
ABSTRACT
Methylation induces epigenetic silencing of tumor suppressor genes in human lung cancer. Inhibition of DNA methyltransferases by decitabine (DAC) can demethylate and activate epigenetically silenced tumor suppressor genes. Epigenetic therapy using DAC should be an attractive strategy for lung cancer therapy. FBW7 is a tumor suppressor that functions as an Mcl-1 E3 ligase to degrade Mcl-1 by ubiquitination. Here we discovered that treatment of various human lung cancer cells with DAC resulted in activation of FBW7 expression, decreased levels of Mcl-1 protein, and growth inhibition. DAC-activated FBW7 expression promoted Mcl-1 ubiquitination and degradation leading to a significant reduction in the half-life of Mcl-1 protein. Mechanistically, treatment of lung cancer cells or lung cancer xenografts with DAC induced the conversion of the FBW7 gene from a methylated form to an unmethylated form, which was associated with the increased expression of FBW7 and decreased expression of Mcl-1 in vitro and in vivo. DAC suppressed lung cancer growth in a dose-dependent manner in vivo. Combined treatment with DAC and a Bcl2 inhibitor, venetoclax, exhibited strong synergistic potency against lung cancer without normal tissue toxicity. These findings uncover a novel mechanism by which DAC suppresses tumor growth by targeting the FBW7/Mcl-1 signaling pathway. Combination of DAC with Bcl2 inhibitor venetoclax provides more effective epigenetic therapy for lung cancer.
Introduction
Methylation of DNA at the 5-position of cytosine plays a key role in transcriptionally suppressing tumor suppressor genesCitation1 in a variety of diseases, including lung cancer.Citation2,Citation3 Epigenetics-based therapies mainly target DNA methyltransferases (DNMTs) and histone-modifying enzymes.Citation3 Targeting DNA methylation as a cancer therapeutic strategy mainly involves DNMT inhibition. Decitabine (5-aza-2ʹ-deoxycytidine, DAC) and azacitidine (AZA) are two major DNMT inhibitors as clinically approved epigenetic drugs for cancer therapy.Citation3 Targeting epigenetic regulation of cell proliferation and cell death is increasingly being investigated as a potent cancer therapy. However, the molecular mechanisms underlying the anticancer effect of these drugs are not fully understood.Citation4 DNA methylation or demethylation in the promoter sequence of genes is an important mechanism to silence or activate tumor suppressor genes in lung cancers.Citation5
Lung cancer is the leading cause of cancer-related death in males and females in the United States and causes one of every three cancer-related deaths. Only 13–17% of lung cancer patients survive more than 5 years.Citation5 A number of aberrantly methylated genes have been identified in lung cancer, such as Wnt, p16, FHIT, RASSFIA, and RARβ2.Citation6,Citation7 However, the role of FBW7 methylation in lung cancer has not been investigated, yet. FBW7 as a tumor suppressor gene is a member of the F-box family of proteins, which functions as interchangeable substrate recognition components of the SCF ubiquitin ligases.Citation8 FBW7 has been characterized as a tumor suppressor protein in the regulation of numerous cellular processes by promoting the degradation of critical regulatory proteins including cyclin E, c-Myc, Notch, c-Jun, and Mcl-1.Citation8–11 Mcl-1 levels are elevated in NSCLC with mutant or low-level FBW7.Citation12 A few prior studies have shown experimental evidence of a role for FBW7 in governing the apoptotic pathway by controlling Mcl-1 destruction.Citation13,Citation14 During mitotic arrest, Mcl-1 protein levels decline markedly, through a post-translational mechanism, potentiating cell death.Citation12 Phosphorylation of Mcl-1 at S159 directs its interaction with the tumor suppressor protein FBW7 and then the polyubiquitylation of Mcl-1 targets it for proteasomal degradation.Citation12
Mcl-1, a pro-survival member of the Bcl-2 family, is frequently increased in various human tumors, suggesting that Mcl-1 is an attractive and potential therapeutic target for human malignancies.Citation15,Citation16 Mcl-1 is distinct from other Bcl-2 family members due to its extremely unstable nature.Citation17 The instability of Mcl-1 allows cells to switch to either survival or apoptotic modes in response to various stresses.Citation18 In addition, prior studies have shown that targeting Mcl-1 is a potentially important therapeutic modality.Citation19,Citation20
In this report, we have discovered that DAC can epigenetically activate FBW7 expression via its demethylation in human lung cancer cells. DAC-enhanced FBW7 directly promotes Mcl-1 ubiquitination and its subsequent degradation leads to suppression of lung cancer growth. Combined treatment with low-dose DAC and Bcl2 inhibitor venetoclax synergistically suppresses lung cancer growth in vitro and in vivo.
Materials and methods
Materials
Decitabine (DAC) was purchased from Carbosynth Limited (Berkshire, UK). Venetoclax (ABT-199) was obtained from MedKoo Biosciences (Morrisville, NC). Anti-FBW7 antibody was obtained from Abcam (Cambridge, MA) and Bethyl Laboratories (Montgomery, TX). Anti-Mcl-1, Bax, HA, and actin antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-Bcl2 antibody was purchased from Calbiochem (Billerica, MA). Anti-Bcl-xL antibody was purchased from Epitomics, Inc. (Burlingame, CA). Anti-Bim antibody was purchased from Cell Signaling Technology (Beverly, MA). Nanojuice transfection Kit was obtained from EMD Chemical, Inc. (Gibbstown, NJ). All other reagents used were obtained from commercial sources unless otherwise stated.
Cell lines and cell culture
H157, H460, and H1299 cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and were maintained with RPMI 1640 containing 10% fetal bovine serum in an incubator with 5% CO2 at 37°C. These cell lines were used for the described experiments without further authentication.
Protein analysis by western blot
Protein expression was detected by Western blotting as previously described.Citation21 Briefly, cells were harvested with a scraper and then washed two times with cold 1× PBS. The cells were then lysed in cold EBC buffer (0.5% N P-40, 50 mM Tris, pH 7.6, 120 mM NaCl, 1 mM EDTA, and 1 mM β-mercaptoethanol) containing protease inhibitor mixture. Following cell lysis by sonication and centrifugation at 14,000 × g for 15 min at 4°C, the resulting supernatant was collected as the total cell lysate. As previously described, Western blot was performed by loading 50 µg of protein per lane on a 10–12% SDS-PAGE, followed by protein transfer to nitrocellulose membrane for analysis of specific proteins.
Measurement of Mcl-1 half-life and ubiquitination
Cells were treated with 100 µg/ml cycloheximide in the absence or presence of decitabine (5µM) for various times up to 300 min, followed by Western blot for analysis of Mcl-1 as we previously described.Citation15 Mcl-1 on Western blot was quantified by Image J software (National Institutes of Health, Bethesda, MD) to calculate the half-life as described.Citation22,Citation23 Ubiquitination of Mcl-1 was carried out as described previously.Citation23 First, the HA-tagged ubiquitin expression plasmid was transfected into H1299 cells. After 24 h, cells were treated with MG132 (20 µM) and replaced with medium in the absence or presence of DAC (5 µM) for 6 h, followed by immunoprecipitation using an anti-Mcl-1 antibody. Mcl-1 ubiquitination was analyzed by Western blot using anti-HA antibody.
Colony formation assay
H157, H460, and H1299 cells were trypsinized and plated in a 6-well-plate (600 cells/well), then treated with DAC or venetoclax. The medium was replaced with fresh medium containing DAC or venetoclax every 2 days. After 10 days of treatment, the medium was removed and cell colonies were stained with crystal violet (0.1% in 20% methanol). Pictures were scanned using a scanner to record the results prior to colony counting as described previously.Citation24
Measurement of FBW7 methylation by methylation-specific PCR (MSP)
MSP for measurement of FBW7 methylation was performed as described.Citation25–27 Briefly, after treatment of cells or mice with DAC, genomic DNA was isolated from cells or tumor tissues using a Wizard DNA purification system (Promega, Madison, WI, USA). Bisulfite modification of the genomic DNA (1 µg) and its purification were performed using a CpGenomeTM DNA Modification kit (Chemicon, Billerica, MA, USA). Primers for methylated FBW7 (M): forward, 5ʹ-GTT CGT GTC GTT AAA TTA GGC-3ʹ and reverse, 5ʹ-CTA AAC GAA CAA AAA CCG CA-3ʹ. Primers for unmethylated FBW7 (U): forward, 5ʹ- GGT GTT TGT GTT GTT AAA TTA GG T- 3ʹ and reverse, 5ʹ-CCC TAA ACA AAC AAA AAC CAC A-3ʹ. PCR was performed using 10 µL bisulfate-modified DNA product, 1 µM of each primer, 1 mM each of dATP, dCTP, dGTP, and dTTP, 5 mM MgSO4, and 0.25 µM DNA polymerase with gold buffer. The amplification method included initial heating at 95°C for 5 min, followed by 45 cycles of 95°C for 30 s, 57°C for 30 s, and 72°C for 30 s, followed by extension at 72°C for 10 min. MSP products were visualized on 1% agarose gel.
Lung cancer xenografts and treatments
Six-week-old Nu/Nu nude mice were purchased from Harlan. Mice were maintained, and animal experiments were conducted, under the Institutional Animal Care and Use Committee of Emory University. Briefly, 3 × 106 of H460 or H1299 cells were injected subcutaneously into the flank. The tumors were allowed to grow to an average volume of 120 mm3 prior to initiation of therapy as described.Citation28 Mice carrying xenografts were treated with DAC intraperitoneally (i.p.) or venetoclax orally. Tumor volume (V) was measured by caliper measurements once every 2 days and calculated with the formula: V = (L × W2)/2 (L: length; W: width). Mice were euthanized by CO2 inhalation at the end of treatment. Harvested tumor tissues were used for further analysis.
Immunohistochemistry (IHC) analysis
Immunohistochemical staining was carried out using the R.T.U. Vectastain kit following procedures previously described.Citation21,Citation29 Briefly, formalin-fixed, paraffin-embedded tissue sections were deparaffinized with xylenes and hydrated by a graded series of ethanol washes. Antigen retrieval was accomplished by heating the slides in citrate buffer (pH 6.0) at 100°C for 20 min. Endogenous peroxidase activity was quenched by 0.3% H2O2 in methanol. Nonspecific binding was blocked with 2.5% normal house serum before incubation with the primary antibody. The slides were incubated with Mcl-1 (1:100), Fbw7 (1:50), Ki67 (1:200), and active caspase 3 (1:200), respectively, overnight at 4°C. The slides were stained with 3,3ʹ-diaminobenzidine (DAB) (Vector Laboratories) and counterstained with hematoxylin (Vector Laboratories), dehydrated, treated with xylene, and mounted. Active caspase 3-positive cells and Ki67 positive cells in tumor tissues were scored at 400× magnification. The average number of positive cells per 0.0625 mm2 area was determined from three separate fields in each of three independent tumor samples as described.Citation16,Citation30,Citation31 The semiquantitative evaluation of IHC staining of FBW7 or Mcl-1 was carried out using an immunoscore based on both the percentage of stained cells and staining intensity as described.Citation21
Mouse blood analysis
Whole blood (250 µL) was collected in EDTA-coated tubes via cardiac puncture of anesthetized mice for hematology studies. Specimens were analyzed for white blood cells (WBC), red blood cells (RBC), platelets (PLT), alanine aminotransferase (ALT), aspartate aminotransferase (AST), and blood urea nitrogen (BUN) in the Clinical Pathology Laboratory at the University of Georgia (Athens, GA).
Statistical analysis
All data are presented as mean ± standard deviation (s.d.) from at least three independent experiments. The statistical significance of differences between groups was analyzed by a two-tailed t test. We chose the sample size to detect a minimum effect size of 1.5 with at least 80% power and a type I error of 0.05 for each comparison. A value of P < .05 was considered to be statistically significant.
Results
Decitabine upregulates FBW7 and downregulates Mcl-1 leading to growth inhibition in human lung cancer cells
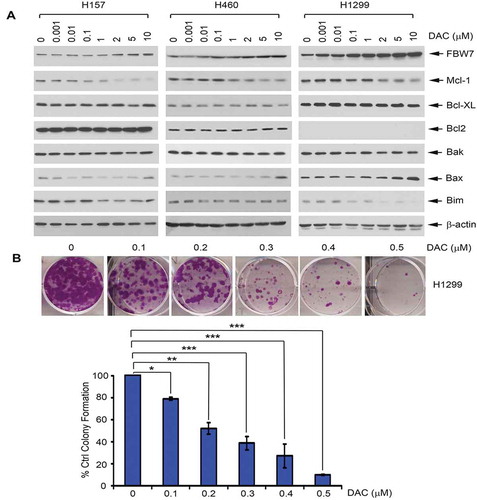
Recent reports reveal that the DNA methylation inhibitor decitabine (DAC) downregulates Mcl-1 protein level in various cancer cells.Citation4,Citation32–34 However, the mechanism(s) involved remain elusive. FBW7 has been identified as an Mcl-1 E3 ligase that promotes Mcl-1 ubiquitination and degradation.Citation13 To investigate whether DAC downregulation of Mcl-1 may occur through its E3 ligase FBW7, H157, H460, and H1299 cells were treated with increasing concentrations (0–10 µM) of DAC for 24 h, followed by Western blot analysis. Treatment with DAC induced a dose-dependent upregulation of FBW7 that is associated with decreased levels of Mcl-1 but had no significant effect on other Bcl-2 family members (i.e. Bcl2, Bcl-XL, etc.) ()), suggesting DAC downregulation of Mcl-1 may occur through activation of FBW7 expression. Functionally, treatment of H1299 cells with DAC resulted in a dose-dependent growth inhibition ()).
Figure 1. Treatment of lung cancer cells with DAC results in downregulation of Mcl-1 and upregulation of FBW7. (a) Human lung cancer H157, H460 and H1299 cells were treated with increasing concentrations of DAC for 24 h, followed by Western blot. (b) H1299 cells were treated with increasing concentrations of DAC for 10 days, followed by colony formation assay
DAC promotes Mcl-1 ubiquitination and degradation
The ubiquitin-proteasome machinery, a common pathway for protein degradation, has been implicated in regulating protein stability, cell viability, and apoptosis.Citation35 Our findings suggest that DAC downregulation of Mcl-1 may occur through activation of its E3 ligase FBW7 (). We next designed experiments to determine whether DAC promotes Mcl-1 ubiquitination and degradation through activation of FBW7. First, H1299 cells were treated with cycloheximide (100 µg/ml) in the absence or presence of DAC (5 µM) for various times up to 300 min, followed by an analysis of half-life of Mcl-1 protein. Treatment with DAC significantly reduced the half-life of Mcl-1 from 55 min to 17.5 min (*P < .05) ()). In addition to H1299 cells, similar results were observed with H157 and H460 cells (Supplementary Fig. S1), suggesting that the inhibitory effect of DAC on Mcl-1 protein stability is not a cell type-specific phenomenon limited to H1299 cells. Second, Mcl-1 ubiquitination was measured following transfection of HA-ubiquitin and treatment with DAC as described previously.Citation23 Intriguingly, treatment of H1299 cells with DAC-enhanced ubiquitination of Mcl-1 ()), suggesting that the reduction in Mcl-1 half-life by DAC may occur through the promotion of Mcl-1 ubiquitination and subsequent degradation.
Figure 2. DAC reduces the half-life of Mcl-1 via increased ubiquitination in human lung cancer cells. (a) H1299 cells were treated with cycloheximide (100 μg/ml) in the absence or presence of decitabine (5uM) for various times. Levels of Mcl-1 were analyzed by Western blot. Mcl-1 protein was quantified by ImageJ software, followed by calculating the half-life of Mcl-1. (b) H1299 cells were transfected with HA-tagged ubiquitin constructs using NanoJuice transfection kit. After 24 h, cells were treated with MG132 for 2 h, then incubated with medium in the absence or presence of decitabine (5uM) for another 24 h, followed by anti-Mcl-1 IP. Mcl-1 ubiquitination was analyzed by Western blot using anti-HA antibody. 50 µg of total lysate was used as input control
DAC activates FBW7 expression through induction of its demethylation
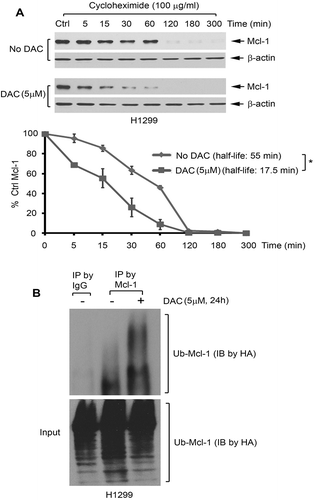
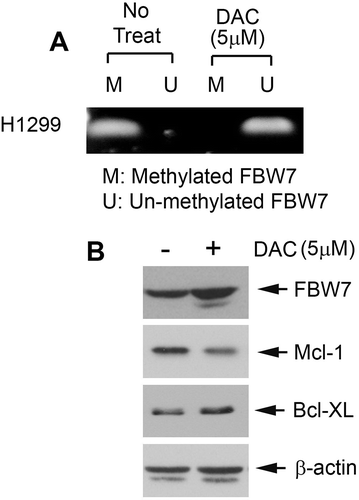
It has been previously reported that methylation could lead to loss of expression of Fbw7 in various cancers because FBW7 promoter hypermethylation causes its gene inactivation.Citation27 To further uncover the mechanism by which DAC upregulates FBW7 and downregulates Mcl-1 in human lung cancer cells, methylation status of the FBW7 promoter was analyzed using methylation-specific PCR following DAC treatment. Treatment of H1299 cells with DAC (5 μM) for 72 h resulted in the conversion of the FBW7 gene from the methylated form to the unmethylated form ()), indicating that DAC can induce demethylation of FBW7 at its promoter in human lung cancer cells. Importantly, DAC-induced demethylation of the FBW7 gene led to increased protein expression of FBW7 (i.e. Mcl-1 E3 ligase) and decreased protein expression of Mcl-1 ()).
Figure 3. DAC induces FBW7 demethylation and activation of FBW7 expression. (a) H1299 cells were treated with DAC (5 μM) for 72 h, followed by MSP for detection of methylated-FBW7 (m) and unmethylated FBW7 (u). (b) H1299 cells were treated with DAC (5 μM) for 72 h, followed by Western blot
DAC suppresses lung cancer growth via apoptosis in lung cancer xenograft models
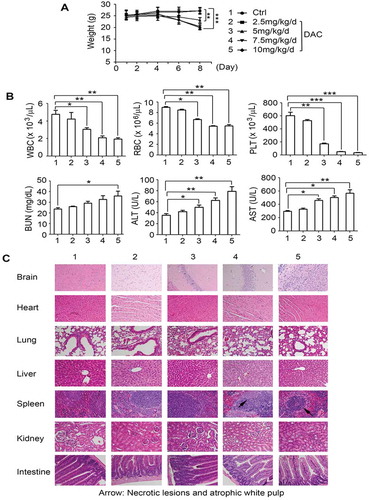
To test the potency of DAC in vivo, Nu/Nu nude mice carrying lung cancer xenografts derived from H460 cells were treated intraperitoneally (i.p.), with increasing doses (0, 2.5, 5, 7.5, 10 mg/kg/d) of DAC for 8 days (n = 6 per group). DAC treatment of lung cancer tumor-bearing mice resulted in a dose-dependent repression of lung cancer growth ()). To determine whether DAC-induced suppression of tumor growth occurs through regulation of FBW7 demethylation, Mcl-1 expression, and apoptosis in vivo, representative samples from harvested tumor tissues were analyzed by methylation-specific PCR. Treatment of mice with DAC resulted in a dose-dependent demethylation of FBW7 (i.e. gradual decrease in methylated form of FBW7 and gradual increase in unmethylated FBW7) ()). Intriguingly, DAC-induced demethylation of the FBW7 gene caused elevated levels of FBW7 protein and reduced levels of Mcl-1 protein, which led to decreased Ki67 and increased active caspase 3 (i.e. apoptosis) in tumor tissues ()). Similar to the results obtained in the mice carrying H460 xenografts, treatment of mice with H1299 xenografts with DAC (5 mg/k/d x 20 days) led to potent growth suppression (Supplementary Fig. S2A). Western blot analysis of tumor lysates confirmed that treatment of mice with DAC also upregulates FBW7 in association with decreased Mcl-1 expression in tumor tissues (Supplementary Fig. S2B). Significant weight loss, decreased white blood cell counts (WBC), decreased red blood cell count (RBCs), decreased platelet counts (PLTs), and increased BUN, ALT, and AST were observed in mice treated with more than 5 mg/kg/d of DAC ()). Histopathology of harvested normal tissues revealed that necrotic lesions were observed in spleen when mice were treated with a dose of more than 7.5 mg/kg/d ()). No other pathological evidence of normal tissue toxicities was observed in heart, liver, lung, brain, kidney, and intestine. These findings suggest that doses less than 5 mg/kg/d should provide the optimal therapeutic index for DAC in vivo experimentation in lung cancer xenograft models.
Figure 4. DAC suppresses lung cancer growth in dose-dependent manner in vivo. (a) Nu/Nu nude mice carrying H460 xenografts were treated with increasing doses of DAC (2.5 ~ 10 mg/kg/d) i.p. for 8 days. Tumor volume was measured once every 2 days. After treatment, mice were sacrificed, and tumors were removed and analyzed. Data represent the mean ± SD, n = 6 per group. *P < .05, ***P < .001, by 2-tailed t test. (b) Methylated FBW7 and unmethylated FBW7 were measured by MSP in tumor tissues at the end of experiments. (c) Mcl-1 and Bcl-XL were analyzed by Western blot in tumor tissues at the end of experiments. (d) FBW7, Mcl-1, Ki67 and active caspase 3 were measured by IHC in tumor tissues at the end of experiments by IHC staining. Data represent the mean ± SD, n = 6 per group. *P < .05, **P < .01, ***P < .001, by 2-tailed t test
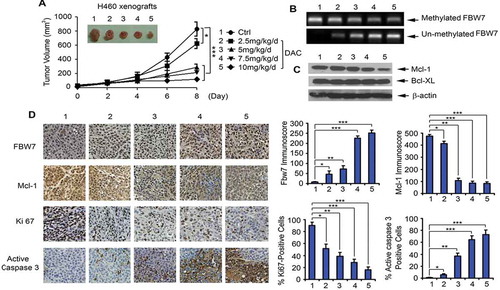
Figure 5. Toxicity of DAC in vivo. (a), (b) and (c) Body weight, blood analysis and H&E histology of various organs from Nu/Nu nude mice carrying H460 xenografts after treatment with increasing doses (0, 2.5, 5, 7.5, 10 mg/kg/d) of DAC for 8 days
DAC synergizes with the Bcl2 inhibitor venetoclax (ABT-199) against NSCLC in vitro and in vivo
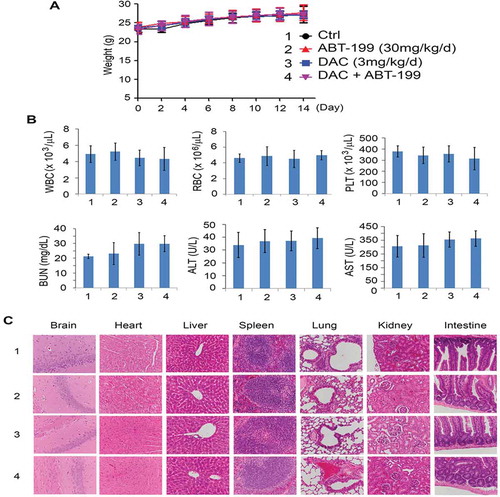
Synergistic drug combinations can be rationalized using knowledge of single-drug action mechanisms. The Bcl2 inhibitor, venetoclax (ABT-199), is FDA-approved for the treatment of CLL and AML.Citation36–42 Mcl-1 expression is a primary mode of resistance to venetoclax.Citation43–46 Since DAC induces downregulation of Mcl-1 via activation of its E3 ligase FBW7 ( and 4), combined treatment with DAC and a Bcl-2 inhibitor may achieve synergistic therapeutic effects in human lung cancer. To investigate this possible synergism, human lung cancer cell lines H460 and Calu-1, which express high levels of endogenous Bcl2 and Mcl-1, were treated with DAC (0.3 μM) and ABT-199 (5 μM) alone or in combination, followed by colony formation assay. The combination of DAC and ABT-199 led to significantly greater growth inhibition than DAC or ABT-199 alone ()), indicating a strong synergistic effect against lung cancer in vitro. To further test this in vivo, nude mice with H460 xenografts (n = 6/group) were treated with a low dose of DAC (3 mg/kg/d), ABT-199 (30 mg/kg/d), or a combination at the same doses for 2 weeks. Intriguingly, the combination of DAC and ABT-199 not only synergistically suppressed lung cancer growth in vivo ()) but was also well tolerated without any toxicity to mice ().
Figure 6. DAC synergizes with Bcl2 inhibitor ABT-199 (venetoclax) against lung cancer in vitro and in vivo. (a) and (b) Human lung cancer H460 and Calu-1 cells were treated with DAC (0.3 μM), ABT-199 (5 μM) or in combination, followed by colony formation assay. Data represent the mean ± SD, n = 3 per group. ***P < .001, by 2-tailed t test. (c) Nu/Nu nude mice carrying H460 xenografts were treated with DAC (3 mg/kg/d) i.p., ABT-199 (30 mg/kg/d) orally, or in combination for 14 days. Tumor volume was measured once every 2 days. After treatment, mice were sacrificed and tumors were removed and analyzed. Data represent the mean ± SD, n = 6 per group. *P < .05, by 2-tailed t test
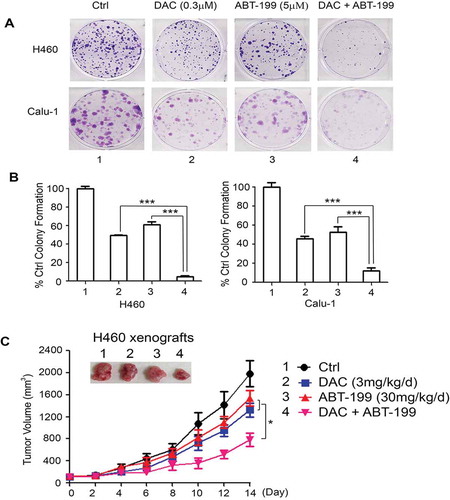
Figure 7. Toxicity of combined treatment with DAC and Bcl2 inhibitor in vivo. (a), (b) and (c), Body weight, blood analysis and H&E histology of various organs from Nu/Nu nude mice carrying H460 xenografts after treatment with DAC (3 mg/kg/d), ABT-199 (30 mg/kg/d), or in combination for 14 days
Discussion
Epigenetic therapy provides a useful treatment option for patients with lung cancer because many tumor suppressor genes are silenced by aberrant hypermethylation in human lung cancer cells.Citation2 Importantly, these silenced tumor suppressor genes can be reactivated by DNMT inhibitor DAC.Citation47 The incorporation of DAC in place of 5-methylcytosine in DNA results in the inactivation of DNMT1 due to covalent bond formation between the 5-azacytosine ring of DAC and this enzyme.Citation47 FBW7 is a critical tumor suppressor and one of the most commonly deregulated ubiquitin-proteasome system proteins in human cancer.Citation48 Importantly, FBW7 is a physiological Mcl-1 E3 ligase that promotes proteasome-mediated degradation of Mcl-1.Citation13 Since high levels of Mcl-1 were observed in NSCLC,Citation16,Citation49 epigenetically targeting the FBW7/Mcl-1 pathway should be an attractive strategy for the treatment of lung cancer. Here we discovered that epigenetic treatment of human lung cancer cells (i.e. H157, H460, and H1299) with DAC activates FBW7 expression leading to a dose-dependent downregulation of Mcl-1 protein and growth inhibition. This suggests that DAC inhibition of lung cancer growth may occur, at least in part, through epigenetic modulation of the FBW7-mediated Mcl-1 degradation pathway. Indeed, treatment of human lung cancers with DAC significantly reduces the half-life of Mcl-1 through ubiquitination. Mechanistically, DAC can convert the methylated form of FBW7 into unmethylated FBW7 in vitro and in vivo, uncovering a novel mechanism by which DAC suppresses lung cancer at least in part through activation of the FBW7/Mcl-1 pathway.
Although our findings demonstrated that DAC has considerable potency in suppressing lung cancer in a dose-dependent manner in a xenograft model, doses at or above 5 mg/kg/d (5, 7.5, 10 mg/kg/d) produced significant myelosuppression and liver toxicity. Doses less than 5 mg/kg/d (i.e. 2.5 mg/kg/d) did not have any toxicities but demonstrated much lower potency compared to that in mice treated with doses of 5 mg/kg/d or above. Similar toxicities, such as myelosuppression, can be seen in human patients treated with higher doses of DAC.Citation47 The use of the epigenetic agent DAC as monotherapy has limited potency for the treatment of patients with NSCLC.Citation2,Citation47,Citation50,Citation51 To enhance the efficacy of DAC and reduce its toxicity by using lower doses, the combination of DAC with another agent could provide a better strategy. It is well known that Mcl-1 upregulation renders several types of cancers resistant to Bcl2 inhibitor.Citation52 Here our findings have demonstrated that DAC can downregulate Mcl-1 via epigenetic modulation of the FBW7/Mcl-1 pathway in lung cancer, suggesting that combination of DAC with the FDA-approved Bcl2 inhibitor venetoclax should produce synergistic activity against lung cancer. Although monotherapy with either a low dose of DAC (3 mg/kg/d) or venetoclax (30 mg/kg/d) has a limited anti-lung cancer effect, combined treatment with DAC and venetoclax at similarly low doses is a potent combination with significant suppression of lung cancer growth in vivo. Importantly, there were no normal tissue toxicities in mice treated with either a single low dose or the combination therapy. These findings suggest that DAC in combination with a Bcl2 inhibitor is an effective approach for lung cancer therapy and overcomes the weaknesses associated with DAC monotherapy for lung cancer.
In conclusion, we have demonstrated that DAC, a DNMT inhibitor, suppresses lung cancer growth through a novel mechanism involving the activation of FBW7-mediated Mcl-1 ubiquitination and degradation pathway. DAC demethylates and thus upregulates FBW7. DAC-induced downregulation of Mcl-1 significantly enhances the sensitivity of lung cancer cells to Bcl-2 inhibitor venetoclax in vitro and in vivo. Combined treatment of DAC with venetoclax represents synergistic and more effective epigenetic therapy for lung cancer.
Author contributions
The project was conceived by X.D. Experiments were performed by M.J.K. and G.C. G.L.S. performed histopathological analysis. X.D. and M.J.K wrote the manuscript.
Supplemental Material
Download MS Word (375.7 KB)Acknowledgments
We thank Anthea Hammond for editing the manuscript.
Disclosure statement
The authors declare that they have no competing interests.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi:10.1016/j.cell.2007.01.029.
- Ansari J, Shackelford RE, El-Osta H. Epigenetics in non-small cell lung cancer: from basics to therapeutics. Transl Lung Cancer Res. 2016;5:155–171. doi:10.21037/tlcr.2016.02.02.
- Schiffmann I, Greve G, Jung M, Lubbert M. Epigenetic therapy approaches in non-small cell lung cancer: update and perspectives. Epigenetics. 2016;11:858–870. doi:10.1080/15592294.2016.1237345.
- Saito Y, Suzuki H, Tsugawa H, Nakagawa I, Matsuzaki J, Kanai Y, Hibi T. Chromatin remodeling at Alu repeats by epigenetic treatment activates silenced microRNA-512-5p with downregulation of Mcl-1 in human gastric cancer cells. Oncogene. 2009;28:2738–2744. doi:10.1038/onc.2009.140.
- Risch A, Plass C. Lung cancer epigenetics and genetics. Int J Cancer. 2008;123:1–7. doi:10.1002/ijc.23605.
- Zochbauer-Muller S, Gazdar AF, Minna JD. Molecular pathogenesis of lung cancer. Annu Rev Physiol. 2002;64:681–708. doi:10.1146/annurev.physiol.64.081501.155828.
- Anglim PP, Alonzo TA, Laird-Offringa IA. DNA methylation-based biomarkers for early detection of non-small cell lung cancer: an update. Mol Cancer. 2008;7:81. doi:10.1186/1476-4598-7-81.
- Tan Y, Sangfelt O, Spruck C. The Fbxw7/hCdc4 tumor suppressor in human cancer. Cancer Lett. 2008;271:1–12. doi:10.1016/j.canlet.2008.04.036.
- Akhoondi S, Sun D, von der Lehr N, Apostolidou S, Klotz K, Maljukova A, Cepeda D, Fiegl H, Dafou D, Marth C, et al. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res. 2007;67:9006–9012. doi:10.1158/0008-5472.CAN-07-1320.
- Kuiper RP, Vreede L, Venkatachalam R, Ricketts C, Kamping E, Verwiel E, Govaerts L, Debiec-Rychter M, Lerut E, van Erp F, et al. The tumor suppressor gene FBXW7 is disrupted by a constitutional t(3;4)(q21;q31) in a patient with renal cell cancer. Cancer Genet Cytogenet. 2009;195:105–111. doi:10.1016/j.cancergencyto.2009.07.001.
- Iwatsuki M, Mimori K, Ishii H, Yokobori T, Takatsuno Y, Sato T, Toh H, Onoyama I, Nakayama KI, Baba H, et al. Loss of FBXW7, a cell cycle regulating gene, in colorectal cancer: clinical significance. Int J Cancer. 2010;126:1828–1837. doi:10.1002/ijc.24879.
- Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, Helgason E, Ernst JA, Eby M, Liu J, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature. 2011;471:110–114. doi:10.1038/nature09779.
- Inuzuka H, Shaik S, Onoyama I, Gao D, Tseng A, Maser RS, Zhai B, Wan L, Gutierrez A, Lau AW, et al. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature. 2011;471:104–109. doi:10.1038/nature09732.
- Inuzuka H, Fukushima H, Shaik S, Liu P, Lau AW, Wei W. Mcl-1 ubiquitination and destruction. Oncotarget. 2011;2:239–244. doi:10.18632/oncotarget.242.
- Liao M, Zhao J, Wang T, Duan J, Zhang Y, Deng X. Role of bile salt in regulating Mcl-1 phosphorylation and chemoresistance in hepatocellular carcinoma cells. Mol Cancer. 2011;10:44. doi:10.1186/1476-4598-10-44.
- Chen G, Park D, Magis AT, Behera M, Ramalingam SS, Owonikoko TK, Sica GL, Ye K, Zhang C, Chen Z, et al. Mcl-1 interacts with Akt to promote lung cancer progression. Cancer Res. 2019;79:6126–6138. doi:10.1158/0008-5472.CAN-19-0950.
- Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell. 2006;21:749–760. doi:10.1016/j.molcel.2006.02.009.
- Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature. 2003;426:671–676. doi:10.1038/nature02067.
- Chen G, Magis AT, Xu K, Park D, Yu DS, Owonikoko TK, Sica GL, Satola SW, Ramalingam SS, Curran WJ, et al. Targeting Mcl-1 enhances DNA replication stress sensitivity to cancer therapy. J Clin Invest. 2018;128:500–516. doi:10.1172/JCI92742.
- Hird AW, Tron AE. Recent advances in the development of Mcl-1 inhibitors for cancer therapy. Pharmacol Ther. 2019;198:59–67. doi:10.1016/j.pharmthera.2019.02.007.
- Liu Y, Sun SY, Owonikoko TK, Sica GL, Curran WJ, Khuri FR, Deng X. Rapamycin induces bad phosphorylation in association with its resistance to human lung cancer cells. Mol Cancer Ther. 2012;11:45–56. doi:10.1158/1535-7163.MCT-11-0578.
- Arnold HK, Sears RC. Protein phosphatase 2A regulatory subunit B56alpha associates with c-myc and negatively regulates c-myc accumulation. Mol Cell Biol. 2006;26:2832–2844. doi:10.1128/MCB.26.7.2832-2844.2006.
- Wang B, Xie M, Li R, Owonikoko TK, Ramalingam SS, Khuri FR, Curran WJ, Wang Y, Deng X. Role of Ku70 in deubiquitination of Mcl-1 and suppression of apoptosis. Cell Death Differ. 2014;21:1160–1169. doi:10.1038/cdd.2014.42.
- Wang X, Yue P, Kim YA, Fu H, Khuri FR, Sun SY. Enhancing mammalian target of rapamycin (mTOR)-targeted cancer therapy by preventing mTOR/raptor inhibition-initiated, mTOR/rictor-independent Akt activation. Cancer Res. 2008;68:7409–7418. doi:10.1158/0008-5472.CAN-08-1522.
- Zhao X, Wang N, Zhang M, Xue S, Shi K, Chen Z. Detection of methylation of the RAR-beta gene in patients with non-small cell lung cancer. Oncol Lett. 2012;3:654–658. doi:10.3892/ol.2011.527.
- Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996;93:9821–9826. doi:10.1073/pnas.93.18.9821.
- Akhoondi S, Lindstrom L, Widschwendter M, Corcoran M, Bergh J, Spruck C, Grandér D, Sangfelt O. Inactivation of FBXW7/hCDC4-beta expression by promoter hypermethylation is associated with favorable prognosis in primary breast cancer. Breast Cancer Res. 2010;12:R105. doi:10.1186/bcr2788.
- Cantor JP, Iliopoulos D, Rao AS, Druck T, Semba S, Han SY, McCorkell KA, Lakshman TV, Collins JE, Wachsberger P, et al. Epigenetic modulation of endogenous tumor suppressor expression in lung cancer xenografts suppresses tumorigenicity. Int J Cancer. 2007;120:24–31. doi:10.1002/ijc.22073.
- Gupta S, Pramanik D, Mukherjee R, Campbell NR, Elumalai S, de Wilde RF, Hong S-M, Goggins MG, De Jesus-Acosta A, Laheru D, et al. Molecular determinants of retinoic acid sensitivity in pancreatic cancer. Clini Cancer Res. 2012;18(1):280–289. doi:10.1158/1078-0432.CCR-11-2165.
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi:10.1038/nature03579.
- Han B, Park D, Li R, Xie M, Owonikoko TK, Zhang G, Sica G, Ding C, Zhou J, Magis A, et al. Small-molecule Bcl2 BH4 antagonist for lung cancer therapy. Cancer Cell. 2015;27:852–863. doi:10.1016/j.ccell.2015.04.010.
- Nishioka C, Ikezoe T, Yang J, Udaka K, Yokoyama A. Simultaneous inhibition of DNA methyltransferase and histone deacetylase induces p53-independent apoptosis via down-regulation of Mcl-1 in acute myelogenous leukemia cells. Leuk Res. 2011;35:932–939. doi:10.1016/j.leukres.2011.04.004.
- Xu J, Liao X, Lu N, Liu W, Wong CW. Chromatin-modifying drugs induce miRNA-153 expression to suppress Irs-2 in glioblastoma cell lines. Int J Cancer. 2011;129:2527–2531. doi:10.1002/ijc.25917.
- Brodská B, Otevřelová P, Holoubek A. Decitabine-induced apoptosis is derived by puma and Noxa induction in chronic myeloid leukemia cell line as well as in PBL and is potentiated by SAJA. Mol Cell Biochem. 2011;350:71–80. doi:10.1007/s11010-010-0683-3.
- Clague MJ, Urbe S. Ubiquitin: same molecule, different degradation pathways. Cell. 2010;143:682–685. doi:10.1016/j.cell.2010.11.012.
- Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19:202–208. doi:10.1038/nm.3048.
- Wei AH, Strickland SA Jr., Hou JZ, Fiedler W, Lin TL, Walter RB, Enjeti A, Tiong IS, Savona M, Lee S, et al. Venetoclax combined with low-dose cytarabine for previously untreated patients with acute myeloid leukemia: results from a phase Ib/II study. J Clin Oncol. 2019;37:1277–1284. doi:10.1200/JCO.18.01600.
- DiNardo CD, Pratz K, Pullarkat V, Jonas BA, Arellano M, Becker PS, Frankfurt O, Konopleva M, Wei AH, Kantarjian HM, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2019;133:7–17. doi:10.1182/blood-2018-08-868752.
- Korycka-Wolowiec A, Wolowiec D, Kubiak-Mlonka A, Robak T. Venetoclax in the treatment of chronic lymphocytic leukemia. Expert Opin Drug Metab Toxicol. 2019;15:353–366. doi:10.1080/17425255.2019.1606211.
- Coutre S, Choi M, Furman RR, Eradat H, Heffner L, Jones JA, Chyla B, Zhou L, Agarwal S, Waskiewicz T, et al. Venetoclax for patients with chronic lymphocytic leukemia who progressed during or after idelalisib therapy. Blood. 2018;131(15):1704–1711. doi:10.1182/blood-2017-06-788133.
- Stilgenbauer S, Eichhorst B, Schetelig J, Hillmen P, Seymour JF, Coutre S, Jurczak W, Mulligan SP, Schuh A, Assouline S, et al. Venetoclax for patients with chronic lymphocytic leukemia with 17p deletion: results from the full population of a phase II pivotal trial. J Clin Oncol. 2018;36(19):1973–1980. doi:10.1200/JCO.2017.76.6840.
- Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, Kipps TJ, Anderson MA, Brown JR, Gressick L, et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374:311–322. doi:10.1056/NEJMoa1513257.
- Choudhary GS, Al-Harbi S, Mazumder S, Hill BT, Smith MR, Bodo J, Hsi ED, Almasan A. MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell Death Dis. 2015;6:e1593. doi:10.1038/cddis.2014.525.
- Ramsey HE, Fischer MA, Lee T, Gorska AE, Arrate MP, Fuller L, Boyd KL, Strickland SA, Sensintaffar J, Hogdal LJ, et al. A novel MCL1 inhibitor combined with venetoclax rescues venetoclax-resistant acute myelogenous leukemia. Cancer Discov. 2018;8(12):1566–1581. doi:10.1158/2159-8290.CD-18-0140.
- Teh TC, Nguyen NY, Moujalled DM, Segal D, Pomilio G, Rijal S, Jabbour A, Cummins K, Lackovic K, Blombery P, et al. Enhancing venetoclax activity in acute myeloid leukemia by co-targeting MCL1. Leukemia. 2018;32:303–312.
- Chen X, Glytsou C, Zhou H, Narang S, Reyna DE, Lopez A, Sakellaropoulos T, Gong Y, Kloetgen A, Yap YS, et al. Targeting mitochondrial structure sensitizes acute myeloid leukemia to venetoclax treatment. Cancer Discov. 2019;9:890–909. doi:10.1158/2159-8290.CD-19-0117.
- Momparler RL. Epigenetic therapy of non-small cell lung cancer using decitabine (5-aza-2ʹ-deoxycytidine). Front Oncol. 2013;3:188. doi:10.3389/fonc.2013.00188.
- Yeh CH, Bellon M, Nicot C. FBXW7: a critical tumor suppressor of human cancers. Mol Cancer. 2018;17:115. doi:10.1186/s12943-018-0857-2.
- Zhang H, Guttikonda S, Roberts L, Uziel T, Semizarov D, Elmore SW, Leverson JD, Lam LT. Mcl-1 is critical for survival in a subgroup of non-small-cell lung cancer cell lines. Oncogene. 2011;30(16):1963–1968. doi:10.1038/onc.2010.559.
- Momparler RL, Ayoub J. Potential of 5-aza-2ʹ-deoxycytidine (Decitabine) a potent inhibitor of DNA methylation for therapy of advanced non-small cell lung cancer. Lung Cancer. 2001;34(Suppl 4):S111–5. doi:10.1016/S0169-5002(01)00397-X.
- Schrump DS, Fischette MR, Nguyen DM, Zhao M, Li X, Kunst TF, Hancox A, Hong JA, Chen GA, Pishchik V, et al. Phase I study of decitabine-mediated gene expression in patients with cancers involving the lungs, esophagus, or pleura. Clin Cancer Res. 2006;12:5777–5785. doi:10.1158/1078-0432.CCR-06-0669.
- Pan R, Ruvolo VR, Wei J, Konopleva M, Reed JC, Pellecchia M, Andreeff M, Ruvolo PP. Inhibition of Mcl-1 with the pan-Bcl-2 family inhibitor (-)BI97D6 overcomes ABT-737 resistance in acute myeloid leukemia. Blood. 2015;126:363–372.