
ABSTRACT
Chemokine (C-X-C motif) ligand 6 (CXCL6), a member of the CXC chemokine family, reportedly mediates several processes such as inflammation, immunoreaction, cell growth, and metastasis through interaction with the chemokine receptors CXCR1 and CXCR2 in humans; further, CXCR1 and CXCR2 can promote repair and regeneration of organs or tissues after ischemia–reperfusion injury (IRI). In this study, we found that HIF-1α, CXCL6, and CXCR2 expression levels were elevated in human brain microvascular endothelial cells (HBMECs) after IRI, whereas silent information regulator of transcription (Sirt) 3 expression level had reduced. HIF-1α inhibition in an IRI model potently promoted HBMEC proliferation, accompanied by increased Sirt3 and decreased CXCL6/CXCR2 expression levels. CXCL6 knockdown in the IRI model significantly decreased HBMEC permeability and promoted HBMEC proliferation, concurrent with a decrease in apoptosis; it also increased Sirt3 expression levels and decreased CXCL6/CXCR2 protein and phosphorylated AKT (p-AKT) and class O of forkhead box (FOXO) 3a (p-FOXO3a) levels. In addition, CXCL6-induced HBMEC permeability and inhibition of HBMEC proliferation were counteracted by Sirt3 overexpression, and the AKT inhibitor LY294002 counteracted the effect of CXCL6 recombinant proteins on Sirt3, p-AKT, and p-FOXO3a expressions. These results suggest that CXCL6 and Sirt3 are downstream of HIF-1α and that CXCL6 regulatesHBMEC permeability, proliferation, and apoptosis after IRI by modulating Sirt3 expression via AKT/FOXO3a activation.
Introduction
In recent years, with the advancement of electroconvulsive therapy and the establishment and promotion of methods such as arterial bypass surgery, thrombolytic therapy, cardiopulmonary resuscitation, limb replantation, and organ transplantation, it is possible to reperfuse many tissues and organs after ischemia. In most cases, ischemia–reperfusion (I/R) can restore the function and structure of tissues and organs and can reverse vascular occlusion in patients by restoring oxygen supply. However, reperfusion after ischemia sometimes aggravates the dysfunction and structural damage of tissues and organs, which is called I/R injury (IRI). The blood–brain barrier (BBB), an anatomical and a functional barrier between blood and the brain, can block harmful substances carried by blood from entering the brain.Citation1 Brain IRI often causes BBB dysfunction, which is an important pathological feature of many neuronal disorders.Citation2 Here, we investigated the mechanisms underlying the regulation of BBB permeability after IRI.
Hypoxia-inducible factor1 (HIF-1), an oxygen-sensitive transcription factor, was originally shown to protect organs such as the brain and heart from ischemic damage after I/R.Citation3 Prolonged activation of intestinal HIF-1 serves as a proximal regulator in intestinal mucosal injury induced by I/R.Citation4 It has been demonstrated that intestinal mucosal response to intestinal I/R is correlated to prolonged increased expression of HIF-1α, an oxygen-labile subunit of HIF-1.Citation5 Chemokine (C-X-C motif) ligand 6 (CXCL6, also known as granulocyte chemotactic protein 2), a member of the CXC chemokine family, is a chemoattractant for neutrophilic granulocytes and initiates chemotaxis through the chemokine receptors CXCR1 and CXCR2.Citation6,Citation7Studies have shown that CXCR1 can promote liver repair and regeneration after IRI;Citation8 on the contrary, CXCR2 is detrimental to liver recovery after IRI.Citation9 Inhibition of CXCR2/CXCR2 ligand interactions is beneficial for IRI after lung transplant.Citation10 Besides, it is found that CCR5 (a chemokine receptor) knockdown significantly improved neurological deficits in global brain IRI.Citation11 Silent information regulator of transcription (Sirt), class III nicotinamide adenine dinucleotide-dependent histone deacetylases present in several subcellular locations such as the nucleus, cytosol, and mitochondria, is associated with gene silencing and is critical to the regulation of cellular stress and metabolic pathways, which are key processes during IRI.Citation12–14 Importantly, it has been reported that Sirt-mediated protein deacetylation is a key post-translational modification that participates in signal transduction in several cell compartments.Citation15 Cumulative reports have shown that nuclear/cytosolic Sirt1 is important in cardioprotection against IRICitation16–18 and that Sirt6 has a role in protecting cardiomyocytes against IRI.Citation19 Mitochondrial Sirt3 is thought to be a responsive deacetylase that regulates metabolism and oxidative stress. Sirt3, with its target proteins, is reported to regulate the response to IRI, and its deficiency prevents the recovery of cardiac function after IRI.Citation20–24 In addition, the Sirt1/Sirt3 axis has been reported to be an important modulator of BBB permeability after ischemic stroke.Citation2 However, the roles of Sirt3 and CXCL6 on BBB permeability after brain IRI as well as the underlying molecular mechanisms remain poorly understood.
In this study, increased HIF-1α, CXCL6, and CXCR2 and decreased Sirt3 expression levels were observed in an IRI model. HIF-1α inhibition in the IRI model potently promoted HBMEC proliferation, accompanied by increased Sirt3 and decreased CXCL6/CXCR2 expression levels. In the IRI model, CXCL6 knockdown significantly promoted HBMEC proliferation and inhibited HBMEC permeability and apoptosis, accompanied by increased Sirt3 expression, decreased CXCL6/CXCR2 expression, and decreased AKT/FOXO3a phosphorylation. In addition, CXCL6-induced HBMEC permeability and inhibition of proliferation was counteracted by Sirt3 overexpression, and an AKT inhibitor counteracted the effect of CXCL6 on Sirt3, p-AKT, and p-FOXO3a expressions. These effects demonstrated that CXCL6 is critical in regulating BBB permeability after IRI, which is probably involved in Sirt3 expression and AKT/FOXO3a activation.
Materials and methods
Cell culture and establishment of the IRI model
Human brain microvascular endothelial cells (HBMECs), purchased from the Cell Bank of the Chinese Academy of Science (Shanghai, China), were cultured at 37°C in a5% CO2 incubator (Thermo Forma 3111; Thermo, USA) with high-glucose Dulbecco’s modified Eagle’s medium (SH30243.01; HyClone) containing 10% fetal bovine serum (16000–044; Gibco) and 1% antibiotic (penicillin and streptomycin; P1400-100; Solarbio, Shanghai, China). Subsequently, HBMECswere applied to establish anIRI model in vitro.Citation25 The IRI model was simulated by inducing oxygen and glucose deprivation in HBMECs, as previously described.Citation26 Details on the modeling process have been reported previously.Citation27
Construction of a lentivirus expression vector
Three shRNAs targeting different sites of CXCL6 () were synthesized to form an shRNA construct, followed by the insertion of apLKO.1-puro vector through Agel I and Ecol I restriction sites to construct the pLKO.1-puro-shCXCL6 plasmid. The primer was designed according to Sirt3 (AF083108.2) gene sequences: 5′-CCCAAGCTTATGGCGTTCTGGGGTTG-3′(HindIII), 5′-CGGAATTCCTATTTGTCTGGTCCATCAAGC-3′ (EcoRI). Then, Sirt3-coding sequences were amplified, followed by insertion of apCDNA3.1 vector through the HindIII and EcoR I restriction sites to construct the pCDNA3.1-Sirt3 plasmid. 293 T cells were transfected by mixing pLKO.1-puro-shCXCL6 or pCDNA3.1-Sirt3 and the viral packaging plasmids psPAX2 and pMD2G (Addgen, USA) with Lipofectamine 2000 (Invitrogen, USA), and the virus was harvested after 48 h.
Table 1. CXCL6 interference target design results
Real-time polymerase chain reaction (RT-PCR)
After extraction of total RNA from treatedHBMECsusing the Trizol reagent (1596–026; Invitrogen), the extracted RNA was subjected to quantification and confirmation of RNA integrity. Following RNA reverse transcription to cDNA using a reverse transcription kit (#K1622; Fermentas, USA), RT-PCR reactions, with cDNA as templates, were performed in triplicates on a real-time PCR system (ABI-7300; Applied Biosystems, USA) using an SYBR Green PCR kit (#K0223; Thermo). RT-PCR reaction procedures were as follows: denaturation at 95°C for 10 min and (annealing at 95°C for 15 s, and extension at 60°C for 45 s) × 40.Citation28 After the procedures, the mRNA expression of HIF-1α, CXCL6, CXCR2, and Sirt3 relative to GAPDH was calculated by the 2−ΔΔCq method.Citation29 Primers are listed in .
Table 2. Primers used for RT-PCR
Western blotting
After extraction of total proteins from treatedHBMECs by RIPA buffer (R0010; Solarbio, Beijing, China), complemented with protease and phosphatase inhibitors, the extracted proteins were quantified using a BCA quantification kit (PICPI23223; Thermo). Following separation of 25 µg protein by 10% and 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis, the proteins were semi-dry transferred onto polyvinylidene fluoride membranes (HATF00010; Millipore, USA). Next, at room temperature, the membranes were blocked for 1 h with 5% skimmed milk (BYL40422; BD Biosciences, USA) and then incubated overnight at 4°C with primary antibodies (). Subsequently, the membranes were washed 5–6 times using Tris-buffered saline and Tween 20 (TBST), followed by 2 h of incubation at room temperature with horseradish peroxidase-labeled secondary antibodies (1:1000; Beyotime, Shanghai, China) of goat anti-rabbit (A0208) and goat anti-mouse (A0216) types. Following TBST washing, the blots were developed for 5 min with chemiluminescent reagent (WBKLS0100; Millipore) and then exposed on an enhanced chemiluminescence imaging system (Tanon-5200; Tanon, Shanghai, China). Subsequently, using ImageJ (version 1.47 v; Bethesda, MD, USA), the protein levels relative to GAPDH were analyzed and calculated.
Table 3. Information for anti-bodies used for WB
Cell proliferation assay
The IRI model was treated with dimethyl sulfoxide (DMSO)/2-methoxyestradiol (2-MeOE2, an HIF-1α inhibitor), shCXCL6, or CXCL6 recombinant proteins+Sirt3 overexpression. The Cell Counting Kit-8 (CCK-8; CP002, SAB, USA) assay was performed to evaluate proliferation of the treated HBMECs. HBMECswere inoculated at a density of 5,000 cells/well in 96-well plates (TR4001; TRUELINE) intriplicatesand cultured overnight. Following 24 h of the treatment, HBMECs were treated with 100 µl of CCK-8 solution (CCK-8:serum-free medium = 1:10) for 1 h at 37°C according to the manufacturer’s instructions. Next, the absorbance value at 450 nm was measured on a microplate reader as an indicator ofHBMEC proliferation.
Cell apoptosis assay
The IRI model was treated with shCXCL6 or CXCL6 recombinant proteins+Sirt3 overexpression; then, HBMECs were collected and subjected to double staining with V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) (C1063; Beyotime). Following the manufacturer’s instructions, 500,000–1,000,000 treatedHBMECswere resuspended in 195 µl of Annexin-V-FITC-binding buffer. Subsequently, the resuspended cells were incubated in the dark at 4°C for 15 min with 5 µl of Annexin-V-FITC, followed by 5 min of incubation in the dark at 4°C with 5 µl of PI; a tube without both Annexin-V-FITC and PI served as the control. On a flow cytometer, the percentages of apoptotic cells were evaluated using BD AccuriTM C6 (version 1.0.264.21; BD Biosciences).
Transepithelial electrical resistance (TEER) and FITC-dextran detection
A well-established method of monitoring HBMEC permeability in polarized endothelial cells is the measurement of TEER. The IRI model was treated with shCXCL6 or CXCL6 recombinant proteins+Sirt3 overexpression. Each cell group was inoculated into the upper chamber of a 24-well Transwell plate using 10,000 cells (100,000 cells for FITC-dextran detection), and 100 and 600 μl of the medium were added to the upper and lower chambers, respectively, followed by culture in a 37°C in a 5% CO2 incubator, with the medium refreshed after 24 h.
Regarding TEER, electrode functionality was tested with a 0.1–0.15 M NaCl solution after calibrating the ohmmeter to a reading of 1000 Ω and 0 mV. The electrode was sterilized in 70% ethanol for 15 min (<30 min), air dried for 15 s, and rinsed with a sterilized electrolyte solution. Subsequently, the transmembrane resistance of each group was measured with the MillicellERS-2 volt ohmmeter (Millipore) after the cells had overgrown at the bottom for 48 h. Meanwhile, blank wells (without cells) were set up and the resistance value was measured. Electric resistance per unit area was calculated according to the following formula: TEER (Ω cm2) = (R1 − R2) (Ω) × effective membrane area (cm2), where R1 is the experimental group and R2 is the blank group.
Regarding FITC-dextran detection, HBMECs in the logarithmic growth phase were inoculated at a density of 1 × 105 cells in the upper chamber of a 24-well Transwell plate (3422; COSTAR). Next, 100 and 600 μl of the medium were added to the upper and lower chambers, respectively, followed by culture at 37°C in a 5% CO2 incubator. After HBMECs were fully cultured, 1 mg/ml of FITC-dextran (Sc-263323; Santa Cruz) was added, followed by incubation at 37°C in a 5% CO2 incubator for 5 min, and 200 μl of the basal medium was used for determining the basic value. After continued incubation of 24 h with supplementary medium, 200 μl of basal medium was collected for examination of FITC fluorescence intensity (excitation wavelength: 490 nm, emission wavelength: 520 nm) with a microplate reader (E8051; Promega), and permeability was calculated according to a standard curve.
Statistical analysis
Statistical analysis was conducted with GraphPad prism 7.0 (San Diego, CA, USA), and graphed results are expressed as mean ± SD for at least three repeated independent experiments. Statistical significance among ≥3 comparisons was determined by one-way analysis of variance with Tukey’s post test. A P-value of <0.05 indicated statistical significance.
Results
IRI model was successfully established in vitro
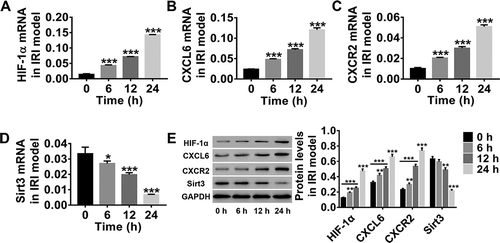
HIF-1α is reported to be a proximal regulator of I/R-induced intestinal mucosal damage, whereas Sirt3 regulates the response to IRI. CXCR2 can promote IRI progression. Here, after the construction of the IRI model using HBMECs, the expression of HIF-1α, CXCL6, CXCR2, and Sirt3 was detected at 0, 6, 12, and 24 h after treatment. shows that HIF-1α, CXCL6, and CXCR2 expression levels, at both mRNA and protein levels, significantly increased in a time-dependent manner in the IRI model (, e), whereas Sirt3 expression level decreased (). These findings indicate that the IRI model was successfully established and that CXCL6, CXCR2, and Sirt3 played crucial roles in IRI progression.
Figure 1. IRI model of HBMECs was successfully established in vitro. After construction of the IRI model of HBMECsin vitro, HBMECs were collected. (a–d) HIF-1α, CXCL6, CXCR2, and Sirt3 mRNA expressions were detected by RT-PCR. (e) HIF-1α, CXCL6, CXCR2, and Sirt3 protein levels were analyzed by western blotting. All results are expressed as mean ± SD with three repeated experiments.*P < .05, **P < .01, and ***P < .001, compared with 0 h
HIF-1α inhibition in the IRI model promoted HBMEC proliferation
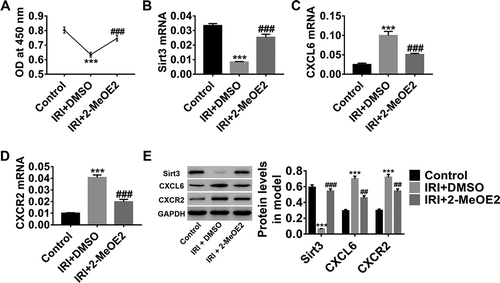
HIF-1α expression and activation have been reported to be involved in protecting organs against IRI,Citation30–32 which is consistent with the result of our study that HIF-1α expression level increased in the IRI model. Here, 2-MeOE2was applied to investigate the function of HIF-1α. As presented in , after IRI, HBMEC proliferation significantly decreased (), and Sirt3 expression level significantly decreased (), whereas CXCL6 () and CXCR2 () expression levels increased. On the contrary, HIF-1α inhibition potently counteracted the induction of IRI. These results indicated that HIF-1α inhibition may contribute to the remission of IRI by promoting HBMECproliferation and that CXCL6/CXCR2 and Sirt3 may be downstream of HIF-1α.
Figure 2. HIF-1α inhibition in IRI model promoted HBMEC proliferation. After construction of the IRI model, HBMECs were divided for treat with DMSO or 10 µM of 2-MeOE2, with medium-treated HBMECs without IRI serving as the control. (a) HBMEC proliferation was assessed by the CCK-8 assay. (b-d) Sirt3, CXCL6, and CXCR2 mRNA levels were detected. (e) Sirt3, CXCL6, and CXCR2 protein levels were also detected. All graphs are presented as mean ± SD with three repeated experiments.***P < .001 compared with control, ##P < .01 and ###P < .001 compared with IRI+2-MeOE2
CXCL6 knockdown in HBMECs by lentiviral infection
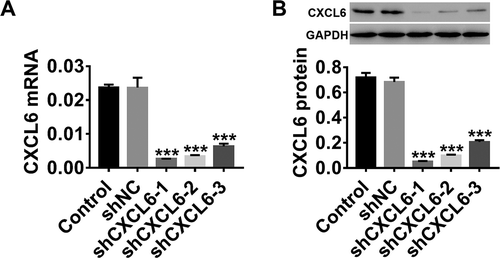
In vitro, HBMECs were grouped for infection with lentiviruses of shNC, shCXCL6-1, shCXCL6-2, and shCXCL6-3, and HBMECs treated with the medium were used as the control. As shown in , both CXCL6 mRNA () and protein () levels were significantly downregulated by all three forms of shCXCL6. Compared with shCXCL6-3, shCXCL6-1 and shCXCL6-2 had better efficiency, so they were selected for the next experiment.
Figure 3. CXCL6 knockdown in HBMECs by lentiviral infection. HBMECs were infected with shNC/shCXCL6 (shCXCL-1, shCXCL6-2, and shCXCL6-3) lentivirus, with medium-treated HBMECs serving as the control. (a and b) The efficiency of shCXCL6-1, shCXCL6-2, and shCXCL6-3 in regulating CXCL6 in HBMECs was determined by RT-PCR and western blotting. Data are expressed as mean ± SD with three repeated experiments.***P < .001 compared with shNC
CXCL6 knockdown inhibited IRI-induced HBMECpermeability and apoptosis
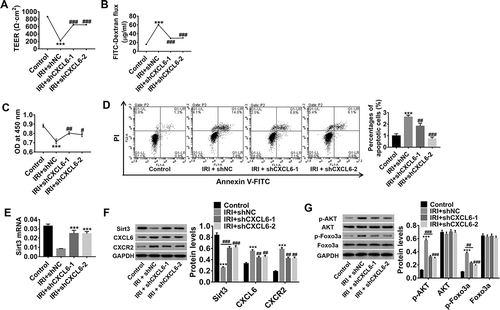
Research has proved that microvascular permeability to plasma proteins is a useful and sensitive indicator for assessing the impact of I/R on microvascular integrity.Citation33 Thus, we performed transmembrane resistance and FITC leakage assays to analyze the effect of CXCL6 interference on HBMECpermeability. As illustrated in , IRI significantly suppressed TEER and inducedHBMECpermeability, which was potently counteracted by CXCL6 knockdown. Furthermore, CXCL6 knockdown significantly promoted IRI-inhibited proliferation () but reduced apoptosis (). In addition, IRI-inhibited Sirt3 expression was significantly increased by CXCL6 knockdown (), whereas IRI-induced CXCL6/CXCR2, p-AKT, and p-FOXO3a levels significantly decreased (). These results demonstrated that CXCL6 knockdown could inhibit IRI-induced HBMECpermeability and apoptosis as well as AKT/FOXO3a activation and that Sirt3 may be downstream of CXCL6.
Figure 4. CXCL6 knockdown inhibited IRI-induced HBMECpermeability and apoptosis. After construction of the IRI model of HBMECsin vitro, CXCL6 expression in the IRI model was knocked down by shCXCL6 infection, with medium-treated HBMECs without IRI serving as the control. (a and b) TEER and FITC leakage assays were performed to analyze the effect of CXCL6 interference on HBMECpermeability. (c and d) HBMEC proliferation and apoptosis after IRI were evaluated. Lower right quadrant presents early apoptotic cells stained with Annexin-V, whereas late apoptotic or necrotic cells stained with Annexin-V and PI are located in the upper right quadrant. At the lower left are living cells. (e) Sirt3 mRNA expression was detected. (f) Sirt3, CXCL6, and CXCR2 protein levels were detected. (g) AKT and FOXO3a expressions and phosphorylation were evaluated. All data are shown as mean ± SD with three repeated experiments.***P < .001 compared with control, ##P < .01 and ###P < .001 compared with IRI+shNC
CXCL6-regulated IRI-induced HBMECpermeability and apoptosis may modulate Sirt3 expression via AKT/FOXO3a activation
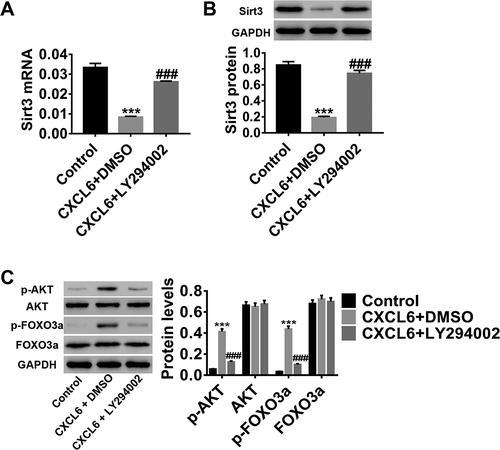
We also investigated the underlying molecular mechanism of CXCL6 regulation of IRI. Studies have shown that the AKT pathway is involved in IRI progression.Citation34 FOXO transcription factors such as FOXO3a can be phosphorylated and inactivated by AKT.Citation35,Citation36 In this study, after the treatment with CXCL6 recombinant proteins and LY294002, an AKT inhibitor, at both mRNA () and protein () levels, Sirt3 expression was significantly inhibited by CXCL6 recombinant proteins; however, AKT and FOXO3a phosphorylation () significantly increased, whereas AKT and FOXO3aexpressions remained unchanged. In addition, LY294002 potently counteracted the effect of CXCL6 recombinant proteins. These results indicated that CXCL6 regulation of IRI-induced HBMECpermeability and apoptosis possibly modulated Sirt3 expression via AKT/FOXO3a activation.
Figure 5. CXCL6-regulated IRI-induced HBMECpermeability and apoptosis may modulate Sirt3 expression AKT/FOXO3a activation. Groups of HBMECs were treated with medium, 10 ng/ml of CXCL6 recombinant proteins+DMSO, or 10 ng/ml of CXCL6 recombinant proteins+10 µM of LY294002 (an AKT inhibitor). (a and b) Sirt3 mRNA and protein expression levels were determined. (c) AKT and FOXO3a expressions and phosphorylation were evaluated. The results are expressed as mean ± SD with three repeated experiments.***P < .001 compared with control, ###P < .001 compared with CXCL6+ DMSO
CXCL6 regulation of IRI-induced HBMECpermeability and apoptosis possibly through modulation of Sirt3 expression
Sirt3 expression in HBMECs was upregulated by Sirt3 lentivirus (). Sirt3 overexpression in HBMECs significantly suppressed CXCL6-induced HBMECpermeability (), whereas CXCL6-induced inhibition of proliferation was also counteracted by Sirt3 upregulation, concurrent with apoptosis reduction (). Moreover, the CXCL6-inhibited levels of claudin-4, occludin, and zonula occludens-1 (ZO-1) which are tight junction proteins, were markedly increased by Sirt3 upregulation (). These results provided further evidence that CXCL6 regulated IRI-induced HBMEC permeability and apoptosis, probably by modulating Sirt3 expression.
Figure 6. CXCL6-regulated IRI-induced HBMECpermeability and apoptosis may act by modulating Sirt3 expression. (a and b) The upregulation efficiency of Sirt3 lentivirus on Sirt3 expression in HBMECs was determined. HBMECs were treated with medium, 10 ng/ml CXCL6 recombinant proteins+vector, or 10 ng/ml of CXCL6 recombinant proteins+Sirt3. (c and d) TEER and FITC leakage assays were performed to analyze HBMECpermeability. (e and f) The proliferation and apoptosis of HBMECs were evaluated. (g) Claudin-4, occludin, and ZO-1 protein levels were determined. All graphs are shown as mean ± SD with three repeated experiments.***P < .001 compared with vector, +++P < .001 compared with control, ##P < .01 and ###P < .001 compared with CXCL6+ vector
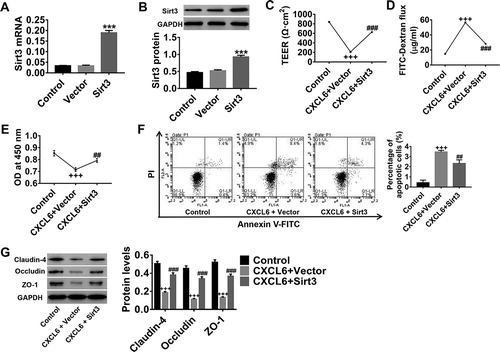
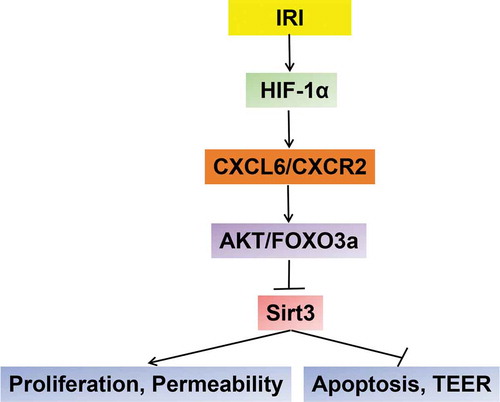
Figure 7. A schematic diagram of the signal pathway A schematic diagram to show the signal pathway that is revealed in this study
Discussion
During the 1970s, IRI was first encountered in coronaryCitation37 and peripheral vascular circulation.Citation38 IRI has become a major problem that interferes with the benefit of reperfusion therapy for ischemic tissues and organs. In this study, high HIF-1α, CXCL6, and CXCR2 expression levels in the IRI model and decreased Sirt3 expression level were observed, indicating that HIF-1α, CXCL6/CXCR2, and Sirt3 possibly play key roles in brain IRI. IRI-inhibited cell proliferation was significantly increased by HIF-1α inhibition, and CXCL6/CXCR2 expression levels decreased, which is consistent with previous studies showing that prolonged increased expression of HIF-1αis involved in IRI.Citation5 Furthermore, CXCL6 knockdown significantly suppressed IRI-induced HBMECpermeability and increased HBMEC proliferation, concurrent with decreased apoptosis, suggesting a critical role of CXCL6 in IRI-induced HBMECpermeability and proliferation. A study showed that HIF-1α could mediate cell functions by modulating CXCL6 expression.Citation39 From these points, it can be inferred that HIF-1α response to brain IRI may occur through the modulation of CXCL6 expression. We also explored the molecular mechanisms of CXCL6 underlyingHBMECpermeability and proliferation after brain IRI. Studies have reported that human CXCL6 could exert a mediating function through CXCR2,Citation7,Citation40 which agrees with the observation that CXCR2 protein level decreased with CXCL6 knockdown. It has been shown that activation of the AKT pathway could prevent adverse remodeling after IRI.Citation34 Moreover, AKT activation can promote cell survival through substrate phosphorylation such as with the FOXO family.Citation41 Recently, the PTEN/AKT/FOXO3a pathway has been reported to participate in neuronal apoptosis in developing rat brain after hypoxia–ischemia.Citation42 In line with these results, we found that CXCL6 recombinant proteins induced HBMECpermeability and inhibition of proliferation, concurrent with reduced apoptosis as well as decreased Sirt3 expression level and increased AKT and FOXO3a phosphorylation, and this induction was potently counteracted by LY294002. These results suggested that CXCL6 regulated IRI-induced HBMECpermeability and proliferation by modulating Sirt3 expression via AKT/FOXO3a activation. This is consistent with previous reports indicating that the AKT-1/FOXO3a pathway is involved in the proliferation of human neural progenitor cells.Citation43 In addition, Sirt3 overexpression potently counteracted the effects of CXCL6 recombinant proteins on HBMECpermeability and proliferation, accompanied by increased claudin-4, occludin, and ZO-1 levels. These tight junction proteins are important in the formation of BBB. Claudin-4 has been confirmed to regulate paracellular ion conductivity, and occludin is localized to tight junctions.Citation44–46 ZO-1, a cytoplasmic protein, forms a scaffold to link tight junction-related transmembrane proteins to the perijunctional actin cytoskeleton.Citation47 ZO-1 is also considered critical in transducing regulatory signals, which control the paracellular barrier.Citation47 Based on these considerations, it is conjectured that CXCL6-regulated HBMEC permeability, proliferation, and apoptosis after IRI occurs through modulation of Sirt3 expression via AKT/FOXO3a activation. Remarkably, higher TEER values were consistently observed compared with the values in the literature (hCMEC/D3 and hBMEC: TEER range: 40–200 Ωcm2).Citation48–50 A possible explanation is that we used a different system for the assessment of TEER. Higher TEER values might also arise from higher Sirt3 expression levels, which has been reported to decrease BBB permeability and promote tight junction formation.Citation2
In conclusion, this study demonstrated the critical roles of HIF-1α, CXCL6, and Sirt3 in regulating HBMEC permeability and proliferation after IRI and that CXCL6 and Sirt3 may be downstream of HIF-1α. Both HIF-1α inhibition and CXCL6 knockdown significantly counteracted IRI-induced HBMECpermeability and inhibition of proliferation, probably by modulating Sirt3 expression via AKT/FOXO3a inactivation (). Therefore, these genes exhibit the potential to be effective therapeutic targets for the treatment of organs or tissues after IRI.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Authors’ contributions
TX and JFW conceived and designed the study. XLW, YQD, XXZ, KP and YD performed the experiments. TX and JFW wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Abbreviations
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Additional information
Funding
References
- Schoknecht K, David Y, Heinemann U. 2015. The blood–brain barrier—Gatekeeper to neuronal homeostasis: clinical implications in the setting of stroke. Semin Cell Dev Biol. 38:35–42. doi:10.1016/j.semcdb.2014.10.004.
- Chen T, Dai S-H, Li X, Luo P, Zhu J, Wang Y-H, Fei Z, Jiang X-F. 2018. Sirt1-Sirt3 axis regulates human blood-brain barrier permeability in response to ischemia. Redox Biol. 14:229–236. doi:10.1016/j.redox.2017.09.016.
- Fraisl P, Aragonés J, Carmeliet P. 2009. Inhibition of oxygen sensors as a therapeutic strategy for ischaemic and inflammatory disease. Nat Rev Drug Discov. 8(2):139. doi:10.1038/nrd2761.
- Feinman R, Deitch EA, Watkins AC, Abungu B, Colorado I, Kannan KB, Sheth SU, Caputo FJ, Lu Q, Ramanathan M, et al. 2010. HIF-1 mediates pathogenic inflammatory responses to intestinal ischemia-reperfusion injury. Am J Physiol. 299(1):833–843.
- Koury J, Deitch EA, Homma H, Abungu B, Gangurde P, Condon MR, Lu Q, Xu D-Z, Feinman R. 2004. Persistent HIF-1α activation in gut ischemia/reperfusion injury: potential role of bacteria and lipopolysaccharidE. Shock. 22(3):270–277. doi:10.1097/01.shk.0000135256.67441.3f.
- Proost P, Wuyts A, Conings R, Lenaerts JP, Billiau A, Opdenakker G, Van Damme J. 1993. Human and bovine granulocyte chemotactic protein-2: complete amino acid sequence and functional characterization as chemokines. Biochemistry. 32(38):10170–10177. doi:10.1021/bi00089a037.
- Wuyts A, Van Osselaer N, Haelens A, Samson I, Herdewijn P, Ben-Baruch A, Oppenheim JJ, Proost P, Van Damme J. 1997. Characterization of synthetic human granulocyte chemotactic protein 2: usage of chemokine receptors CXCR1 and CXCR2 and in vivo inflammatory properties. Biochemistry. 36(9):2716–2723. DOI:10.1021/bi961999z.
- Clarke C, Kuboki S, Sakai N, Kasten KR, Tevar AD, Schuster R, Blanchard J, Caldwell CC, Edwards MJ, Lentsch AB, et al. 2011. CXC chemokine receptor-1 is expressed by hepatocytes and regulates liver recovery after hepatic ischemia/reperfusion injury. Hepatology. 53(1):261–271. DOI:10.1002/hep.24028.
- Kuboki S, Shin T, Huber N, Eismann T, Galloway E, Schuster R, Blanchard J, Edwards MJ, Lentsch AB. 2010. Hepatocyte signaling through CXC chemokine receptor-2 is detrimental to liver recovery after ischemia/reperfusion in mice. Hepatology. 48(4):1213–1223. doi:10.1002/hep.22471.
- Belperio JA, Keane MP, Burdick MD, Gomperts BN, Xue YY, Hong K, Mestas J, Zisman D, Ardehali A, Saggar R, et al. 2005. CXCR2/CXCR2 ligand biology during lung transplant ischemia-reperfusion injury. J Immunol. 175(10):6931–6939. DOI:10.4049/jimmunol.175.10.6931.
- Victoria EC, Ec DBT, Ac DSC, Da SD, de Miranda AS, Da SBL, Sugimoto MA, Sousa LP, de Assis Lima IV, de Oliveira ACP, et al. 2017. Knockdown of C-C chemokine receptor 5 (CCR5) is protective against cerebral ischemia and reperfusion injury. Curr Neurovasc Res. 14:999. doi:10.2174/1567202614666170313113056.
- Baur JA, Ungvari Z, Minor RK, Couteur DGL, Cabo RD. Are sirtuins viable targets for improving healthspan and lifespan? Nat Rev Drug Discov. 2012;11(6):443.
- Dali‐Youcef N, Lagouge M, Froelich S, Koehl C, Schoonjans K, Auwerx J. 2007. Sirtuins: the “magnificient seven”, function, metabolism and longevity. Ann Med. 39(5):335–345. doi:10.1080/07853890701408194.
- Park S, Mori R, Shimokawa I. 2013. Do sirtuins promote mammalian longevity?: A Critical review on its relevance to the longevity effect induced by calorie restriction. Mol Cells. 35(6):474–480. doi:10.1007/s10059-013-0130-x.
- Libert S, Guarente L. 2013. Metabolic and Neuropsychiatric Effects of Calorie Restriction and Sirtuins. Annu Rev Physiol. 75(1):669–684. doi:10.1146/annurev-physiol-030212-183800.
- Hsu CP, Zhai P, Yamamoto T, Maejima Y, Matsushima S, Hariharan N, Shao D, Takagi H, Oka S, Sadoshima J, et al. 2010. Silent Information regulator 1 protects the heart from ischemia/reperfusion. Circulation. 122(21):2170–2182. DOI:10.1161/CIRCULATIONAHA.110.958033.
- Nadtochiy SM, Redman E, Rahman I, Brookes PS. 2011. Lysine deacetylation in ischaemic preconditioning: the role of SIRT1. Cardiovasc Res. 89(3):643. doi:10.1093/cvr/cvq287.
- Nadtochiy SM, Yao H, Mcburney MW, Gu W, Guarente L, Rahman I, Brookes PS. 2011. SIRT1-mediated acute cardioprotection. Am J Physiol Heart Circ Physiol. 301(4):H1506. doi:10.1152/ajpheart.00587.2011.
- Wang XX, Wang XL, Tong MM, Gan L, Chen H, Wu SS, Chen J-X, Li R-L, Wu Y, Zhang H-Y, et al. 2016. SIRT6 protects cardiomyocytes against ischemia/reperfusion injury by augmenting FoxO3α-dependent antioxidant defense mechanisms. Basic Res Cardiol. 111(2):13. DOI:10.1007/s00395-016-0531-z.
- Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, et al. 2007. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol. 27(24):8807–8814. DOI:10.1128/MCB.01636-07.
- Murphy E, Steenbergen C. 2008. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 88(2):581–609. doi:10.1152/physrev.00024.2007.
- Sack MN. 2011. Emerging characterization of the role of SIRT3-mediated mitochondrial protein deacetylation in the heart. Am J Physiol Heart Circ Physiol. 301(6):2191–2197. doi:10.1152/ajpheart.00199.2011.
- Jin L, Galonek H, Israelian K, Choy W, Morrison M, Xia Y, Wang X, Xu Y, Yang Y, Smith JJ, et al. 2010. Biochemical characterization, localization, and tissue distribution of the longer form of mouse SIRT3. Pro Sci. 18(3):514–525. DOI:10.1002/pro.50.
- Koentges C, Pfeil K, Meyer-Steenbuck M, Lother A, Hoffmann MM, Odening KE, Hein L, Bode C, Bugger H. 2015. Preserved recovery of cardiac function following ischemia–reperfusion in mice lacking SIRT3. Can J Physiol Pharmacol. 94(1):72–80. doi:10.1139/cjpp-2015-0152.
- Shi Y, Zhang L, Pu H, Mao L, Hu X, Jiang X, Xu N, Anne Stetler R, Zhang F, Liu X, et al. Rapid endothelial cytoskeletal reorganization enables early blood brain barrier disruption and long-term ischemic/reperfusion brain injury. Nature Communications. 2016;7:10523. doi:10.1038/ncomms10523.
- Zhang W, Smith C, Shapiro A, Monette R, Hutcheon B, Stanimirovic D. 1999. Increased expression of bioactive chemokines in human cerebromicrovascular endothelial cells and astrocytes subjected to simulated ischemia in vitro. J.Neuroimmunol. 101(2):148–160. doi:10.1016/S0165-5728(99)00137-X.
- Zhao Z, Zhang X, Dai Y, Pan K, Deng Y, Meng Y, Xu T. 2019. PPAR-γ promotes p38 MAP kinase-mediated endothelial cell permeability through activating Sirt3. BMC Neurol. 19(1):289. doi:10.1186/s12883-019-1508-y.
- Hong JY, Kang B, Kim A, Hwang S, Ahn J, Lee S, Kim J, Park J-H, Cheon D-S. 2011. Development of a highly sensitive real-time one step RT-PCR combined complementary locked primer technology and conjugated minor groove binder probe. Virol J. 8(1):330. doi:10.1186/1743-422X-8-330.
- Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 25(4):402–408. doi:10.1006/meth.2001.1262.
- Loor G, Schumacker PT. 2008. Role of hypoxia-inducible factor in cell survival during myocardial ischemia–reperfusion. Cell Death Different. 15(4):686. doi:10.1038/cdd.2008.13.
- Imamura R, Moriyama T, Isaka Y, Namba Y, Ichimaru N, Takahara S, Okuyama A. 2007. Erythropoietin protects the kidneys against ischemia reperfusion injury by activating hypoxia inducible factor-1α. Transplantation. 83(10):1371–1379. doi:10.1097/01.tp.0000264200.38926.70.
- Du F, Zhu L, Qian Z-M, Wu X-M, Yung W-H KY. 2010. Hyperthermic preconditioning protects astrocytes from ischemia/reperfusion injury by up-regulation of HIF-1 alpha expression and binding activity. BBA. 1802(11):1048–1053. doi:10.1016/j.bbadis.2010.06.013.
- Granger DN, Sennett M, Mcelearney P, Taylor AE. 1980. Effect of local arterial hypotension on cat intestinal capillary permeability. Gastroenterology. 79(3):474–480. doi:10.1016/0016-5085(80)90372-8.
- Arslan F, Lai RC, Smeets MB, Akeroyd L, Choo A, Aguor ENE, Timmers L, van Rijen HV, Doevendans PA, Pasterkamp G, et al. 2013. Mesenchymal stem cell-derived exosomes increase ATP levels, decrease oxidative stress and activate PI3K/Akt pathway to enhance myocardial viability and prevent adverse remodeling after myocardial ischemia/reperfusion injury. Stem Cell Res. 10(3):301–312. DOI:10.1016/j.scr.2013.01.002.
- Accili D, Arden KC. 2004. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell. 117(4):421–426. doi:10.1016/S0092-8674(04)00452-0.
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME, et al. 1999. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell. 96(6):857–868. DOI:10.1016/S0092-8674(00)80595-4.
- Ginks WR, Sybers HD, Maroko PR, Covell JW, Sobel BE, Ross J. 1972. Coronary artery reperfusion. J Clin Invest. 51(10):2717–2723. doi:10.1172/JCI107091.
- Haimovici H. Muscular, renal, and metabolic complications of acute arterial occlusions: myonephropathic-metabolic syndrome. Surgery. 1979;85(4):461.
- Tian H, Huang P, Zhao Z, Tang W, Xia J. 2014. HIF-1α plays a role in the chemotactic migration of hepatocarcinoma cells through the modulation of CXCL6 expression. Cell Physiol Biochem. 34(5):1536–1546. doi:10.1159/000366357.
- Catusse J, Struyf S, Wuyts A, Weyler M, Loos T, Gijsbers K, Gouwy M, Proost P, Damme JV. 2004. Rabbit neutrophil chemotactic protein (NCP) activates both CXCR1 and CXCR2 and is the functional homologue for human CXCL6. Biochem Pharmacol. 68(10):1947–1955. doi:10.1016/j.bcp.2004.07.003.
- Downward J. 2004. PI 3-kinase, Akt and cell survival. Semin Cell Dev Biol. 15(2):177–182. doi:10.1016/j.semcdb.2004.01.002.
- Li D, Qu Y, Mao M, Zhang X, Li J, Ferriero D, Mu D. 2009. Involvement of the PTEN–AKT–FOXO3a pathway in neuronal apoptosis in developing rat brain after hypoxia–ischemia. J Cerebral Blood Flow Meta. 29(12):1903–1913. doi:10.1038/jcbfm.2009.102.
- Wu Y, Peng H, Cui M, Whitney NP, Huang Y, Zheng JC. 2010. CXCL12 increases human neural progenitor cell proliferation through Akt-1/FOXO3a signaling pathway. J Neurochem. 109(4):1157–1167. doi:10.1111/j.1471-4159.2009.06043.x.
- Van Itallie C, Rahner C, Anderson JM. 2001. Regulated expression of claudin-4 decreases paracellular conductance through a selective decrease in sodium permeability. J Clin Invest. 107(10):1319–1327. doi:10.1172/JCI12464.
- Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S, Tsukita S. 1993. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol. 123(6):1777. doi:10.1083/jcb.123.6.1777.
- González-Mariscal L, Betanzos A, Nava P, Jaramillo BE. 2003. Tight junction proteins. Prog Biophys Mol Biol. 81(1):1–44. doi:10.1016/S0079-6107(02)00037-8.
- Matter K, Balda MS. 2003. Signalling to and from tight junctions. Nat Rev Mol Cell Biol. 4(3):225–236. doi:10.1038/nrm1055.
- Förster C, Burek M, Romero IA, Weksler B, Couraud PO, Drenckhahn D. 2008. Differential effects of hydrocortisone and TNFα on tight junction proteins in an in vitro model of the human blood–brain barrier. J Physiol. 586(7):1937–1949. doi:10.1113/jphysiol.2007.146852.
- Weksler B, Subileau E, Perriere N, Charneau P, Holloway K, Leveque M, Tricoire‐Leignel H, Nicotra A, Bourdoulous S, Turowski P, et al. 2005. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. Faseb J. 19(13):1872–1874. DOI:10.1096/fj.04-3458fje.
- Stins MF, Badger J, Kim KS. 2001. Bacterial invasion and transcytosis in transfected human brain microvascular endothelial cells. Microb Pathog. 30(1):19–28. doi:10.1006/mpat.2000.0406.