01
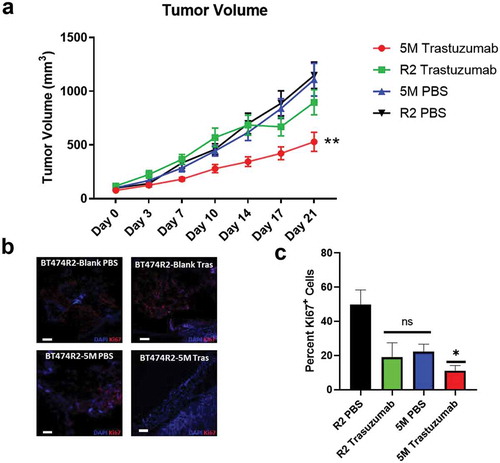
Trastuzumab therapy significantly slowed tumor growth for the BT474R2-5M tumors by day 14, when compared with the PBS-treated control BT474R2-5M tumors, and the BT474R2-Blank tumors treated with PBS or trastuzumab (p < .05). This finding was most pronounced by 21 days of treatment (p < .01) ()). After 21 days of trastuzumab therapy, BT474R2-5M tumor volume was 529 mm3 on average, significantly lower than PBS-treated BT474R2-Blank tumors (1148 mm3, p < .0001), trastuzumab-treated BT474R2-Blank tumors (897 mm3, p < .01), and PBS-treated BT474R2-5M tumors (1108 mm3, p < .0001). Ki67 immunostaining of BT474R2-5M tumors from day 21 of trastuzumab treatment ()) showed a significantly decreased proportion of proliferative cells, relative to BT474R2-Blank tumors treated with PBS (11% v. 50% Ki67+ cells, respectively, p < .05, )). BT474R2-Blank tumors treated with trastuzumab and BT474R2-5M tumors treated with PBS tumors both had a higher percentage of Ki67+ cells relative to the BT474R2-5M trastuzumab treated tumors (19% and 22% Ki67+ cells, respectively), however these differences were not statistically significant when compared to either the BT474R2-Blank PBS group (p = .06, p = .1) or the BT474R2-5M trastuzumab group (p = .99, 0.85). Together, these results indicated CCNE-overexpressing BT474R2 cells could be sensitized to trastuzumab therapy through the blockade of noncanonical CDK SMAD3 phosphorylation sites and the subsequent restoration of canonical SMAD3 signaling ().
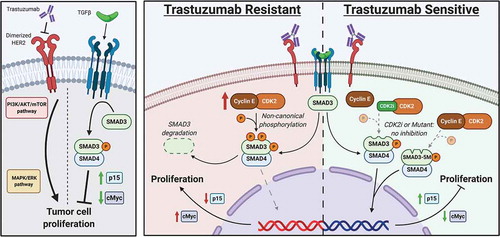
Figure 6. Role of noncanonical SMAD3 phosphorylation in mediating resistance to trastuzumab. Left panel shows canonical signaling pathway activity. Middle panel shows role of trastuzumab resistance in the setting of CCNE overexpression. Right panel shows restoration of trastuzumab sensitivity after blockade of noncanonical SMAD3 phosphorylation though either expression of a mutant SMAD3 construct or treatment with a CDK2 inhibitor
Discussion
Trastuzumab therapy in the adjuvant setting, for patients with HER2+ breast cancer, has been associated with a 30% increase disease-free survival.Citation3,Citation43 This therapeutic benefit, however, is not universal, and approximately 70% of patients, in the primary or metastatic setting, are found to be resistant to trastuzumab.Citation44,Citation45 Thus, there is a vital need to understand the mechanisms associated with resistance to trastuzumab treatment. By uncovering mechanisms associated with treatment resistance, therapeutic strategies will be identified to greatly expand the number of HER2+ patients that may benefit from trastuzumab. The results of this study suggest a potential approach to improve trastuzumab sensitivity in HER2+, CCNE-high, breast cancer cells through the restoration of canonical SMAD3 function and implementation of CDK2 inhibition.
The transcription factor SMAD3 is canonically activated by TGFβ, which results in cell regulatory and tumor suppressant events.Citation15–17,Citation19 The results of this study indicated that noncanonical SMAD3 phosphorylation, mediated by CCNE/CDK2, contributes to trastuzumab resistance in CCNE-overexpressing HER2+ breast cancer cells. Prior work showed that fadraciclib treatment could improve sensitivity to trastuzumab in treatment resistant CCNE overexpressing, HER2+ breast cancer cells.Citation22 In triple negative (estrogen, progesterone, and HER2 receptor negative) breast cancer cells, noncanonical phosphorylation of SMAD3 by CCNE/CDK2 promoted cycle progression and cell migration.Citation16,Citation21 Conversely, inhibition of noncanonical CDK2 phosphorylation of SMAD3, specifically at the T179 site,Citation16 resulted in reduced invasion and migration for the triple negative breast cancer cells, due, in part, to inhibition of the Pin1-SMAD3 interaction.Citation17 Notably, Pin1 has also been implicated in trastuzumab resistance in the HER2+ cell line SK-BR-3, indicating a possible connection to the CCNE-mediated resistance we observed in these cells (Figure S2).Citation46 In the current study, inhibition of CDK2 mediated phosphorylation by fadraciclib in BT474R2 cells was also observed to result in decreased phosphorylation at the SMAD3 S204, S213, and T179 sites (–c)). Additionally, recent work in HER2+ SKBR3 breast cancer cells identified an association between TGFβ and HER2/AKT signaling through phosphorylation of SMAD3 at the S208 linker region site, leading to loss of tumor suppression and increased invasiveness of these cells.Citation47 Similar associations among noncanonical SMAD3 phosphorylation, deregulation of normal tumor suppressant SMAD3 actions, and aggressive cancer biology have been identified in colorectal,Citation48,Citation49 lung,Citation50 and gastricCitation51 cancer models, providing further evidence for a connection between noncanonical SMAD3 phosphorylation, and oncogenic progression.
This study found an increase in relative phosphorylation of SMAD3 at the linker region in BT474R2 cells relative to the parental BT474 cell line (). Increased pSMAD3/total SMAD3 ratio was observed at the T179 and S204 phosphorylation sites, however was not present at S213 or in the canonical C-terminus S423/425 location. Phosphorylation of SMAD3 by CDK2 occurs primarily at the T179 and S213 locations in vivo,Citation41 and S204 in vitro, which agreed with the phosphorylation sites observed in the in vivo-derived BT474R2 cell line. Previous work suggested phosphorylation at these linker region sites leads to degradation of SMAD3,Citation19 which was consistent with the identified decrease in SMAD3 protein abundance in the BT474R2 study cells. The impact of CCNE on SMAD3 phosphorylation and function was shown through inhibition of CDK2, and the subsequent decrease in the pSMAD3/total SMAD3 ratio at linker region phosphorylation sites. To this end, CDK2 inhibition by fadraciclib resulted in the restoration of downstream canonical TGFβ/SMAD3 associated signaling events, demonstrated through decreased cMyc and increased p15 protein expression ().
The BT474R2 cell line was previously reported to have constitutively upregulated cyclin dependent kinase inhibitor p27, and this cell line was found to be insensitive to treatment with trastuzumab.Citation22 CDK2 and p27 have been associated with regulation of the response to anti-HER2 antibodies in HER2+ breast cancer cells.Citation52 As stated, high CCNE expression has also been correlated with HER2 overexpression and trastuzumab resistance in HER2+ breast cancers.Citation13,Citation22 Deregulation of p27 has been actively studied as a link to CCNE, HER2 overexpression, and trastuzumab resistance,Citation22,Citation53 yet crosstalk with other signaling events has also been proposed. Under physiologic cell cycle regulation, p27 acts an inhibitor of CDK activity and p27 is phosphorylated for degradation by CCNE/CDK2.Citation18 The finding that BT474R2 cells can maintain high levels of p27 protein, and yet are relatively insensitive to p27 function, indicates that p27 activity alone is insufficient to inhibit cell cycle progression in this cancer cell line.Citation22 Canonical TGFβ/SMAD3 action contributes to physiologic cell cycle progression and the fidelity of normal cell cycle checkpoints, including controlled expression of p15, inhibition of cMyc, and the subsequent regulation of CCND/CDK4, which precede activity of CCNE/CDK2. In the current study of BT474R2 cells, treatment with trastuzumab alone did not impact expression of p15 and cMyc. We found that treatment with fadraciclib, blocking noncanonical phosphorylation of the SMAD3, led to upregulation of p15 and downregulation of cMyc expression, and that this effect on p15 and cMyc expression was not altered through the combination of fadraciclib and trastuzumab treatment (,e)). Thus, our prior work examining the impact of noncanonical SMAD3 phosphorylation, along with the results in the current study, indicate that inhibiting noncanonical CDK2 phosphorylation of SMAD3, may be a viable strategy to restore physiologic cell regulatory control in vitro, and sensitivity to trastuzumab in vivo, for HER2+ breast cancer cells that overexpress CCNE.Citation15,Citation16
The TRACER network analysis identified SMAD3 as having a central role in the response of BT474R2 cells to trastuzumab (), mediating the observed downregulation of FOXC2 and GATA3 activity, and upregulation of NFκB activity.Citation54–57 Among several proposed actions, trastuzumab treatment blocks dimerization of the HER2 receptor with HER1/3/4, decreasing activation of the PI3K/Akt and Ras/MAK pathways, and subsequently downregulating NFκB and FOXC2 activity, and leading to reduced tumor growth.Citation4,Citation58,Citation59 Thus, some degree of downregulation of FOXC2 activity could be expected after exposure to trastuzumab treatment. SMAD3 activity was also inferred to be associated with the downregulation of GATA3 activity for BT474R2 cells treated with trastuzumab ()). Decreased GATA3 expression has been suggested as a negative prognostic marker in breast cancer.Citation60 GATA3 has been shown to interact directly with SMAD3, conferring TGFβ-responsiveness to GATA3 target genes in helper T cells.Citation61 Additionally, overexpression of GATA3 in the CCNE-high triple-negative breast cancer cell line MDA-MB-231 resulted in restoration of tumor suppression by TGFβ signaling,Citation62 potentially indicating synergy between elevated GATA3 and canonical SMAD3 activity. Conversely, the relationship among SMAD3 and decreased GATA3 activity, and trastuzumab resistance in BT474R2 cells, may reveal a further connection between CCNE/CDK2 noncanonical SMAD3 phosphorylation, and the potential for CDK2 inhibition to restore sensitivity to trastuzumab in the setting of cyclin E upregulation.
TRACER network modeling linked SMAD3 activity to the treatment resistant BT474R2 cells and also identified upregulation of NFκB activity after exposure to trastuzumab ()). Our study showed alterations in TGFβ signaling in this BT474R2 cell line were associated with the loss of canonical SMAD3 function through CCNE/CDK2 mediated phosphorylation of SMAD3 linker region sites. Notably, canonical TGFβ/SMAD3 action also leads to inhibition of PI3K/Akt.Citation63 Therefore the CCNE/CDK2-mediated decrease in canonical SMAD3 action ()), may also contribute to a concurrent increase in PI3K/Akt signaling, the observed changes in NFKB activity for the BT474R2 cell line, and resistance to trastuzumab. We expect this proposed CCNE/CDK2-mediated mechanism of trastuzumab resistance, associated with noncanonical SMAD3 phosphorylation, could also be treated with a targeted strategy implementing fadraciclib or other CDK2 inhibitor therapy. This treatment plan would extend to combination therapies using trastuzumab, along with other anti-HER2-targeted treatments, including pertuzumab.Citation64
The functional effects of SMAD3 on BT474R2 cells were also directly associated with sensitivity to trastuzumab therapy in vitro and in in vivo ( and ). Overexpressing either a wild-type SMAD3 or a linker-region mutant SMAD3 (5M) both significantly decreased proliferation of BT474R2 cells in vitro. Importantly, the 5M SMAD3-expressing cells, resistant to noncanonical CDK phosphorylation, were significantly more susceptible to trastuzumab therapy in vitro ()), as evidenced by decreased proliferation in these cells relative to both the SMAD3 overexpressing cell line and the blank BT474 transfected cell line. Overexpression of the wild-type SMAD3 construct was not sufficient to alter the expression of p15, suggesting CDK mediated noncanonical phosphorylation in the expressed WT SMAD3 construct. Expression of the CDK phosphorylation-resistant 5M SMAD3 construct led to a significant upregulation of p15 protein expression (,d)) following trastuzumab therapy, indicating that this construct was active in altering the transcriptome of these cells, favoring the restoration of canonical SMAD3 tumor suppressant behaviors. These findings also supported the TRACER results, indicating that the canonical, tumor-suppressant functions of SMAD3 signaling in CCNE overexpression BT474 cells played a role in promoting the response to trastuzumab. To this end, inhibition of CDK2 phosphorylation and restoration of canonical SMAD3 function were also shown to support sensitivity to trastuzumab in vivo. PBS treated BT474R2-5M tumors grew at the same rate as BT474R2-Blank tumors, while only the BT474R2-5M cells, transduced with CDK phosphorylation resistant 5M SMAD3 construct, responded robustly to treatment with trastuzumab, observed through both a decrease in tumor volume ()), as well as a decrease in Ki67 immunostaining (,c)). Together, these results provide evidence that CCNE overexpression confers resistance to trastuzumab, in part, through aberrant noncanonical CDK mediated phosphorylation of SMAD3. This data further supports that restoring SMAD3 function through treatment with CDK2 inhibitors such as fadraciclib or introduction of a mutant, CDK phosphorylation resistant 5M SMAD3 construct (), may be possible strategies to management patients presenting with treatment resistant HER2+ breast cancer.
In summary, our study provides further evidence regarding a treatment strategy that includes trastuzumab, along with a potential therapeutic role for CDK2 inhibitors to target noncanonical SMAD3 phosphorylation, for patients with trastuzumab resistant CCNE-high HER2+ breast cancer. CDK2 inhibitors are currently being implemented in clinical trials targeting several types of cancer including, leukemia, non-small cell lung cancer, and melanoma.Citation23 Importantly, preclinical studies using CDK2 inhibitors such as fadraciclib therapy are also showing promise for the management of aggressive or treatment resistant breast cancer.Citation65–67 We expect the rational implementation of CDK inhibition would also extend the therapeutic potential combination of trastuzumab/pertuzumab therapy.Citation64 Taken together, these results suggest both a promising avenue of treatment for patients with CCNE-high, HER2+ breast cancer, as well as a potential mechanism to explore in other aggressive, treatment-resistant, HER2+ cancer models.
Disclosure of potential conflicts of interest
The authors have nothing to disclose relative to this work.
Supplemental Material
Download MS Word (331.1 KB)Acknowledgments
We thank Rohit Maramraju for his thoughtful contribution to the manuscript. PK is a recipient of the NIH Institutional Research Training Grant T32CA009672. JTD is supported by NIH K01EB028877.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. doi:10.3322/caac.21332. PubMed PMID: 26742998.
- Nahta R, Yu D, Hung MC, Hortobagyi GN, Esteva FJ. Mechanisms of disease: understanding resistance to HER2-targeted therapy in human breast cancer. Nat Clin Pract Oncol. 2006;3(5):269–280. doi:10.1038/ncponc0509. PubMed PMID: 16683005.
- Piccart-Gebhart MJ, Procter M, Leyland-Jones B, Goldhirsch A, Untch M, Smith I, Gianni L, Baselga J, Bell R, Jackisch C, et al. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med. 2005;353(16):1659–1672. Epub 2005/ 10/21. doi: 10.1056/NEJMoa052306. PubMed PMID: 16236737.
- Hudis CA. Trastuzumab — mechanism of action and use in clinical practice. N Engl J Med. 2007;357(1):39–51. doi:10.1056/NEJMra043186. PubMed PMID: 17611206.
- Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344(11):783–792. doi:10.1056/NEJM200103153441101. PubMed PMID: 11248153.
- Stern HM. Improving treatment of HER2-positive cancers: opportunities and challenges. Sci Transl Med. 2012;4(127):127rv2. doi:10.1126/scitranslmed.3001539. PubMed PMID: 22461643.
- Baselga J. Clinical trials of herceptin(trastuzumab). Eur J Cancer. 2001;37(Suppl 1):S18–24. PubMed PMID: 11167087.
- Nahta R, Esteva FJ. HER2 therapy: molecular mechanisms of trastuzumab resistance. Breast Cancer Res. 2006;8(6):215. doi:10.1186/bcr1612. PubMed PMID: 17096862; PMCID: PMC1797036.
- Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, Linn SC, Gonzalez-Angulo AM, Stemke-Hale K, Hauptmann M. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12(4):395–402.
- Nagata Y, Lan K-H, Zhou X, Tan M, Esteva FJ, Sahin AA, Klos KS, Li P, Monia BP, Nguyen NT. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6(2):117–127.
- Nahta R, Yuan LX, Zhang B, Kobayashi R, Esteva FJ. Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Res. 2005;65(23):11118–11128.
- Arteaga CL, Engelman JA. ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell. 2014;25(3):282–303.
- Mittendorf EA, Liu Y, Tucker SL, McKenzie T, Qiao N, Akli S, Biernacka A, Liu Y, Meijer L, Keyomarsi K, et al. A novel interaction between HER2/neu and cyclin E in breast cancer. Oncogene. 2010;29(27):3896–3907. doi:10.1038/onc.2010.151. PubMed PMID: 20453888; PMCID: PMC2900397.
- Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015;14(2):130–146. doi:10.1038/nrd4504. PubMed PMID: 25633797; PMCID: PMC4480421.
- Tarasewicz E, Hamdan R, Straehla J, Hardy A, Nunez O, Zelivianski S, Dokic D, Jeruss JS. CDK4 inhibition and doxorubicin mediate breast cancer cell apoptosis through Smad3 and survivin. Cancer Biol Ther. 2014;15(10):1301–1311. doi:10.4161/cbt.29693. PubMed PMID: 25006666; PMCID: PMC4130723.
- Tarasewicz E, Rivas L, Hamdan R, Dokic D, Parimi V, Bernabe BP, Thomas A, Shea LD, Jeruss JS. Inhibition of CDK-mediated phosphorylation of Smad3 results in decreased oncogenesis in triple negative breast cancer cells. Cell Cycle. 2014;13(20):3191–3201. doi:10.4161/15384101.2014.950126. PubMed PMID: 25485498; PMCID: PMC4614520.
- Thomas AL, Lind H, Hong A, Dokic D, Oppat K, Rosenthal E, Guo A, Thomas A, Hamden R, Jeruss JS. Inhibition of CDK-mediated Smad3 phosphorylation reduces the Pin1-Smad3 interaction and aggressiveness of triple negative breast cancer cells. Cell Cycle. 2017;16(15):1453–1464. doi:10.1080/15384101.2017.1338988.
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13(12):1501–1512. PubMed PMID: 10385618.
- Tarasewicz E, Jeruss JS. Phospho-specific Smad3 signaling: impact on breast oncogenesis. Cell Cycle. 2012;11(13):2443–2451. doi:10.4161/cc.20546. PubMed PMID: 22659843; PMCID: PMC3404875.
- Roberts AB, Tian F, Byfield SD, Stuelten C, Ooshima A, Saika S, Flanders KC. Smad3 is key to TGF-β-mediated epithelial-to-mesenchymal transition, fibrosis, tumor suppression and metastasis. Cytokine Growth Factor Rev. 2006;17(1–2):19–27.
- Matsuura I, Denissova NG, Wang G, He D, Long J, Liu F. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature. 2004;430(6996):226–231. doi:10.1038/nature02650. PubMed PMID: 15241418.
- Scaltriti M, Eichhorn PJ, Cortes J, Prudkin L, Aura C, Jimenez J, Chandarlapaty S, Serra V, Prat A, Ibrahim YH, et al. Cyclin E amplification/overexpression is a mechanism of trastuzumab resistance in HER2+ breast cancer patients. Proc Natl Acad Sci U S A. 2011;108(9):3761–3766. doi:10.1073/pnas.1014835108. PubMed PMID: 21321214; PMCID: PMC3048107.
- Chohan TA, Qian H, Pan Y, Chen JZ. Cyclin-dependent kinase-2 as a target for cancer therapy: progress in the development of CDK2 inhibitors as anti-cancer agents. Curr Med Chem. 2015;22(2):237–263. Epub 2014/ 11/12. PubMed PMID: 25386824.
- Dull T, Zufferey R, Kelly M, Mandel R, Nguyen M, Trono D, Naldini L. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72(11):8463–8471.
- Decker JT, Hall MS, Penalver-Bernabe B, Blaisdell RB, Liebman LN, Jeruss JS, Shea LD. Design of large-scale reporter construct arrays for dynamic, live cell systems biology. ACS Synth Biol. 2018;7:2063–2073.
- Decker JT, Hobson EC, Zhang Y, Shin S, Thomas AL, Jeruss JS, Arnold KB, Shea LD. Systems analysis of dynamic transcription factor activity identifies targets for treatment in olaparib resistant cancer cells. Biotechnol Bioeng. 2017;114(9):2085–2095.
- Bernabé BP, Shin S, Rios PD, Broadbelt LJ, Shea LD, Seidlits SK. Dynamic transcription factor activity networks in response to independently altered mechanical and adhesive microenvironmental cues. Integr Biol. 2016;8(8):844–860.
- Smyth GK. Limma: linear models for microarray data. Bioinformatics and computational biology solutions using R and Bioconductor. Springer; 2005. p. 397–420.
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B (Methodological). 1995;57:289–300.
- Decker JT, Hall MS, Blaisdell RB, Schwark K, Jeruss JS, Shea LD. Dynamic microRNA activity identifies therapeutic targets in trastuzumab‐resistant HER2+ breast cancer. Biotechnol Bioeng. 2018;115:2613–2623.
- Mevik B-H, Wehrens R. The pls package: principal component and partial least squares regression in R. J Stat Softw. 2007;18(2):1–24.
- Siletz A, Schnabel M, Kniazeva E, Schumacher AJ, Shin S, Jeruss JS, Shea LD. Dynamic transcription factor networks in epithelial-mesenchymal transition in breast cancer models. PloS One. 2013;8:4.
- Haury A-C, Mordelet F, Vera-Licona P, Vert J-P. TIGRESS: trustful inference of gene regulation using stability selection. BMC Syst Biol. 2012;6(1):1.
- Breiman L. Random forests. Mach Learn. 2001;45(1):5–32.
- Margolin AA, Nemenman I, Basso K, Wiggins C, Stolovitzky G, Favera RD, Califano A. ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinform. 2006;7(Suppl 1):S7.
- Faith JJ, Hayete B, Thaden JT, Mogno I, Wierzbowski J, Cottarel G, Kasif S, Collins JJ, Gardner TS. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 2007;5(1):e8.
- Meyer PE, Kontos K, Lafitte F, Bontempi G. Information-theoretic inference of large transcriptional regulatory networks. EURASIP J Bioinform Syst Biol. 2007;2007:8.
- Csardi G, Nepusz T. The igraph software package for complex network research. Int J Complex Syst. 2006;1695(5):1–9.
- Frame, S, Saladino C, MacKay C, Atrash B, Sheldrake P, McDonald E, Clarke PA, Worman P, Blake D, Zheleva D. Fadraciclib (CYC065), a novel CDK inhibitor, targets key pro-survival and oncogenic pathways in cancer. Plos one. 2020;15(7):e0234103.
- Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, McQuaid S, Gray RT, Murray LJ, Coleman HG. QuPath: open source software for digital pathology image analysis. Sci Rep. 2017;7(1):1–7.
- Matsuura I, Denissova NG, Wang G, He D, Long J, Liu F. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature. 2004;430(6996):226.
- Alarcón C, Zaromytidou A-I, Xi Q, Gao S, Yu J, Fujisawa S, Barlas A, Miller AN, Manova-Todorova K, Macias MJ. Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF-β pathways. Cell. 2009;139(4):757–769.
- Romond EH, Perez EA, Bryant J, Suman VJ, Geyer CE Jr., Davidson NE, Tan-Chiu E, Martino S, Paik S, Kaufman PA, et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med. 2005;353(16):1673–1684. Epub 2005/ 10/21. doi: 10.1056/NEJMoa052122. PubMed PMID: 16236738.
- Chung A, Cui X, Audeh W, Giuliano A. Current status of anti–human epidermal growth factor receptor 2 therapies: predicting and overcoming herceptin resistance. Clin Breast Cancer. 2013;13(4):223–232.
- Santa-Maria CA, Nye L, Mutonga MB, Jain S, Gradishar WJ. Management of metastatic HER2-positive breast cancer: where are we and where do we go from here? Oncology. 2016;30:2.
- Sajadimajd S, Yazdanparast R. Sensitizing effect of juglone is mediated by down regulation of Notch1 signaling pathway in trastuzumab-resistant SKBR3 cells. Apoptosis. 2017;22(1):135–144.
- Huang F, Shi Q, Li Y, Xu L, Xu C, Chen F, Wang H, Liao H, Chang Z, Liu F, et al. HER2/EGFR–AKT signaling switches TGFβ from inhibiting cell proliferation to promoting cell migration in breast cancer. Cancer Res. 2018;78(21):6073–6085. doi:10.1158/0008-5472.can-18-0136.
- Matsuzaki K, Kitano C, Murata M, Sekimoto G, Yoshida K, Uemura Y, Seki T, Taketani S, Fujisawa J-I, Okazaki K. Smad2 and Smad3 phosphorylated at both linker and COOH-terminal regions transmit malignant TGF-β signal in later stages of human colorectal cancer. Cancer Res. 2009;69(13):5321–5330.
- Yamagata H, Matsuzaki K, Mori S, Yoshida K, Tahashi Y, Furukawa F, Sekimoto G, Watanabe T, Uemura Y, Sakaida N. Acceleration of Smad2 and Smad3 phosphorylation via c-Jun NH2-terminal kinase during human colorectal carcinogenesis. Cancer Res. 2005;65(1):157–165.
- Jo E, Park SJ, Choi YS, Jeon W-K, Kim B-C. Kaempferol suppresses transforming growth factor-β1–induced epithelial-to-mesenchymal transition and migration of A549 lung cancer cells by inhibiting Akt1-mediated phosphorylation of Smad3 at threonine-179. Neoplasia. 2015;17(7):525–537.
- Li L, Cao F, Liu B, Luo X, Ma X, Hu Z. TGF-β induces fascin expression in gastric cancer via phosphorylation of smad3 linker area. Am J Cancer Res. 2015;5(6):1890.
- Lane HA, Beuvink I, Motoyama AB, Daly JM, Neve RM, Hynes NE. ErbB2 potentiates breast tumor proliferation through modulation of p27(Kip1)-Cdk2 complex formation: receptor overexpression does not determine growth dependency. Mol Cell Biol. 2000;20(9):3210–3223. PubMed PMID: 10757805; PMCID: PMC85615.
- Nahta R, Takahashi T, Ueno NT, Hung MC, Esteva FJ. P27(kip1) down-regulation is associated with trastuzumab resistance in breast cancer cells. Cancer Res. 2004;64(11):3981–3986. doi:10.1158/0008-5472.CAN-03-3900. PubMed PMID: 15173011.
- Bitzer M, von Gersdorff G, Liang D, Dominguez-Rosales A, Beg AA, Rojkind M, Böttinger EP. A mechanism of suppression of TGF–β/SMAD signaling by NF-κB/RelA. Genes Dev. 2000;14(2):187–197.
- Borthwick LA, Gardner A, De Soyza A, Mann DA, Fisher AJ. Transforming growth factor-β1 (TGF-β1) driven epithelial to mesenchymal transition (EMT) is accentuated by tumour necrosis factor α (TNFα) via crosstalk between the SMAD and NF-κB pathways. Cancer Microenvironment. 2012;5(1):45–57. doi:10.1007/s12307-011-0080-9.
- Chou J, Lin JH, Brenot A, Kim J-W, Provot S, Werb Z. GATA3 suppresses metastasis and modulates the tumour microenvironment by regulating microRNA-29b expression. Nat Cell Biol. 2013;15:201. doi:10.1038/ncb2672.
- Fujita H, Kang M, Eren M, Gleaves LA, Vaughan DE, Kume T. Foxc2 is a common mediator of insulin and transforming growth factor β signaling to regulate plasminogen activator inhibitor type I gene expression. Circ Res. 2006;98(5):626–634.
- Triulzi T, Forte L, Regondi V, Di Modica M, Ghirelli C, Carcangiu ML, Sfondrini L, Balsari A, Tagliabue E. HER2 signaling regulates the tumor immune microenvironment and trastuzumab efficacy. Oncoimmunology. 2019;8(1):e1512942.
- Cui Y-M, Jiang D, Zhang S-H, Wu P, Ye Y-P, Chen C-M, Tang N, Liang L, Li -T-T, Qi L. FOXC2 promotes colorectal cancer proliferation through inhibition of FOXO3a and activation of MAPK and AKT signaling pathways. Cancer Lett. 2014;353(1):87–94.
- Mehra R, Varambally S, Ding L, Shen R, Sabel MS, Ghosh D, Chinnaiyan AM, Kleer CG. Identification of GATA3 as a breast cancer prognostic marker by global gene expression meta-analysis. Cancer Res. 2005;65(24):11259–11264.
- Blokzijl A, Ten Dijke P, Ibáñez CF. Physical and functional interaction between GATA-3 and Smad3 allows TGF-β regulation of GATA target genes. Curr Biol. 2002;12(1):35–45.
- Chu IM, Lai W-C, Aprelikova O, El Touny LH, Kouros-Mehr H, Green JE. Expression of GATA3 in MDA-MB-231 triple-negative breast cancer cells induces a growth inhibitory response to TGFss. PLoS One. 2013;8:4.
- Zhang L, Zhou F, Ten Dijke P. Signaling interplay between transforming growth factor-β receptor and PI3K/AKT pathways in cancer. Trends Biochem Sci. 2013;38(12):612–620. doi:10.1016/j.tibs.2013.10.001.
- Scheuer W, Friess T, Burtscher H, Bossenmaier B, Endl J, Hasmann M. Strongly enhanced antitumor activity of trastuzumab and pertuzumab combination treatment on HER2-positive human xenograft tumor models. Cancer Res. 2009;69(24):9330–9336.
- Rao SS, Stoehr J, Dokic D, Wan L, Decker JT, Konopka K, Thomas AL, Wu J, Kaklamani VG, Shea LD, et al. Synergistic effect of eribulin and CDK inhibition for the treatment of triple negative breast cancer. Oncotarget [Internet]. 2017 2017/10//; 8 48:[83925-39].
- Doostan I, Karakas C, Kohansal M, Low KH, Ellis MJ, Olson JA Jr., Suman VJ, Hunt KK, Moulder SL, Keyomarsi K. Cytoplasmic cyclin E mediates resistance to aromatase inhibitors in breast cancer. Clin Cancer Res. 2017;23(23):7288–7300. Epub 2017/ 09/28. doi: 10.1158/1078-0432.ccr-17-1544. PubMed PMID: 28947566; PMCID: PMC5768442.
- Alexander A, Karakas C, Chen X, Carey JP, Yi M, Bondy M, Thompson P, Cheung KL, Ellis IO, Gong Y, et al. Cyclin E overexpression as a biomarker for combination treatment strategies in inflammatory breast cancer. Oncotarget. 2017;8(9):14897–14911. Epub 2017/ 01/21. doi: 10.18632/oncotarget.14689. PubMed PMID: 28107181; PMCID: PMC5362453.