
ABSTRACT
BRAF mutations are present in over half of all melanoma tumors. Although BRAF inhibitors significantly improve survival of patients with metastatic melanoma, recurrences occur within several months. We previously reported that BRAF mutant melanoma cells are more sensitive to a novel arylmethyl-polyamine (AP) compound that exploits their increased polyamine uptake compared to that of BRAF wildtype cells. Using an animal model of BRAF inhibitor-resistant melanoma, we show that co-treatment with the BRAF inhibitor, PLX4720, and AP significantly delays the recurrence of PLX4720-resistant melanoma tumors and decreases tumor-promoting macrophages. Development of BRAF inhibitor-resistance enriches for metastatic cancer stem cells (CSC) and increases tumor-promoting macrophages. In vitro studies demonstrated that CD304+, CXCR4+ spheroid cultures of BRAF mutant melanoma cells are resistant to PLX4720 but are more sensitive to AP compared to monolayer cultures of the same cells. AP significantly inhibited YUMM1.7 melanoma cell invasiveness across a Matrigel-coated filter using the CXCR4 ligand, SDF-1α, as the chemoattractant. AP also blocked the chemotactic effect of SDF-1α on CXCR4+ macrophages and inhibited M2 polarization of macrophages. In melanoma-macrophage co-cultures, AP prevented the PLX4720-induced release of pro-tumorigenic growth factors, such as VEGF, from macrophages and prevented the macrophage rescue of BRAF mutant melanoma cells treated with PLX4720. Our study offers a novel therapy (AP) to treat chemo-resistant melanoma. AP is unique because it targets the polyamine transport system in BRAF inhibitor-resistant CSCs and also blocks CXCR4 signaling in invasive melanoma cells and pro-tumorigenic macrophages.
Introduction
Despite being a rare type of skin cancer, metastatic melanoma is one of the most highly mutated and lethal types of cancer.Citation1,Citation2 Multiple oncogenic mutations (including BRAF, NRAS, KIT) drive this highly heterogeneous disease, with BRAF mutations detected in half of all melanoma tumors.Citation3 Although targeting the V600E-mutant BRAF kinase with BRAF inhibitors significantly improves survival of patients with metastatic melanoma, recurrences often occur within several months.Citation1,Citation2 Recent studies have shown that BRAF resistance includes multiple molecular changes in the tumor involving either reactivation of the MAPK kinase pathway via B-raf/c-Raf heterodimerization or concurrent activation of alternative growth factor signaling pathways such as PI3K/AKTCitation4 or (HGF)/MET signaling.Citation5,Citation6 Whereas preexisting or acquired genetic mutations are well documented to cause drug resistance, non-genetic mechanisms involving chromatin remodeling or activation of stress-related pathways may also play a role in establishing a population of drug-tolerant tumor cells that regain stem-like properties during their dormant state.Citation7,Citation8 For instance, development of BRAF inhibitor-resistance enriches for highly tumorigenic and metastatic slow-cycling tumor cells which have many characteristics of cancer stem cells (CSC).Citation9–11
Oncogenes such as MYC, RAS, and BRAFV600E upregulate polyamine biosynthesis and increase cellular uptake of polyamines by inducing the polyamine transport system (PTS).Citation12–14 Thus, polyamine levels are dramatically elevated in tumor cells compared to normal cells. Polyamines (putrescine, spermidine, and spermine) are amino acid-derived polycations that play key roles in cellular proliferation, gene expression, immunomodulation, and autophagic states that all contribute to tumor survival.Citation15 Previously, we reported that PTS activity is significantly increased in mutant BRAFV600E melanoma cells compared to BRAFWT melanoma cells, resulting in greater sensitivity to a novel PTS-targeted, cytotoxic, arylmethyl-polyamine (AP) compound.Citation14,Citation16 By utilizing the PTS for cell entry, the AP compound can attack mutant BRAFV600E melanoma cells via one of their key modes of survival. Because polyamines are essential for the survival of melanoma tumor cells, we explored the therapeutic use of AP to overcome drug resistance in models of mutant BRAFV600E melanoma. Interestingly, we found that AP inhibits melanoma progression by targeting not only polyamine uptake via the PTS, but also CXCR4 signaling in both BRAFV600E resistant melanoma CSCs and pro-tumorigenic macrophages.
Results
AP therapy reduces tumor growth in BRAF inhibitor-resistant melanomas
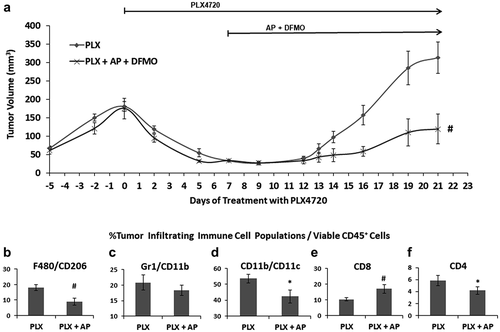
Because polyamine biosynthesis and uptake via the PTS is dramatically elevated in tumors compared to normal tissue, we exploited the oncogene-induced PTS activity in BRAFV600E metastatic melanoma cells by targeting the PTS with a novel arylmethyl-polyamine (AP) compound which is cytotoxic upon cell entry ().Citation16 The anti-tumor effects of AP were evaluated using the V600E-mutant BRAF melanoma model YUMM1.7, in which YUMM1.7 cells were orthotopically injected in syngeneic C57Bl/6 mice. When tumors were approximately 40 mm3, treatment was initiated with AP (1 mg/kg, i.p.) and the BRAF inhibitor PLX4720. Treatment of YUMM1.7 tumors with either AP or PLX4720 was very effective at inhibiting tumor growth when administered when the tumors were small, resulting in almost complete inhibition of tumor progression compared to untreated controls (Supplemental ). However, if treatment was initiated in mice with larger, more established YUMM1.7 tumors (approximately 150 mm3), AP significantly slowed YUMM1.7 tumor growth compared to vehicle control-treated mice (), but was not able to completely inhibit or regress growth of larger tumors as seen with PLX4720 ().
Figure 1. Inhibition of YUMM1.7 tumor growth by AP. A. Chemical structures of AP, DFMO and the Trimer PTI. B. C57Bl/6 mice were subcutaneously injected with 4 × 105 YUMM1.7 cells in the right flank. After two weeks, when the tumors were approximately 150 mm3 in size, the mice were separated into groups and treatment was initiated with AP (1 mg/kg, i.p, daily) or the BRAF inhibitor PLX4720 (417 mg/kg mouse chow). Graph shows YUMM1.7 tumor growth under different treatments (mean tumor volume ± SEM). C. C57Bl/6 mice were subcutaneously injected with 2 × 105 YUMM1.7 cells in the right flank. After two weeks, when the tumors were approximately 40 mm3, mice were separated into groups and treatment was initiated with the polyamine transport inhibitor Trimer44NMe PTI (3 mg/kg, i.p., daily) plus 0.25% DFMO (w/v) in the drinking water ± PLX4720 (25 mg/kg by oral gavage twice daily during the week and once daily on the weekends). Graph shows YUMM1.7 tumor growth under different treatments (mean tumor volume ± SEM). D. C57Bl/6 mice were subcutaneously injected with 4 × 105 YUMM1.7 cells in the right flank. After two weeks when the tumors were between 150–250 mm3, the mice were separated into treatment groups and treatment with PLX4720 (417 mg/kg in mouse chow) was initiated. One day later, co-treatment with AP (1 mg/kg, i.p., daily) and DFMO (0.5% w/v in the drinking water) was started in PLX4720 + (AP + DFMO) group concurrent with continued PLX4720 treatment. Co-treatment with AP and DFMO was initiated in a third group of PLX4720 treated mice 9 days after PLX4720 was started treatment (termed the PLX4720 + [AP + DFMO: delayed] treatment group). n = 4–6 mice per group; * = p ≤ 0.05 compared to vehicle treated mice
![Figure 1. Inhibition of YUMM1.7 tumor growth by AP. A. Chemical structures of AP, DFMO and the Trimer PTI. B. C57Bl/6 mice were subcutaneously injected with 4 × 105 YUMM1.7 cells in the right flank. After two weeks, when the tumors were approximately 150 mm3 in size, the mice were separated into groups and treatment was initiated with AP (1 mg/kg, i.p, daily) or the BRAF inhibitor PLX4720 (417 mg/kg mouse chow). Graph shows YUMM1.7 tumor growth under different treatments (mean tumor volume ± SEM). C. C57Bl/6 mice were subcutaneously injected with 2 × 105 YUMM1.7 cells in the right flank. After two weeks, when the tumors were approximately 40 mm3, mice were separated into groups and treatment was initiated with the polyamine transport inhibitor Trimer44NMe PTI (3 mg/kg, i.p., daily) plus 0.25% DFMO (w/v) in the drinking water ± PLX4720 (25 mg/kg by oral gavage twice daily during the week and once daily on the weekends). Graph shows YUMM1.7 tumor growth under different treatments (mean tumor volume ± SEM). D. C57Bl/6 mice were subcutaneously injected with 4 × 105 YUMM1.7 cells in the right flank. After two weeks when the tumors were between 150–250 mm3, the mice were separated into treatment groups and treatment with PLX4720 (417 mg/kg in mouse chow) was initiated. One day later, co-treatment with AP (1 mg/kg, i.p., daily) and DFMO (0.5% w/v in the drinking water) was started in PLX4720 + (AP + DFMO) group concurrent with continued PLX4720 treatment. Co-treatment with AP and DFMO was initiated in a third group of PLX4720 treated mice 9 days after PLX4720 was started treatment (termed the PLX4720 + [AP + DFMO: delayed] treatment group). n = 4–6 mice per group; * = p ≤ 0.05 compared to vehicle treated mice](/cms/asset/436d34b6-c12c-4025-997b-a5d238755fae/kcbt_a_1883185_f0001_b.gif)
We then compared the effect of AP with another polyamine targeting therapy that includes the Trimer44NMe PTI (), Citation17 another polyamine-containing compound that competitively inhibits cellular uptake of endogenous polyamines. AP is designed to be a cytotoxic ligand of the PTS.Citation14,Citation16 In contrast, co-treatment with both the Trimer44NMe PTI (to inhibit polyamine uptake via the PTS) as well as α-difluoromethylornithine (DFMO, to inhibit polyamine biosynthesis) is designed to lower intracellular polyamine levels.Citation17,Citation18 Previous animal tumor experiments have shown that the anti-tumor efficacy of this polyamine blocking therapy (PBT) with the Trimer44NMe PTI and DFMO is significantly better than treatment with the Trimer44NMe PTI alone or DFMO alone.Citation17 YUMM1.7 tumor-bearing mice were administered PLX4720 with and without PBT. Unlike the effective anti-tumor efficacy demonstrated with AP alone (Supplemental and ), treatment with PBT (Trimer44NMe PTI + DFMO) did not inhibit YUMM1.7 tumor growth (). Moreover, PBT did not significantly slow the growth of PLX4720-resistant tumors () suggesting that AP is inhibiting YUMM1.7 tumor growth via a different mechanism than simply blocking polyamine transport in melanoma tumors.
Treatment of BRAF mutant melanomas with a BRAF inhibitor results in dramatic reductions in tumor burden; however, over time tumors develop resistance to BRAF inhibition resulting in tumor relapse. It is thought that the development of BRAF inhibitor-resistant tumors enriches a subpopulation of slow-cycling stem-like tumor cells that can survive anti-proliferative chemotherapies, such as a BRAF inhibitor, and eventually leads to tumor regrowth.Citation9–11 AP and DFMO co-treatment with PLX4720, when started 7 days after PLX4720 initiation (point at which tumors had regressed the most), delayed the recurrence of YUMM1.7 tumors more than AP + DFMO treatment when initiated at the same time as PLX4720 treatment (), although not statistically significant.
It has been reported that the initial period of remission produced by a BRAF inhibitor (PLX4720) enhances tumor T-cell infiltrates, decreases immunosuppressive cytokines, increases melanoma antigen expression, and enhances reactivity to antigen-specific T-cells.Citation19 Thus, the early, but transient, immune response in the initial period of tumor remission following PLX4720 treatment may provide an opportunity to activate an immune attack that can prevent the recurrence of BRAF inhibitor-resistant melanoma. Because we previously showed that lowering tumor polyamine levels with a combination treatment of DFMO and the Trimer44NMe PTI enhances the anti-tumor immune responseCitation17, we predicted that a similar polyamine blocking therapy with AP + DFMO may also activate an immune response to suppress recurrence of aggressive malignant melanoma following treatment with a BRAF inhibitor. We investigated to what extent co-treatment with AP + DFMO will not only increase the anti-tumor effects of PLX4720, but also diminish infiltrating tumor-associated myeloid cells.
YUMM1.7 tumors were allowed to grow to approximately 200 mm3 in size, at which point PLX4720 treatment was initiated. One week of PLX4720 treatment resulted in a 10-fold reduction in tumor volume (), at which point half of the mice began co-treatment with AP and DFMO. As expected, mice treated with only PLX4720 had a pronounced relapse of the YUMM1.7 tumors. However, mice treated with PLX4720 + AP + DFMO demonstrated a significantly slower rate of tumor regrowth, indicating that treatment with AP + DFMO delayed the development of BRAF inhibitor-resistant tumor cells. Analysis of tumor immune cell infiltrates showed that treatment with AP and DFMO decreased levels of pro-tumorigenic F480+/CD206+ M2-like macrophages, Gr1+/CD11b+ MDSCs, and CD11b+/CD11c+ immunosuppressive immature dendritic cells while also increasing levels of cytotoxic INF-γ+CD8+ T-cells, including those with markers of tissue-resident memory CD8 T-cells (, Supplemental ). The addition of DFMO in the drinking water did not appear to enhance the anti-tumor efficacy of AP (Supplemental ). However, the addition of DFMO made AP more tolerable to the mice and eliminated adverse side effects, such as weight loss, in the AP groups. Previous studies have shown that DFMO increases PTS activity in BRAF mutant melanoma cellsCitation14, targeting more of the AP to the tumor instead of healthy tissue as we have previously observed with the Trimer PTI.Citation17
Figure 2. AP slows the progression of BRAF inhibitor resistant YUMM1.7 tumors. A. C57Bl/6 mice were subcutaneously injected with 4 × 105 YUMM1.7 cells in the right flank. When tumors were approximately 200 mm3 in size, treatment with PLX4720 (417 mg/kg PLX4720 mouse chow) was initiated in all mice. After one week of PLX4720 therapy, mice with similar-sized tumors were organized into two treatment groups and treatment with AP (0.5 mg/kg, i.p. daily) plus DFMO (0.5% w/v in the drinking water) was initiated in half the mice. Treatment with PLX4720 was maintained in both treatment groups until the mice were sacrificed. Graph shows YUMM1.7 tumor growth under different treatments (mean tumor volume ± SEM). Upon sacrifice, tumors were excised from YUMM1.7-tumor bearing mice and equal numbers of cells were analyzed by flow cytometry. Infiltrating CD45+ leukocytes were analyzed for (b) M2 macrophages (F480+/CD206+) ± SEM, (c) MDSCs (Gr1+/CD11b+) ± SEM, (d) immature dendritic cells (CD11b+/CD11c+) ± SEM, (e) CD8+ T-cells ± SEM and (f) CD4+ T-cells ± SEM. n = 7–10 mice per group; * = p ≤ 0.05 and # = p ≤ 0.01 compared to vehicle treated mice
AP targets stem-like cells and inhibits tumor invasiveness
To investigate whether AP is preferentially targeting CSC-like cells, we compared YUMM1.7 cells grown in a monolayer in adherent tissue culture plates with YUMM1.7 cells grown as spheroid cultures using the low adherent NanoCulture plate (NCP)-based 3D cell culture system.Citation20 Studies have shown that culture in the 3D nanoenvironment of NCP plates with a defined stem cell medium more closely models in vivo melanoma tumors that contain hypoxic regions and enables cells to form hypoxic, slower growing stem cell aggregates that are more resistant to chemotherapy including BRAF inhibitors.Citation20,Citation21 YUMM1.7 monolayer and spheroid cultures were treated with 20 µM AP and 5 µM PLX4720, individually, for 48 h and 72 h, respectively, and then assayed for cell viability. YUMM1.7 cells grown in a monolayer were resistant to 20 µM AP with no significant cell death detected after 48 h (). However, YUMM1.7 spheroids were significantly more susceptible to AP with only 40% surviving cells after 48 h. The inverse was true following 72 h treatment with the BRAF inhibitor PLX4720 where monolayer tumor cells fared worse than the YUMM1.7 spheroid cultures.
Figure 3. AP preferentially targets stem-cell like cells. A. YUMM1.7 cells were cultured as a monolayer in adherent tissue culture plates or as spheroids in low binding NCP plates to enrich for YUMM1.7 cells with stem-cell like properties. YUMM1.7 cells in a monolayer (at less than 50% confluency) and spheroids were treated with 20 µM AP and 5 µM PLX4720 individually for 48 h and 72 h, respectively, and then assayed for cell viability using the CellTiter-Glo® Luminescent Cell Viability Assay (Fisher). Data were normalized to control samples (mean ± SD) for comparison. B. WM3734 cells were cultured as a monolayer in adherent tissue culture plates or as spheroids in ultra-low attachment plates. WM3734 cells in a monolayer (at less than 50% confluency) and spheroids were treated with 20 µM AP and 5 µM PLX4720 individually for 48 h and 72 h, respectively, and then assayed for cell viability using the CellTiter-Glo® Luminescent Cell Viability Assay (Fisher). Data were normalized to control samples (mean ± SD) for comparison. C. YUMM1.7 cells grown in a monolayer or as spheroids were treated with 5 µM PLX4720 for 72 h. After 24 h of PLX4720 treatment, 20 µM AP was added to the appropriate samples for the remaining 48 h and then all samples were assayed for CD304 (mean ± SD) expression by flow cytometry. n = 3–5 per group; * = p ≤ 0.05 and # = p ≤ 0.01 compared to indicated group
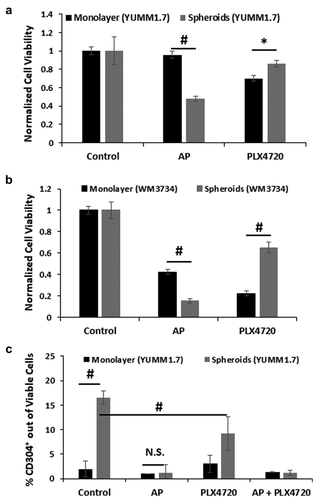
Our data indicate that AP may specifically target slower cycling stem-like cells more than proliferative tumor cells, while the inverse is true for PLX4720. These results were similar to what was observed previously with 1205Lu human melanoma cells grown as monolayer and NCP spheroid cultures.Citation14 To further verify these findings, WM3734 and FS4 human melanoma spheroids were grown in Costar ultra-low adherence tissue culture plates and treated with 20 µM AP and 5 µM PLX4720 and then compared to monolayer cultures. Similar to YUMM1.7 cells, WM3734 cells grown in monolayer cultures were less susceptible to killing by AP compared to WM3734 spheroids, while the inverse was true for PLX4720 with WM3734 monolayer cultures more sensitive to PLX4720 than WM3734 spheroids (). Similarly, spheroid cultures of human FS4 melanoma cells were also more susceptible to AP killing than FS4 cells grown as monolayer cultures (Supplemental ). Both the mutant BRAFV600E FS4 melanoma spheroid and monolayer cultured cells were resistant to PLX4720 cytotoxicity. However, FS4 cells have been shown to be particularly resistant to BRAF inhibitors due to high WNT5A expression.Citation22
YUMM1.7 cells were also assayed for CD304 (also known as neuropilin-1) expression, a transmembrane protein that serves as a co-receptor for VEGF and other growth factors and has been implicated in the tumor immune response, the epithelial to mesenchymal transition in cancer, and has been identified as a putative CSC marker.Citation23–25 Expression of CD304 was enriched 5-fold in cultures of YUMM1.7 spheroids compared to monolayer cells (). Treatment with AP lowered levels of CD304+ cells in the spheroids to a level similar to the monolayer cells, almost completely eliminating the CD304+ subpopulation in the YUMM1.7 spheroids. Incubation with PLX4720 reduced the percentage of CD304+ cells in the YUMM1.7 spheroids, but not to the same degree as AP, with no effect on the CD304+ monolayer cells. Human melanoma cell lines were also assayed for expression of another putative CSC marker, Jarid1B protein, a histone lysine demethylase identified as an oncogene in multiple cancer types including melanoma.Citation26,Citation27 We observed that Jarid1B protein expression was dramatically upregulated in spheroid cultures compared to monolayer cultures for WM88, WM983B, WM989 and FS4 human melanoma cell lines (Supplemental ).
Both monolayer and spheroid cultures of YUMM1.7 cells were then assayed for the expression of CXCR4, a transmembrane receptor that is often aberrantly expressed on the surface of cancer cells, particularly CSCCitation28, and has been linked to metastasis to tissues with high levels of SDF-1α, the ligand for CXCR4.Citation29 YUMM1.7 spheroids had dramatically elevated levels of CXCR4 protein expression compared to YUMM1.7 cells in monolayer (). Treatment with a very low concentration of AP (0.05 µM) significantly reduced CXCR4-expressing spheroids. Similar analyses of human melanoma cell lines revealed that CXCR4 was also elevated in spheroid cultures of WM989, WM983B and FS5 melanoma cells (Supplemental ).
Figure 4. AP reduces YUMM1.7 invasiveness. A. YUMM1.7 cells grown in a monolayer or as spheroids were treated with 20 µM AP or 5 µM PLX4720 for 48 h and then assayed for CXCR4 (mean ± SD) protein expression by flow cytometry. B. Boyden chamber inserts with 8 µm porous filters were coated with growth factor-reduced Matrigel and then seeded with 1 × 105 YUMM1.7 melanoma cells in serum-free media containing either 0.05 µM AP or 2.5 µM AMD3100 in the well of each insert. A gradient was then initiated with the addition of serum-free media supplemented with the chemoattractant (200 ng/ml SDF-1α) in the lower compartment of the invasion chamber. After a 24-hour incubation, cells that had invaded to the underside of the filter were fixed, stained, and counted using a microscope. Data expressed as mean number of cells ± SD C. Boyden chamber inserts with 8 µm porous filters were coated with growth factor-reduced Matrigel and then seeded with 1.5 × 105 WM989 melanoma cells in serum-free media in the well of each insert with some cells also cultured with 0.2 µM AP. A gradient was then initiated with the addition of serum-free media supplemented with the chemoattractant (250 ng/mL SDF-1α) in the lower compartment of the invasion chamber. After a 24-hour incubation, cells that had invaded to the underside of the filter were fixed, stained, and counted using a microscope. Data expressed as mean number of cells ± SD. D. Boyden chamber inserts with 8 µm porous filters were coated with growth factor-reduced Matrigel and then seeded with 1.0 × 105 FS5 melanoma cells in serum-free media containing either 0.25 µM AP or 2.5 µM AMD3100 in the well of each insert. A gradient was then initiated with the addition of serum-free media supplemented with the chemoattractant (250 ng/ml SDF-1α) in the lower compartment of the invasion chamber. After a 24-hour incubation, cells that had invaded to the underside of the filter were fixed, stained, and counted using a microscope. Data expressed as mean number of cells ± SD. n = 3 per group; * = p ≤ 0.05 and # = p ≤ 0.01 compared to indicated group
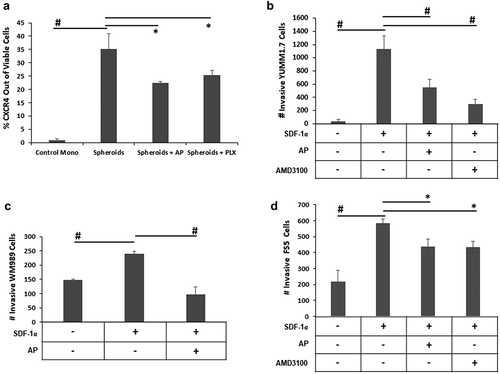
Because of the elevated levels of CXCR4 observed in the YUMM1.7 spheroid cultures, YUMM1.7 cells were evaluated for invasiveness using Boyden chambers with Matrigel-coated inserts and SDF-1α as the chemoattractant in the lower chamber. Compared to starving media, SDF-1α resulted in a 20-fold increase in invasive cells over a 24 h period (). Incubation with 0.05 µM AP significantly reduced the number of invasive YUMM1.7 cells to almost the same degree as incubation with 2.5 µM AMD3100, a CXCR4 inhibitor.Citation30 This observation was not due to cell death as YUMM1.7 cell viability was not affected by incubation with AP or AMD3100 at these concentrations (Supplemental ). Furthermore, these non-cytotoxic, low concentrations of AP or AMD3100 (Supplemental ) inhibited the invasiveness of human WM989 and FS5 melanoma cells similarly to that of YUMM1.7 cells using only SDF-1α as a chemoattractant (). Whereas SDF-1α-induced invasion was inhibited by AP, there was no effect of AP on MMP-2, MMP-9, or CXCR7 (another SDF-1α receptor) expression in the melanoma cells (Supplemental ), suggesting that the AP inhibition of YUMM1.7 cell invasiveness was due more to inhibition of CXCR4/SDF-1α signaling than to a decreased ability to degrade the extracellular matrix or CXCR7/SDF-1α signaling.
AP inhibits M2 macrophage polarization
SDF-1α regulates the trafficking of CXCR4+ hematopoietic cells, their homing/retention in major hemato/lymphopoietic organs, and the accumulation of CXCR4+ immune cells in tissues affected by inflammation.Citation28 Indeed, SDF-1α is elevated in the tumor microenvironment of many cancers and is involved in recruiting pro-tumorigenic M2 macrophages.Citation29 M2-like tumor-associated macrophages (TAMs), which are the predominant cells expressing CXCR4 in chemotherapy-treated melanomas, accumulate around blood vessels in tumors following chemotherapy and promote chemoresistance.Citation31–33 We tested the ability of AP to inhibit macrophage migration across a fibronectin-coated transwell insert (5 μm pore sized membrane) toward the chemoattractant SDF-1α in the bottom well. FACS analysis revealed that RAW 264.7 macrophages strongly express CXCR4. The number of migrated cells in response to SDF-1α was blocked by 85% when RAW 264.7 macrophages were pre-incubated for 30 min with 5 μM AMD3100 and blocked by 55% with 5 μM AP (dose which led to no cytotoxic effects over the 3 h chemotaxis assay) (). Similar results were obtained with CXCR4+ Jurkat cells (data not shown). Thus, in addition to targeting the PTS, AP appears to also block SDF-1-mediated migration of CXCR4+ leukocytes.
Figure 5. AP inhibits macrophage migration and reduces M2 polarization. A. Boyden chamber inserts with 5 µm porous filters were coated with fibronectin and then seeded with RAW 264.7 macrophages in serum-free media containing either 5 µM AP or 5 µM of the CXCR4 inhibitor AMD3100 on the interior of each insert. A gradient was then initiated with the addition of serum-free media supplemented with the chemoattractant (200 ng/ml SDF-1α) to the lower compartment of the invasion chamber. After a 24-hour incubation, cells that had migrated to the underside of the filter were fixed, stained and counted using a microscope (mean ± SD). B. Primary bone marrow macrophage cultures were established from C57Bl/6 mice. Macrophages were polarized to the M2 phenotype using IL-4 (10 ng/mL) ± 0.5 µM AP. To test for M2 polarization, an arginase assay (mean ± SD) was performed. C. Primary bone marrow macrophage cultures were established from C57Bl/6 mice. Macrophages were polarized to the M2 phenotype using IL-4 (10 ng/mL) ± 0.5 µM AP. Polyamine transport activity (3H-spermidine uptake/µg protein ± SD) was measured via an 3H-spermidine uptake assay. D. CXCR4 ± SD expression on replicates of bone marrow macrophages from panel B were determined by flow cytometry. n = 3 per group; * = p ≤ 0.05 and # = p ≤ 0.01 compared to indicated group
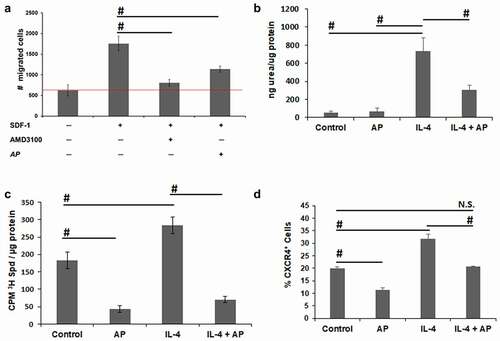
Because previous studies indicate that polyamines regulate the formation of pro-inflammatory M1-like or pro-tumorigenic M2-like macrophages, Citation34,Citation35 we next investigated the effect of AP on the polarization of macrophages to a pro-tumorigenic M2-like phenotype. Murine bone marrow derived macrophages were treated with IL-4 using arginase activity as a marker for polarization to a M2-like phenotype. At a low concentration of AP that did not affect the viability of macrophages (data not shown), AP inhibited IL-4-induced arginase activity indicating that AP inhibits the polarization of macrophages to the M2 phenotype (). Polyamine uptake is increased with macrophage M2 polarization (). As expected, low non-cytotoxic treatment with AP inhibited polyamine uptake in conjunction with its inhibition of M2 polarization (). In agreement with previous reports that formation of pro-tumorigenic M2-like macrophages is dependent on elevated CXCR4/SDF-1α signaling, Citation33,Citation36 polarization to the M2 phenotype with IL-4 increased the percentage of CXCR4+ cells by over 50% (). Incubation with AP reduced the percentage of CXCR4+ macrophages in both the unpolarized control macrophages as well as in macrophages treated with IL-4 ().
AP inhibits the release of macrophage-derived growth factors that promote tumor growth
Tumor-associated macrophages have been shown to reduce the effectiveness of BRAF inhibition in BRAF mutant melanomas. BRAF inhibitors paradoxically activate the MAPK pathway in macrophages to stimulate the release of growth factors, including VEGF, from tumor-associated macrophages that can stimulate melanoma cell growth.Citation32 We hypothesized that AP may block this potent PLX4720-induced macrophage response.
YUMM1.7 melanoma cells were co-cultured with murine bone marrow-derived macrophages that were seeded on collagen-coated Boyden chamber inserts with 0.4 µM pores to allow the diffusion of media ± AP overnight at a concentration that did not affect macrophage cell viability (data not shown). The next morning, the AP-containing medium was removed from the macrophage cultures, and the inserts plated with macrophages were placed in 24-well dishes containing YUMM1.7 cells ± PLX4720 (). After a 72-h incubation, PLX4720 resulted in 50% reduction in YUMM1.7 cell viability; however, co-culture with macrophages that were not pretreated with AP rescued the melanoma cells from PLX4720-mediated cell death (). Pre-treatment of the macrophages with AP restored the effectiveness of the BRAF inhibitor. These results were replicated using macrophages generated from human peripheral blood monocytes and the BRAF mutant human melanoma cell line WM983B. AP pre-treatment prevented the macrophage- rescue of the WM983B cells from PLX4720-mediated cell death ().
Figure 6. AP reverses macrophage inhibition of PLX4720 killing of melanoma cells. A. Macrophage and melanoma co-culture system. Melanoma cells were seeded on the bottom of tissue culture plates. Collagen type I was coated onto Transwell inserts (pore size 0.4 µm). Macrophages were then seeded onto the collagen layer. B. YUMM1.7 melanoma cells were co-cultured with bone marrow derived macrophages from C57Bl/6 mice polarized to the M2 phenotype with IL-4 in the presence of PLX4720 (1 µM) for 72 h. Macrophages treated with AP (0.1 µM) were pretreated with AP 18 h before co-culture with YUMM1.7 melanoma cells. Inserts were washed and then the media containing PLX4720 was added. Cell viability of the melanoma cells was determined and used to calculate the relative viability (treated cells to untreated cells). Data shown as mean ± SD. C. WM983B human melanoma cells were co-cultured with M2-polarized macrophages derived from human peripheral blood monocytes and in the presence of PLX4720 (1 µM) for 72 h. Groups treated with AP (0.1 µM) received an 18 h pretreatment with AP. Inserts were washed and then media containing PLX4720 was added. Cell viability of the melanoma cells was determined and used to calculate the relative viability (treated cells to untreated cells). Data shown as mean ± SD. D. Human macrophages derived from human peripheral blood monocytes were polarized to the M2 phenotype with M-CSF (10 ng/ml). AP samples were pretreated with 0.1 µM AP for 18 h. AP containing media was removed and fresh media containing PLX4720 (3 µM) was added. After 72 h media was collected and analyzed for VEGF protein by ELISA assay (R&D Systems). Data shown as mean ± SD. n = 3 per group; * = p ≤ 0.05 and # = p ≤ 0.01 compared to indicated group
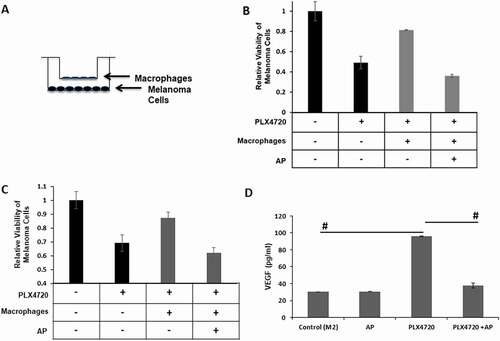
VEGF levels were analyzed in conditioned medium from both human and murine macrophages treated with the BRAF inhibitor PLX4720 ± AP. PLX4720 increased VEGF secretion from both human and murine macrophages but the PLX4720-induction of VEGF was significantly inhibited by pre-treatment with AP ( and Supplemental ). In vivo, VEGF can also be released from extracellular matrix stores by metalloproteinase activity in the tumors.Citation37 The alternatively activated M2-like phenotype of macrophages is associated with increased metalloproteinase expression.Citation38,Citation39 IL-4 polarization of bone marrow-derived macrophages resulted in a 2-fold increase in the activity of secreted MMP-2 and MMP-9 (Supplemental ). Interestingly, PLX4720 increased MMP-9, but not MMP-2 in IL-4-polarized macrophages, whereas AP inhibited this PLX4720 induction of MMP-9 activity. Altogether, these data indicate that AP inhibits the release of macrophage-derived growth factors, such as VEGF, that rescue melanoma cells from PLX4720-mediated cell death.
Discussion
The complexity of melanoma tumors and their ability to develop resistance to a variety of therapeutic regimens suggests that a combination therapy that targets not only BRAF/MEK, but also common pathways that are heavily relied upon for tumor survival is needed. The basis for our study was that the oncogene addiction and BRAF inhibitor-resistance of melanoma tumors correspond to a high demand for polyamine growth factors and greatly upregulated polyamine uptake via the PTS. Utilizing the PTS for delivery of the novel AP compound, we have attacked the tumor by means of one of the key modes of survival for oncogene-addicted melanoma tumor cells. Our data indicate that AP retards the development of BRAF inhibitor-resistant melanoma via two separate, but complementary mechanisms. First, AP targets slow-cycling stem-like cells that regenerate melanomas after debulking proliferating tumor cells with PLX4720 and, second, AP prevents the release of pro-tumorigenic growth factors from macrophages that are induced by BRAF inhibition.
Whereas cytotoxic agents, including conventional chemotherapy and BRAF inhibitors, effectively eliminate rapidly dividing tumor cells, quiescent, slow-cycling CSC subpopulations are enriched. This drug-resistant CSC-like subpopulation of melanoma cells can continuously repopulate the tumor and is most likely responsible for tumor relapse after chemotherapy and formation of metastases.Citation9,Citation10 Solid tumors contain poorly vascularized, hypoxic regions that contribute to tumor progression by activating a hypoxia stress response that promotes cell survival, tumor angiogenesis, and metastasis. It is thought that CSCs exist in this hypoxic microenvironment.Citation40,Citation41 Levels of endogenous reactive oxygen species are increased in melanoma stem-like cells as a result of increased mitochondrial respiration and oxidative phosphorylation.Citation10 This high oxygen consumption contributes to hypoxic conditions that have been shown to favor the slow-cycling CSC-like phenotype.Citation11 Hypoxia also stimulates expression of the chemokine receptor CXCR4 in CD133+ CSCs enriched following chemotherapy, and has been shown to drive metastatic spread.Citation28 Indeed there is a strong correlation between SDF-1α/CXCR4 signaling with SDF-1α expression that is highly increased in preferred metastatic sites such as lymph nodes and lung.Citation29
Polyamines play an essential role in the ability of tumor cells to adapt to hypoxic stress. Svensson et al.Citation42 reported that polyamine biosynthesis and cellular uptake are induced in hypoxic regions of tumors and in tumor spheroids, and depletion of polyamines during hypoxia resulted in increased apoptosis. Indeed, polyamines are well known to exert anti-oxidant functions.Citation43 Polyamines may also contribute to tumor survival by inducing an autophagic state. Autophagy is induced by cellular stress such as nutrient deprivation, withdrawal of growth factors, and hypoxia, Citation44 and it has recently emerged as a common survival process that tumors undergo following chemotherapy and radiation.Citation45 Spermidine has also been shown to induce autophagy in multiple systems including yeast cells, C. elegans, Drosophila, human tumor cellsCitation46 and to increase survival of pluripotent stem cells in culture.Citation47 Increased levels of the polyamine, spermidine, have been shown to induce autophagy via decreasing the acetyltransferase activity of EP300Citation46 and by increasing the activity of the translation factor eIF5A via hypusination, leading to increased translation of the transcription factor, TFEB, that controls autophagy.Citation48 In established tumors, autophagy is also a resistance mechanism to many therapeutic modalities including BRAF inhibitors .Citation49 We have shown that increased polyamine uptake provides a mechanism for BRAF inhibitor-resistant melanoma cells to survive,Citation14 and AP treatment targets and kills this resistant melanoma subpopulation resulting in significant delay of tumor progression.
The plasticity of the melanoma CSC subpopulation is governed in part by microenvironmental cues from stromal cells (including immune cells such as macrophages, cancer-associated fibroblasts, and endothelial cells) which participate in a dynamic interaction with the CSC tumor cells.Citation50 Moreover, tumor-associated macrophages (TAMs) demonstrate a considerable degree of plasticity and functional heterogeneity, and they undergo a phenotypic “switch” during tumor progression, eventually exhibiting the alternatively activated, “M2”, phenotype that is associated with immunosuppression, promotion of tumor angiogenesis and metastasis.Citation51 Recruitment of these TAMs is dependent on the HIF-1-induction of the chemoattractant SDF-1α in tumorsCitation52, which is not only a critical regulator of monocyte migration, but also responsible for promoting polarization to a M2 phenotype.Citation33,Citation36 In all, CXCR4/SDF-1α signaling appears to regulate not only the metastatic potential of chemotherapy-resistant melanoma CSC subpopulations, but also the recruitment and polarization of TAMs that promote chemotherapy-resistance and tumor progression.
Our data support that AP both inhibits uptake of polyamines and also inhibits CXCR4/SDF-1α signaling that is a critical regulator of melanoma-stromal interactions driving metastasis. The two cationic polyamine arms of AP provide this compound with PTS hyper-selectivity, particularly in targeting tumor cells with high polyamine transport rates via a specific uptake system.Citation16 Moreover, AP appears to block CXCR4/SDF-1α signaling, perhaps in a similar way as the structurally related CXCR4 antagonist AMD3100, wherein the appended cationic arms bind to the predominantly anionic extracellular domain of CXCR4.Citation30 With their evenly spaced cationic charges, polyamines are known to affect multiple biologic pathways via their electrostatic interactions with anionic sites on a wide variety of macromolecules including DNA. Interestingly, another polyamine transport inhibitor, the 1,3,5-trisubstituted design of Trimer PTICitation17, does not block SDF-1α induced invasiveness of melanoma cells and does not delay BRAF inhibitor-resistant melanoma progression, suggesting a novel structural specificity of AP for targeting both polyamine uptake via the PTS and also CXCR4. From this comparison, we speculate that the 1,4-substitution pattern of the polyamine arms in AP and AMD3100 may play a role in their ability to target CXCR4. Armed with this dual activity, AP can both accumulate selectively in BRAF inhibitor-resistant CSCs as a result of induced PTS activity and also block CXCR4 signaling in metastatic tumor cells and stromal macrophages, thus inhibiting melanoma progression. In addition, AP inhibits polarization of CXCR4+ M2 macrophages and their secretion of tumor survival factors such as VEGF that contribute to BRAF inhibitor resistance in melanoma cells.
Most cancer chemotherapeutic agents are strong cytotoxic drugs that very effectively kill rapidly proliferating tumor cells. However, slow-cycling cancer stem cell subpopulations, that comprise only a very small part of the pre-treatment melanoma tumor, can survive attack from cytotoxic chemotherapeutic agents including BRAF inhibitors, leading to tumor relapse and metastasis. Here we describe an innovative treatment approach that targets not only the chemo-resistant CSC subpopulation, but also tumor-associated macrophages that contribute to the acquisition of BRAF inhibitor resistance. Our study indicates that co-treatment with the novel AP compound may provide new hope to treat recurrent melanomas that are resistant to current therapies such as BRAF inhibitors.
Materials and methods
Drugs
Working solutions of the ornithine decarboxylase (ODC) inhibitor α-difluoromethylornithine (DFMO, kindly donated by Dr. Patrick Woster at the University of South Carolina) were prepared in sterile water. AP (Me44Nap44NMe)Citation16 and the Trimer44NMe PTICitation18 were synthesized as previously described, and 10 mM stock solutions were prepared in sterile PBS and stored at −20°C. Chemical authentication of AP and the Trimer PTI included 1H NMR, 13C NMR, mass spectrometry, melting point, and elemental analysis of the final compound used in this study. Subsequent dilutions AP and the Trimer PTI were made in PBS to generate the desired working solutions. PLX4720, (S1152; Selleckchem, Houston, TX, USA) a derivative related to PLX4032/Vemurafenib (Plexxikon, Berkeley, CA, USA), was prepared as a 50 mM stock solution in dimethyl sulfoxide and stored at −20°C. For mouse tumor experiments, PLX4720 was supplemented in the chow (417 mg/kg mouse chow) with PLX4720 provided by Plexxikon.
Animals
Female C57Bl/6 mice were obtained from Charles Rivers/NCI. Protocols for the use of animals in these studies were reviewed and approved by the Institutional Animal Care and Use Committee of the Lankenau Institute for Medical Research in accordance with the current U.S. Department of Agriculture, Department of Health and Human Service regulations and standards.
Cell culture
The BRAFV600E, PTEN-null YUMM1.7 melanoma cell lineCitation53 (kindly provided by Marcus Bosenburg, Yale University, New Haven, CT) was maintained in DMEM/F12 (Invitrogen) medium supplemented with 10% fetal bovine serum and 100 U/ml Penicillin/Streptomycin. Human WM88, WM983B and WM989 melanoma cells were obtained from the Wistar Institute collection as kind gifts from Dr. Meenhard Herlyn. These cells were maintained in MCDB153 (Sigma)/Leibovitz’s L-15 (Cellgro) medium (4:1 ratio) supplemented with 2% fetal calf serum and 2 mmol/L CaCl2. Human FS4 and FS5 melanoma cells were obtained from the Lankenau Institute of Medical Research as kind gifts from Dr. Marie Webster .Citation22 FS4 and FS5 cells were maintained in RPMI media supplemented with 10% fetal calf serum, 2 mM glutamine and 1% penicillin/streptomycin. All cells were freshly thawed from early passage cells and cultured for no more than 2 months. Cell line authentication by chromosomal analysis and short tandem repeat (STR)-based DNA profiling as well as IMPACT tests to verify the absence of murine pathogens and mycoplasma (IDEXX BioAnalytics) were performed on these cells before use in animal experiments.
3D spheroid culture
Low binding NanoCulture plates (NCP) (MH pattern from SCIVAX) were used for spheroid cultures in YUMM1.7 cells .Citation20 YUMM1.7 melanoma cells were seeded in 96-well NCPs at 8 × 103 cells/well in DMEM/F12 (Invitrogen) basal medium supplemented with 2% B-27, 20 ng/mL EGF, 20 ng/mL FGF, 0.4 U/mL heparin, 1% Penicillin/Streptomycin, and 0.1% gentamycin, and incubated in a conventional cell incubator at 37°C in an atmosphere of 5% CO2 and normal O2 levels. When visible spheroids began to form on the NCPs, treatment with PLX4720 and/or AP was initiated. After drug treatment for 48 (AP) or 72 (PLX4720) hours, the spheroid cultures were assayed for cell viability using a CellTiter – Glo Luminescent cell viability assay (Promega). Ultra-low attachment plates (Costar® 96-well plates from Corning) were used for spheroid cultures of human melanoma cell lines (WM88, WM983B, WM989, FS4 and FS5). WM88, WM983B and WM989 cells were seeded in MCDB153 basal media with 20% Leibovitz’s L15, 0.005 mg/mL bovine insulin, 1% Penicillin/Streptomycin, 1.68 mM CaCl2, 1x B27 supplement, 20 ng/mL EGF, 20 ng/mL bFGF and 4 µg/mL heparin. FS4 and FS5 cells were seeded in RPMI basal media with 2 mM glutamine, 1% Penicillin/Streptomycin, 1x B27 supplement, 20 ng/mL EGF, 20 ng/mL bFGF and 4 µg/mL heparin. When visible human melanoma spheroids began to form in the ultra-low attachment plates, treatment with PLX4720 and/or AP was initiated.
Bone marrow derived macrophages
Bone marrow was flushed from the femurs C57BL/6 mice with RPMI under sterile conditions, red blood cells lysed in lysis buffer (0.17 M Tris-HCl, 0.16 M NH4Cl) for 3 minutes, and then resuspended in bone marrow media (RPMI, 10% heat-inactivated FBS and 1% penicillin/streptomycin). Cells were plated in 100 mm uncoated Petri dishes with 10 mL of bone marrow media containing 20 ng/mL macrophage colony-stimulating factor (M-CSF) (R&D Systems, 216MC025). On day two, an additional 10 mL of bone marrow media containing 20 ng/mL M-CSF was added. On day five, 10 mL of media was gently removed and 10 mL of bone marrow media containing 20 ng/mL M-CSF (R&D Systems, 216MC025) was added. By day 7 the cells had differentiated into macrophages. Macrophages were polarized to M2 with 10 ng/mL IL-4 (R&D Systems, 404-ML-010) overnight.
Differentiation of human monocytes to macrophages
Enriched monocytes were obtained from healthy volunteers by leukapheresis followed by countercurrent elutriation (Immunology Cell Core at the University of Pennsylvania). To generate M2 macrophages, 2 × 106 monocytes were seeded in tissue-culture treated 6-well plates in monocyte media (RPMI, 10% FBS, 10 mM HEPES, 100 µM 2-mercaptoethanol and 1% penicillin/streptomycin) plus M-CSF (10 ng/mL) for 7 days at 37°C in a humidified tissue culture incubator at 5% CO2. On day 3, 50% of the media was removed from each plate and replaced with fresh media.
Arginase assay
Differentiated but unpolarized bone marrow derived macrophages were plated in 96-well dishes at 5 × 105 cells per well in bone marrow media for 24 h. Cells were rinsed with bone marrow media and refed with treatment media containing IL-4 (10 ng/mL) ± 0.5 µM AP and incubated for 24 h. To test for M2 polarization an arginase assay was performed. Treatment media was removed, and the cells were washed with PBS and then lysed (0.1% Triton X-100 + Halt protease/phosphatase cocktail (Fisher Scientific, PI78441)) with shaking for 30 minutes. Once the cells were lysed, 100 µL of 50 mM Tris-HCl was added to each well. 100 µL of cell lysate was removed and placed into Eppendorf tubes, 10 µL of 10 mM MnCl2 was added and the samples were incubated at 55°C for 10 minutes. Once heated, 100 µL of 0.5 M L-arginine was added to each sample and incubated for 2 h at 37°C. The reaction was stopped by the addition of 800 µL of stop buffer (7H2O:3 H2PO4:1H2SO4) and 40 µl of 9% isonitrosopropiophenone in ethanol. The lysates were then heated at 100°C for 45 minutes, the absorbance read at 540 nm, and the results normalized to protein levels in the lysates.
In Vivo tumor models
In initial studies, tumors were established by subcutaneous injection of 2 × 105 YUMM1.7 cells in the right flank of C57Bl/6 mice. Treatment with PLX4720 (25 mg/kg, oral gavage, 2X daily during the week and 1X daily on weekends) or AP (1 mg/kg, i.p. daily) was initiated when tumors were between 40 and 60 mm3 in size. For experiments with polyamine blocking therapy (PBT), administration of the Trimer 44NMe PTI (3 mg/kg, i.p. daily) ± DFMO (0.25% in the drinking water) was initiated 2 weeks after tumor injection when the tumors were between 40 and 60 mm3 in size. In regression and relapse experiments, tumors were established by subcutaneous injection of 4 × 105 YUMM1.7 cells in the right flank of C57Bl6 mice. Treatment with PLX4720 (417 mg/kg PLX4720 chow or placebo chow) was initiated when tumors were between 100 and 200 mm3 in size. Seven days later, once tumors had regressed to approximately 40 mm3, treatment with AP (0.5 mg/kg or 1.0 mg/kg, i.p. daily) and DFMO (0.5% in the drinking water) was started concurrent with continued PLX4720 treatment. In all experiments, before treatments were initiated, mice were randomized into groups with similar mean tumor volumes and standard deviations. Tumor growth was assessed morphometrically using calipers twice a week, and tumor volumes were calculated using the formula V (mm3) = π/6 x A x B2 (A is the larger diameter and B is the smaller diameter).Citation54
Zymography
Equal number of cells were grown to 80% confluency, washed twice with PBS and then changed to serum-free RPMI medium. After 48 h the conditioned media was collected and centrifuged at 600 x g to remove cell debris, and then loaded on a 10% zymography gel containing 0.1% gelatin (Novex, ZY00100BOX). MMP-2 and MMP-9 activity was detected by incubating the gel in 1x zymography renaturing buffer (Novex, LC2670) for 30 min at room temperature followed by 1x developing buffer (Novex, LC2671) 24 h at 37°C. After Coomassie staining, the clear bands of proteolytic degradation were quantified.
Flow cytometry analysis
Tumor tissue was digested in a 0.3% collagenase/0.1% hyaluronidase solution, pressed through a nylon mesh filter to obtain a single cell suspension and incubated in red cell lysis buffer (0.17 M Tris-HCl, 0.16 M NH4Cl) for 3 minutes, spun down and resuspended in FACS buffer (PBS + 1.5% FBS). Equal numbers of viable cells were stained with a viability dye and the indicated antibodies: CD8a-PECy7 (eBioscience, 25–0081-82) CD4-PE (eBioscience, 12–0041-83), F4/80-PECy7(eBioscience, 25–4801-82), CD206-FITC (Biolegend, 141704), Gr1-APC (eBioscience, 17–5931-81), CD11b-PE (eBioscience, 12–0112-82), CD45-PE-Cy5 (eBioscience, 15–0451-83). Melanoma cells lines were analyzed using equal numbers of viable cells stained with CXCR4-PECy5 (eBioscience, 46–9991-82), CXCR4-PECy7 (eBioscience, 15–9999-42) and Jarid1B (SDIX 2226.00.02) Flow-cytometric data were acquired on a BD FACSCanto II cytometer and analyzed using FACSDiva software (BD Biosciences).
Invasion assay
Boyden chamber inserts (for a 24 well plate) with 8 µm porous filters were coated with 50 μg of growth factor-reduced Matrigel and incubated at 37°C for 3 h. Yumm1.7 cells were seeded at densities of 1 × 105 in serum-free DMEM/F12 media containing either 0.05 µM AP or 2.5 µM of the CXCR4 inhibitor AMD3100 in the interior of each insert. After 1 hr, a gradient was initiated with the addition of serum-free DMEM/F12 media supplemented with the chemoattractant (200 ng/mL, SDF-1α) to the lower compartment of the invasion chamber. After 24-hour incubation at 37°C, cells that had not migrated through the Matrigel-coated filter were removed with a cotton-tipped applicator and migrated cells on the underside of the filter were fixed in 10% neutral buffered formalin, stained with 1% crystal violet, and then mounted on a slide and counted using a microscope. Each condition was performed in triplicate.
Macrophage-melanoma co-culture survival assays
Boyden chamber inserts (for a 24 well plate) with 0.4 µm porous filters were coated with 10 μg rat tail collagen and incubated 37°C for 3 h. Human macrophages differentiated from peripheral blood monocytes or bone marrow derived macrophages from C57BL6 mice were plated in complete media ± 0.1 µM AP and incubated overnight. At the same time YUMM1.7 or WM983B melanoma cells were plated at a density of 5.0 × 103 and 2.0 × 104 cells, respectively, in 24-well plates and allowed to plate down overnight in 24-well dishes separate from the macrophages. The next day media was removed from both the inserts with the macrophages and from the dishes seeded with the melanoma cells and the cells were gently washed. The media was replaced with complete media plus 1 µM PLX4720 and the chamber inserts containing the macrophages were placed into the wells with the melanoma cells. After 72 h, cell viability of the melanoma cells was determined via the EZQuant cell quantifying kit (Alstem CQ01).
Statistical analysis
All in vitro experiments were performed at least in triplicate, and data were compiled from two to three separate experiments. Analyses were done using a 1-way ANOVA with a Tukey test for statistical significance or a Students t-test. In vivo studies were carried out using minimal cohort sizes of animals (n = 7–10 per treatment group based on previous repeated murine tumor experiments). All analyses were performed in Stata/MP 15.1 (StataCorp LP., Texas, USA). All tests were two-sided, and the statistical significance level was set to 0.05. Since tumor volume grew exponentially over time, all volumes were transformed and analyzed on the logarithmic (base 10) scale. All means and standard errors presented were back-transformed to represent geometric means and standard errors .Citation55 Since tumor volume was measured longitudinally for each mouse, linear mixed-effects models were used to evaluate change in tumor volume over time. In the models, log tumor volume was the response and time, treatment, and the interaction between time and treatment were the fixed effects, and each mouse was considered a random effect. Two-sample t-tests were used to compare log responses at each timepoint.
Abbreviations
AP, arylmethyl polyamine compound; CXCL12, C-X-C motif chemokine ligand-12; CXCR4, C-X-C chemokine receptor type 4; CXCR7, C-X-C chemokine receptor type 7; DFMO, α-difluoromethylornithine; ELISA, enzyme-linked immunosorbent assay; H3K4, histone 3, lysine 4; IL-4, interleukin 4; LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinase; M-CSF, macrophage colony-stimulating factor; MMP-2 and MMP-9, matrix metalloprotease-2 and −9; MDSCs, myeloid-derived suppressor cells; ODC, ornithine decarboxylase; SDF-1α, stromal-derived factor-1 alpha; TAM, tumor-associated macrophages; VEGF, vascular endothelial growth factor.
Author contributions
S.K.G. conceived and designed the experiments. E.T.A., O.E.N., E.F., K.M., J.D., and K.W. performed most of the experiments and analyzed the data. E.T.A., O.E.N., E.F., K.M., J.D., and S.K.G. performed animal experiments. O.P. produced and provided AP and Trimer PTI. E.T.A. and S.K.G. wrote the manuscript. All authors have read and agreed to this version of the manuscript.
Disclosure of potential conflicts of interest
The authors report no conflict of interest.
Supplemental Material
Download Zip (4 MB)Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Solit DB, Rosen N. Resistance to BRAF inhibition in melanomas. N Engl J Med. 2011;364(8):772–774. doi:10.1056/NEJMcibr1013704.
- Haq R, Fisher DE. Targeting melanoma by small molecules: challenges ahead. Pigment Cell Melanoma Res. 2013;26(4):464–469.
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. 2002. Mutations of the BRAF gene in human cancer. Nature. 417(6892):949–954.
- Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, Wubbenhorst B, Xu X, Gimotty PA, Kee D, et al. 2010. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 18(6):683–695.
- Jilaveanu LB, Zito CR, Aziz SA, Conrad PJ, Schmitz JC, Sznol M, Camp RL, Rimm DL, Kluger HMC. Raf is associated with disease progression and cell proliferation in a subset of melanomas. Clin Cancer Res. 2009;15(18):5704–5713.
- Bucheit AD, Davies MA. Emerging insights into resistance to BRAF inhibitors in melanoma. Biochem Pharmacol. 2014;87(3):381–389.
- De Angelis ML, Francescangeli F, La Torre F, Zeuner A. Stem Cell Plasticity and Dormancy in the Development of Cancer Therapy Resistance. Front Oncol. 2019;9:626.
- Rossari F, Zucchinetti C, Buda G, Orciuolo E. Tumor dormancy as an alternative step in the development of chemoresistance and metastasis - clinical implications. Cell Oncol (Dordr). 2020;43(2):155–176.
- Zubrilov I, Sagi-Assif O, Izraely S, Meshel T, Ben-Menahem S, Ginat R, Pasmanik-Chor M, Nahmias C, Couraud PO, Hoon DS, et al. 2015. Vemurafenib resistance selects for highly malignant brain and lung-metastasizing melanoma cells. Cancer Lett. 361(1):86–96.
- Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D, Korbel C, Laschke MW, Gimotty PA, Philipp SE, et al. Overcoming Intrinsic Multidrug Resistance in Melanoma by Blocking the Mitochondrial Respiratory Chain of Slow-Cycling JARID1Bhigh Cells. Cancer Cell. 2013;23(6):811–825. doi:10.1016/j.ccr.2013.05.003.
- Roesch A, Fukunaga-Kalabis M, Schmidt EC, Zabierowski SE, Brafford PA, Vultur A, Basu D, Gimotty P, Vogt T, Herlyn M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell. 2010;141(4):583–594.
- Bello-Fernandez C, Packham G, Cleveland JL. The ornithine decarboxylase gene is a transcriptional target of c-Myc. Proc Natl Acad Sci USA. 1993;90:7804–7808.
- Origanti S, Shantz LM. Ras transformation of RIE-1 cells activates cap-independent translation of ornithine decarboxylase: regulation by the Raf/MEK/ERK and phosphatidylinositol 3-kinase pathways. Cancer Res. 2007;67(10):4834–4842.
- Peters MC, Minton A, Iv O P, Gilmour SK. A Novel Polyamine-Targeted Therapy for BRAF Mutant Melanoma Tumors. Med Sci (Basel). 2018;6(1):3.
- Casero RA Jr., Murray Stewart T, Pegg AE. Polyamine metabolism and cancer: treatments, challenges and opportunities. Nat Rev Cancer. 2018;18(11):681–695.
- Muth A, Kamel J, Kaur N, Shicora AC, Ayene IS, Gilmour SK, Phanstiel O. Development of polyamine transport ligands with improved metabolic stability and selectivity against specific human cancers. J Med Chem. 2013;56(14):5819–5828.
- Alexander ET, Minton A, Peters MC, Phanstiel O, Gilmour SK. A novel polyamine blockade therapy activates an anti-tumor immune response. Oncotarget. 2017;8(48):84140–84152.
- Muth A, Madan M, Archer JJ, Ocampo N, Rodriguez L, Phanstiel O. Polyamine transport inhibitors: design, synthesis, and combination therapies with difluoromethylornithine. J Med Chem. 2014;57(2):348–363.
- Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, Mitra D, Boni A, Newton LP, Liu C, et al. 2013. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res. 19(5):1225–1231.
- Eguchi T, Sogawa C, Okusha Y, Uchibe K, Iinuma R, Ono K, Nakano K, Murakami J, Itoh M, Arai K, et al. Organoids with cancer stem cell-like properties secrete exosomes and HSP90 in a 3D nanoenvironment. PLoS One. 2018;13:2.
- Qin Y, Roszik J, Chattopadhyay C, Hashimoto Y, Liu C, Cooper ZA, Wargo JA, Hwu P, Ekmekcioglu S, Grimm EA. Hypoxia-Driven Mechanism of Vemurafenib Resistance in Melanoma. Mol Cancer Ther. 2016;15(10):2442–2454.
- Webster MR, Xu M, Kinzler KA, Kaur A, Appleton J, O’Connell MP, Marchbank K, Valiga A, Dang VM, Perego M, et al. 2015. Wnt5A promotes an adaptive, senescent-like stress response, while continuing to drive invasion in melanoma cells. Pigment Cell Melanoma Res. 28(2):184–195.
- Prud’homme GJ, Glinka Y. Neuropilins are multifunctional coreceptors involved in tumor initiation, growth, metastasis and immunity. Oncotarget. 2012;3(9):921–939.
- Jimenez-Hernandez LE, Vazquez-Santillan K, Castro-Oropeza R, Martinez-Ruiz G, Munoz-Galindo L, Gonzalez-Torres C, Cortes-Gonzalez CC, Victoria-Acosta G, Melendez-Zajgla J, Maldonado V. NRP1-positive lung cancer cells possess tumor-initiating properties. Oncol Rep. 2018;39(1):349–357.
- Grun D, Adhikary G, Eckert RLNRP. 1 interacts with GIPC1 and SYX to activate p38 MAPK signaling and cancer stem cell survival. Mol Carcinog. 2019;58(4):488–499.
- Kuo YT, Liu YL, Adebayo BO, Shih PH, Lee WH, Wang LS, Liao YF, Hsu WM, Yeh CT, Lin CM. JARID1B Expression Plays a Critical Role in Chemoresistance and Stem Cell-Like Phenotype of Neuroblastoma Cells. PLoS One. 2015;10(5):e0125343.
- Held M, Bosenberg M. A role for the JARID1B stem cell marker for continuous melanoma growth. Pigment Cell Melanoma Res. 2010;23(4):481–483.
- Kawaguchi N, Zhang TT, Nakanishi T. Involvement of CXCR4 in Normal and Abnormal Development. Cells. 2019;8:2.
- Kim M, Koh YJ, Kim KE, Koh BI, Nam DH, Alitalo K, Kim I, Koh GY. CXCR4 signaling regulates metastasis of chemoresistant melanoma cells by a lymphatic metastatic niche. Cancer Res. 2010;70(24):10411–10421.
- Debnath B, Xu S, Grande F, Garofalo A, Neamati N. Small molecule inhibitors of CXCR4. Theranostics. 2013;3(1):47–75.
- Gazzaniga S, Bravo AI, Guglielmotti A, van Rooijen N, Maschi F, Vecchi A, Mantovani A, Mordoh J, Wainstok R. Targeting tumor-associated macrophages and inhibition of MCP-1 reduce angiogenesis and tumor growth in a human melanoma xenograft. J Invest Dermatol. 2007;127(8):2031–2041.
- Wang T, Xiao M, Ge Y, Krepler C, Belser E, Lopez-Coral A, Xu X, Zhang G, Azuma R, Liu Q, et al. 2015. BRAF Inhibition Stimulates Melanoma-Associated Macrophages to Drive Tumor Growth. Clin Cancer Res. 21(7):1652–1664.
- Hughes R, Qian BZ, Rowan C, Muthana M, Keklikoglou I, Olson OC, Tazzyman S, Danson S, Addison C, Clemons M, et al. Perivascular M2 Macrophages Stimulate Tumor Relapse after Chemotherapy. Cancer Res. 2015 Sep 1;75(17):3479–91.
- Van den Bossche J, Lamers WH, Koehler ES, Geuns JM, Alhonen L, Uimari A, Pirnes-Karhu S, Van Overmeire E, Morias Y, Brys L, et al. 2012. Pivotal Advance: arginase-1-independent polyamine production stimulates the expression of IL-4-induced alternatively activated macrophage markers while inhibiting LPS-induced expression of inflammatory genes. J Leukoc Biol. 91(5):685–699.
- Latour YL, Gobert AP, Wilson KT. The role of polyamines in the regulation of macrophage polarization and function. Amino Acids. 2020 Feb;52(2):151–160.
- Beider K, Bitner H, Leiba M, Gutwein O, Koren-Michowitz M, Ostrovsky O, Abraham M, Wald H, Galun E, Peled A, et al. 2014. Multiple myeloma cells recruit tumor-supportive macrophages through the CXCR4/CXCL12 axis and promote their polarization toward the M2 phenotype. Oncotarget. 5(22):11283–11296.
- Lee S, Jilani SM, Nikolova GV, Carpizo D, Iruela-Arispe ML. Processing of VEGF-A by matrix metalloproteinases regulates bioavailability and vascular patterning in tumors. J Cell Biol. 2005;169(4):681–691.
- Allavena P, Sica A, Solinas G, Porta C, Mantovani A. The inflammatory micro-environment in tumor progression: the role of tumor-associated macrophages. Crit Rev Oncol Hematol. 2008;66(1):1–9.
- Liu L, Ye Y, Zhu XMMP. 9 secreted by tumor associated macrophages promoted gastric cancer metastasis through a PI3K/AKT/Snail pathway. Biomed Pharmacother. 2019;117:109096.
- Mohyeldin A, Garzon-Muvdi T, Quinones-Hinojosa A. Oxygen in stem cell biology: a critical component of the stem cell niche. Cell Stem Cell. 2010;7(2):150–161.
- Mathieu J, Zhang Z, Zhou W, Wang AJ, Heddleston JM, Pinna CM, Hubaud A, Stadler B, Choi M, Bar M, et al. 2011. HIF induces human embryonic stem cell markers in cancer cells. Cancer Res. 71(13):4640–4652.
- Svensson KJ, Welch JE, Kucharzewska P, Bengtson P, Bjurberg M, Pahlman S, Ten Dam GB, Persson L, Belting M. Hypoxia-mediated induction of the polyamine system provides opportunities for tumor growth inhibition by combined targeting of vascular endothelial growth factor and ornithine decarboxylase. Cancer Res. 2008;68(22):9291–9301.
- Mozdzan M, Szemraj J, Rysz J, Stolarek RA, Nowak D. Anti-oxidant activity of spermine and spermidine re-evaluated with oxidizing systems involving iron and copper ions. Int J Biochem Cell Biol. 2006;38(1):69–81.
- Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40(2):280–293.
- Strohecker AM, White E. Targeting mitochondrial metabolism by inhibiting autophagy in BRAF-driven cancers. Cancer Discov. 2014;4(7):766–772.
- Eisenberg T, Knauer H, Schauer A, Buttner S, Ruckenstuhl C, Carmona-Gutierrez D, Ring J, Schroeder S, Magnes C, Antonacci L, et al. 2009. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol. 11(11):1305–1314.
- Chen T, Shen L, Yu J, Wan H, Guo A, Chen J, Long Y, Zhao J, Pei G. Rapamycin and other longevity-promoting compounds enhance the generation of mouse induced pluripotent stem cells. Aging Cell. 2011;10(5):908–911.
- Zhang H, Alsaleh G, Feltham J, Sun Y, Napolitano G, Riffelmacher T, Charles P, Frau L, Hublitz P, Yu Z, et al. 2019. Polyamines Control eIF5A Hypusination, TFEB Translation, and Autophagy to Reverse B Cell Senescence. Mol Cell. 76(1):110–125.
- Ma XH, Piao SF, Dey S, McAfee Q, Karakousis G, Villanueva J, Hart LS, Levi S, Hu J, Zhang G, et al. 2014. Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J Clin Invest. 124(3):1406–1417.
- Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell. 2012;10(6):717–728.
- Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG, Rimoldi M, Biswas SK, Allavena P, Mantovani A. Macrophage polarization in tumour progression. Semin Cancer Biol. 2008;18(5):349–355.
- Riboldi E, Porta C, Morlacchi S, Viola A, Mantovani A, Sica A. Hypoxia-mediated regulation of macrophage functions in pathophysiology. Int Immunol. 2013;25(2):67–75.
- Obenauf AC, Zou Y, Ji AL, Vanharanta S, Shu W, Shi H, Kong X, Bosenberg MC, Wiesner T, Rosen N, et al. 2015. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature. 520(7547):368–372.
- Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, Viollet B, Thompson CB. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007;67(14):6745–6752.
- R Ms S, Curwen J, Dymond M. Design, analysis and reporting of tumor models. Lab Anim (NY). 2017;46(5):207–211.