
ABSTRACT
Enhancer of zeste homolog 2 (EZH2) is a histone methyltransferase and a catalytic subunit of the polycomb repressive complex 2 (PRC2) that catalyzes the mono-, di-, and tri-methylation of histone H3 at Lys 27 (H3K27me3) to facilitate chromatin-remodeling and gene-silencing functions. Previous reports showed a significant association of EZH2 aberrations in pediatric cancers, such as soft tissue sarcomas and glioblastoma. Recent reports in human subjects and animal models have also suggested a central role of EZH2 in the induction and progression of acute myeloid leukemia. In this study, we aimed to investigate the molecular status of EZH in cell lines derived from distinct pediatric leukemia to assess the efficacy of targeting EZH2 to suppress cancer cell survival and proliferation. Our results showed that EZH2 protein is overexpressed in the pediatric monocytic cell-line THP-1, but not in other leukemia-derived cell lines MV4;11 and SEM. Screening a panel of methyltransferase inhibitors revealed that three inhibitors; GSK126, UNC1999 and EPZ-5687 are the most potent inhibitors that suppressed EZH2 activity selectively on lysine 27 which resulted in increased apoptosis and inhibition of AKT and ERK protein phosphorylation in THP-1 cells. Our data demonstrated a significant increase in apoptosis in cells treated with drug combination (EZH2i and selinexor) compared to EZH2i inhibitors alone. Taken together, our data provide initial evidence that targeting EZH2 is a promising therapeutic strategy for the treatment of subtypes of pediatric AML. Also, combining EZH2 inhibitors with selinexor may increase the treatment efficacy in these patients.
KEYWORDS:
INTRODUCTION
Leukemia is the most common childhood malignancy, and it represents about 30% of all cancers diagnosed in this age group. Most pediatric leukemias are acute and consist of two major types, acute lymphoblastic leukemia (ALL) and acute myeloid leukemia (AML). In the recent past, significant improvements have been achieved in ALL with current survival rates close to 85–90%. However, the prognosis of AML and certain other subgroups such as infant and adolescent leukemias, blasts with high risk molecular abnormalities and relapsed leukemia, still remains poor.Citation1 Among childhood leukemias, AML represents a clonal malignant transformation of early hematopoietic progenitor cells that leads to the production of nonfunctional myeloid blasts. Although the overall survival rate has increased during the past 25 years, children with AML continue to endure an unacceptable relapse rate of about 30%. Hence, new treatment and therapeutic approaches are urgently needed to address this unmet need in pediatric oncology.Citation2 Acute monocytic leukemia, AML‐M5 in the French‐American‐British classification, is a subtype of AML.Citation3 The complete remission rate of AML-M5 was reported to be 52% with a 26% three-year disease-free survival.Citation4 Poorer outcomes in AML-M5 patients have been noted in children who are less than three years of age, who present with hyperleukocytosis and molecular alterations, such as MLL-R and FLT3-ITD.Citation5 In patients with such high-risk characteristics conventional chemotherapy treatments remain highly inadequate.Citation5 The THP-1 cell line was established by Tsuchiya and colleagues from cells isolated from the peripheral blood of a one-year-old male child with AML-M5.Citation6 These cells exhibit distinct monocytic markers and functional properties and their molecular and cytogenetic characteristics have been described.Citation7 The complete gene expression profile of activated THP-1 cells has been published and it has been considered to be an applicable experimental system to study immune-modulatory functions.Citation8,Citation9 Recently, a number of studies have used THP-1 cells as a model system to investigate epigenetically regulated pathways in various disease conditions.Citation10–13
Polycomb group proteins (PcGs) are key regulators of the epigenetic dynamics that play an important role in cell proliferation, stem cell differentiation, and aberrant gene expression during tumorigenesis of several cancers.Citation14,Citation15 The PcG machinery contains two core complexes: Polycomb repressive complex 1 and 2 (PRC1 and PRC2) .Citation16 PRC1 catalyzes the mono-ubiquitination of the histone H2A at Lys 119 through RING1A and RING1B ubiquitin ligases, while PRC2 catalyzes the mono-, di-, and tri-methylation of histone H3 at Lys 27 (H3K27me3) to modulate gene transcription.Citation17 Enhancer of zeste homolog 2 (EZH2), the catalytic subunit of PRC2 complex is frequently overexpressed and associates with high levels of H3K27me3 in various tumor types.Citation18–20 Overexpression of EZH2 has been linked to various types of solid tumors including glioblastoma, soft tissue sarcoma, melanoma and breast and prostate cancers.Citation21–23 Previous reports have shown that EZH2 is highly integrated into various signaling pathways in cancer, including and not limited to, cell proliferation, cell survival, invasion, migration, epithelial to mesenchymal transition and drug resistance.Citation24–28 Current evidence suggests a central role of EZH2 in the development of pediatric soft tissue sarcomas and glioblastoma.Citation29,Citation30 Importantly, targeting EZH2 by small-molecule inhibitors resulted in a significant reduction of cancer cell viability and increased apoptosis, suggesting that targeting EZH2 is a potential therapeutic strategy for treating a number of malignancies.Citation31 In addition, a comprehensive mutational analysis performed on a cohort of AML patients revealed that mutated EZH2 is associated with AML progression and it can be used as a potential prognostic marker and a possible target for therapeutic applications development.Citation32 In this study, we aimed to investigate the molecular status of EZH in cell lines derived from distinct pediatric leukemia to assess the efficacy of targeting EZH2 to suppress cancer cell survival and proliferation. Our data showed higher levels of EZH2 protein in the AML-M5 cell-line THP-1 which suggests a potential involvement of EZH2 in the development and progression of these cells. We further analyzed the ability to target EZH2 using specific pharmacological agents and drug combinations.
METHODS
Cell lines and cell culture
The pediatric monocyte cell-line THP-1 (ATCC, TIB-202), the childhood B-cell precursor leukemia cell line SEM, and pediatric AML cell line MV4;11 (ATCC, CRL-9591) were maintained in Opti-MEM media (Gibco, Invitrogen Corporation, Burlington, ON) supplemented with 5% fetal bovine serum and 100 units/ml penicillin and 100 units/ml streptomycin (Gibco). Immortalized primary fibroblast cells (hTERT) (ATCC, CRL-2846) were used as a control in this study. All cell cultures were maintained at 37°C in a humidified incubator with 5% CO2. Inhibitors for EZH2 were purchased from Selleck (Cedarlane, Burlington, Ontario, Canada), selinxor was purchased from Karyopharm Therapeutics. Stock solutions were prepared in DMSO at 10 mM concentrations and stored in aliquots at −20°C.
Cytotoxicity assay
THP-1, hTERT and primary cells were cultured in 1 × 104 cells in 100 µl of Opti-MEM per well in 96 well plates. Cells were treated with each EZH2 inhibitor (EZH2i) to a final concentration ranging from 1 nM to 100 µM. Cultured cells were incubated in the presence or absence of the tested drug for 96 hours. Total cell viability (% cytotoxicity) was measured using Alamar blue assay as described previously.Citation33 Briefly, cells were incubated with 10% Alamar blue for 4 hours, then the absorbance at 570–620 nm was measured (Opsys MR Plate Reader, Dynex Technologies, Chantilly, Virginia). Cell viability (%) for THP-1 cells and control (hTERT) cells were calculated by normalizing the absorbance ratio of the treated cells to the vehicle control (DMSO). IC50 was calculated by GraphPad Prism 6.0 program (GraphPad Software, Inc; CA, USA).
Western blot analyses
Whole-cell extracts were prepared using radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl (pH 8), 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate; 0.1% sodium dodecyl sulfate (SDS)) supplemented with 1% phosphatase inhibitor (Sigma-Aldrich), 1% protease inhibitor (Sigma-Aldrich) and 1% sodium orthovanadate (Alfa Aesar, Ward Hill, Massachusetts). Cellular extracts were resolved by SDS-PAGE and transferred to nitrocellulose membranes in a Tris/glycine transfer buffer containing 10% (v/v) methanol. Nonspecific binding sites were blocked with 5% (w/v) nonfat dry milk in Tris-buffered saline with Tween (TBST, 25 mM Tris-HCl, 137 mM NaCl, 3 mM KCl, and 0.05% (v/v) Tween-20). Membranes were washed and probed overnight with primary antibody at 1:1,000 dilution: histone 3, methylation-histone 3 [K4, K9, K27, K36, and K79], PARP, caspase3, caspase7, caspase9, ERK1/2, Anti-pERK1/2 (pThr202/pTyr204), Akt, pS308-Akt, β-actin, P27, P21 (Cell Signaling, Danvers, MA), EZH2 and pS21-EZH2 (Abcam, Cambridge, United Kingdom). Membranes were then incubated for 50 minutes with horseradish peroxidase (HRP)-conjugated secondary antibody (dilution 1:7500) in TBST and developed with enhanced chemiluminescence (Amersham Biosciences).
DNA extraction and sequencing
DNA was extracted from THP-1, SEM, and MV4;11 cell lines using DNeasy Blood & Tissue extraction kit from Omega Bio-tek (GA, United States). DNA was amplified using the following primers; Forward: 5’- GAGAGTCAGTGAGATGCCCAG −3’ and Reverse: R: 5`-TTTGCCCCAGCTAAATCATC-3`. Sequences were confirmed by the University of Calgary’s Core DNA Services (www.sequencing.ucalgary.ca/) using an Applied Biosystems 3730xl (96 capillary) genetic analyzer.
Immunocytochemistry
THP-1 cells were plated at 5 × 103 in Opti-MEM media with 5% (v/v) FBS at 37°C with 5% CO2. Cells were fixed for 30 min in 4% (v/v) paraformaldehyde in PBS and then permeabilized with 0.5% Tween-20 for 10 min. Cells were then incubated at 4°C overnight with primary antibody diluted 1:250 in blocking serum (0.3% BSA, 5% goat serum, 0.25% Triton X-100 in PBS, pH 7.4). Cells were washed three times with PBS before adding Alexa Fluor 568-conjugated secondary antibody (1:500) for one hour at room temperature. Cells were rinsed with PBS, counterstained with DAPI for 5 min to detect nuclei, and then visualized with an InCell 6000 Imaging System (GE Healthcare) .Citation34
Statistical analysis
Data are presented as the mean ± S.E.M from at least three independent experiments. Statistical significance was assessed using Student’s t-test, and P < .05 was considered to indicate statistical significance. All statistical analyses were performed using the GraphPad Prism 6.0 program.
RESULTS
EZH2 protein is upregulated in THP-1 cells
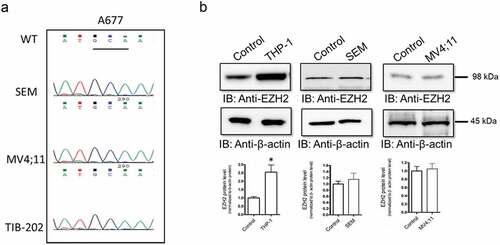
To study the molecular status of EZH2 in different types of pediatric leukemia, we examined the molecular status of EZH2 in three different pediatric leukemia cells lines; THP-1, SEM and, MV4;11. Several earlier studies have shown that EZH2 mutation in alanine 677 to a glycine (A677G) is implicated in pediatric refractory tumors such as follicular lymphoma, Citation35 and diffuse large B-cell lymphoma.Citation36 To determine whether EHZ2 is mutated in these cell lines, we completed mutational profiling analysis. DNA was extracted from the cells, amplified and visualized in agarose gels to confirm the size (~350 bp) of the band that corresponds to the size of the amplicon at exon 15 of EZH2. DNA sequencing chromatogram confirmed the sequence and showed no changes of the DNA sequence (no mutations) of the A677 codon in all three cell lines compared to the reference sequence (). We have further characterized the molecular status of EZH2 in these pediatric leukemia cell lines by assessing the protein expression levels compared to immortalized primary fibroblast cells (hTERT) (ATCC, CRL-2846) that were used as a control. Western blot analysis showed a significant increase of EZH2 protein levels in THP-1 cells, but not in SEM and MV4;11 (). EZH2 protein signal was normalized to total β-actin protein levels.
Figure 1. Analysis of EZH molecular status in pediatric leukemia cell lines. (a) DNA was extracted from THP-1, SEM, and MV4;11 cell lines. Following PCR amplification, size of the DNA fragments was detected on agarose gel, then subjected for DNA sequencing to study to examine the presence of mutation at A677 for all cell lines as showed in the DNA chromatogram. (b) EZH2 protein levels were analyzed by western blot. Protein levels in THP-1, SEM, and MV4;11 cell lines were evaluated using a monoclonal antibody against EZH2. Bands were quantified by scanning densitometry and normalized to total β-actin (loading control). Values represent means ± S.E.M. for n = 4 independent experiments. *Significantly different from the vehicle control (Student’s t test, p < .05)
Targeting EZH-2 activity by a panel of selective small-molecule inhibitors induces apoptosis in THP-1 cells
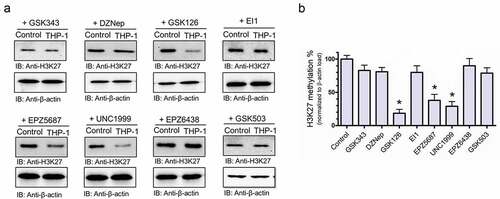
To assess if targeting EZH2 in THP-1 cells has a potential therapeutic value, we then profiled the cytotoxic activity of a panel of EZH2-selcetive inhibitors; GSK343, Citation37 DZNep, Citation38 GSK126, Citation39 EI1, Citation40 EPZ-5687, Citation41 UNC1999, Citation42 EPZ-6438, Citation43 and GSK503.Citation44 In vitro cell cytotoxicity of THP-1 and hTERT (control) cells were evaluated by treating the cells with an increased concentration [1 × 10−9 to 1 × 10−Citation4, M] of each compound (). Cell cytotoxicity assays demonstrated three compounds (GSK 126, UNC 1999, and EPZ-5687) to show IC50 values in the nanomolar range with a minimal cytotoxicity level in the control hTERT cells (). Other compounds, EI1, GSK 503, and DZNep induced similar cytotoxicity effects in both THP-1 cells and the control hTERT cells at 0.9, 1.3, and 2.1 µM, respectively, while GSK343 and EPZ-6438 displayed poor inhibitory effects. We further characterized the activity of this panel of EZH2-selective inhibitors by treating THP-1 cells with 1 µM of each inhibitor to profile the putative methylation site at histone 3 lysine 27 (). Western blot analysis showed a significant reduction of H3K27 methylation of THP-1 upon treatment cells with GSK126, UNC1999, and EPZ-5687 (). However, no significant reduction of H3K27 methylation was observed when treated with other molecules within the selected panel (). The total methylation at H3K27 was normalized to total β-actin loading control. These findings suggest that GSK126, UNC1999, and EPZ-5687 hold the potential to be effective therapeutic agents to target specified AML-M5 cells via the suppression of histone methylation at lysine 27 residue.
Table 1. List of all tested EZH-2 inhibitors with their calculated IC50s in THP-1 and control (hTERT) cell lines
Figure 2. GSK126, UNC1999, and EPZ-5687 induced cell cytotoxicity in THP-1 cells. THP-1 cells or hTERT cells were treated with increasing concentration [1 × 10−Citation4 to 100, μM] of GSK343, DZNep, GSK126, EI1, EPZ-5687, UNC1999, EPZ-6438, EPZ-5687, and GSK503 for 96 hours. Cellular viability was calculated as (%) by comparing the absorbance ratio (percentage) of the treated cells normalized to the control (DMSO) treated cells
![Figure 2. GSK126, UNC1999, and EPZ-5687 induced cell cytotoxicity in THP-1 cells. THP-1 cells or hTERT cells were treated with increasing concentration [1 × 10−Citation4 to 100, μM] of GSK343, DZNep, GSK126, EI1, EPZ-5687, UNC1999, EPZ-6438, EPZ-5687, and GSK503 for 96 hours. Cellular viability was calculated as (%) by comparing the absorbance ratio (percentage) of the treated cells normalized to the control (DMSO) treated cells](/cms/asset/26ef56c4-45ca-436f-8d15-12c7ff7e065b/kcbt_a_1902913_f0002_oc.jpg)
Figure 3. GSK126, UNC1999, and EPZ-5687 suppressed H3K27 methylation in THP-1 cells. (a) western blot analysis of H3K27 methylation in THP-1 cells treated with 1 µM of GSK343, DZNep, GSK126, EI1, EPZ-5687, UNC1999, EPZ-6438, EPZ-5687, and GSK503 compounds. H3K27 methylation signal was normalized to total β-actin loading control. Bands were quantified by scanning densitometry and normalized to total β-actin (loading control) (b). Values represent means ± S.E.M. for n = 4 independent experiments. *Significantly different from the vehicle control (Student’s t test, p < .05)
GSK126, UNC1999, and EPZ-5687 selectively suppressed methylation of H3K27
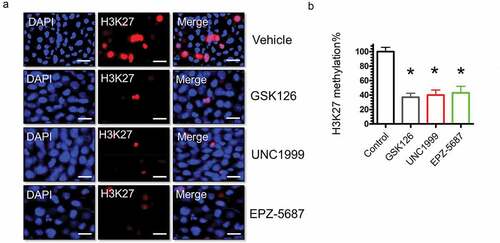
Histone methylation has been shown to occur on different lysine residues.Citation45 The most well-characterized histone methylation sites include, H3K4, H3K9, H3K27, H3K36, and H3K79. We aimed to identify the lysine residue of histone 3 that is selectively methylated by EZH2 and to study the potentials of suppressing histone 3 methylation using EZH2i We profiled the methylation levels of H3K4, H3K9, H3K27, H3K36, and H3K79 by western blots (). Our data demonstrated that GSK 126, UNC 1999, and EPZ-5687 are exclusively inhibit H3 at lysine 27 (). No significant changes on other methylation sites were observed following treatment with the EZH2i. Methylation levels of histone 3 was normalized to total histone protein levels to the β-actin loading control. To further confirm this observation, we performed immunocytochemical analysis to visualize any changes on histone methylation in THP-1 cells. In agreement with western blot analysis results, immunocytochemical analysis showed a significant reduction of H3K27 protein signal following treatment with 1 µM of GSK 126, UNC 1999, or EPZ-5687 compared with the vehicle control (DMSO) treatment ().
Figure 4. H3K27 methylation was exclusively targeted by GSK126, UNC1999, and EPZ-5687 in THP-1 cells. (a) western blot analysis of H3K27, H3K4, H3K9, H3K36, and H3K79 methylation in THP-1 cells treated with 1 µM of GSK126, UNC1999, and EPZ-5687. Methylation signal was normalized to total β-actin loading control. Bands were quantified by scanning densitometry and normalized to total β-actin protein (loading control) (b). Values represent means ± S.E.M. for n = 3 independent experiments. *Significantly different from the vehicle control (Student’s t test, p < .05). ns; no significant difference
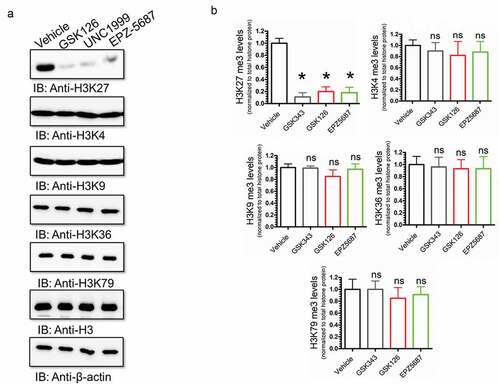
Figure 5. Immunocytochemistry of THP-1 cells for H3K27 methylation following treatment with GSK126, UNC1999, and EPZ-5687. THP-1 cells fixed and stained with anti-H3K27 (red channel) and nuclear DAPI staining (blue channel) to examine methylation levels of H3 at lysine residue number 27 following vehicle control (DMSO) and 1 μM of GSK126, UNC1999, and EPZ-5687 (a). For each independent plate of cells, five random visual fields were acquired from each whole-well scan, and cells in eight images were quantified from each field. Scale bars = 30 μm. (b) the total cellular immunofluorescence was calculated and then normalized to the DAPI nuclear stain. Values represent means ± S.E.M. for n = 3 independent experiments. *Significantly different from the vehicle control (Student’s t test, p < .05)
H3K27 methylation is attenuated by GSK 126, UNC 1999, and EPZ-5687 in a time-dependent manner
To evaluate the efficacy of GSK 126, UNC 1999, and EPZ-5687 in suppressing histone 3 methylation at Lysine 27 residue, THP-1 cells were treated with 1 µM of each compound for 0, 30, 60, 90, and 120 hours. Western blot analysis showed a gradual decrease of lysine 27 methylation of histone 3 over time increase (). Total H3K27 methylation level (%) was quantified following treatment with GSK 126, UNC 1999, and EPZ-5687 at all selected time points ().
Figure 6. GSK126, UNC1999, and EPZ-5687 attenuated H3K27 methylation in a time-dependent manner in THP-1 cells. (a) western blot analysis of H3K27 methylation in THP-1 cells treated with 1 µM of GSK126, UNC1999, and EPZ-5687. Methylation signal was analyzed in a time course [0–120 hours] and normalized to total H3 protein. Total methylation was calculated as (%) and quantified by scanning densitometry and normalized to total β-actin (loading control) (b). Values represent means ± S.E.M. for n = 3 independent experiments
![Figure 6. GSK126, UNC1999, and EPZ-5687 attenuated H3K27 methylation in a time-dependent manner in THP-1 cells. (a) western blot analysis of H3K27 methylation in THP-1 cells treated with 1 µM of GSK126, UNC1999, and EPZ-5687. Methylation signal was analyzed in a time course [0–120 hours] and normalized to total H3 protein. Total methylation was calculated as (%) and quantified by scanning densitometry and normalized to total β-actin (loading control) (b). Values represent means ± S.E.M. for n = 3 independent experiments](/cms/asset/e655ee28-7fae-41d9-8e6e-1039591c1011/kcbt_a_1902913_f0006_b.gif)
Activation of pro-apoptotic protein markers in THP-1 cells following GSK 126, UNC 1999, and EPZ-5687 treatments
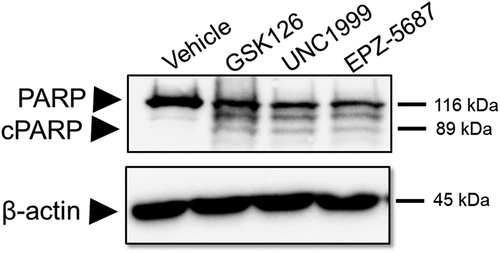
Previous reports showed the activation of pro-apoptotic (PARP-1) signals in different types of cancer cell lines following treatment with GSK 126Citation46,Citation47 and UNC1999.Citation48 We showed earlier that THP-1 cells exhibited cytotoxic effects when treated with different EZH2i. To confirm the cytotoxic activity of the EZH2i, we examined for the activation of pro-apoptotic signals upon treating THP-1 cells with these compounds. It was noted that exposure to 1 µM of GSK 126, UNC 1999, or EPZ-5687 induced THP-1 cell apoptosis via the activation of PARP-1 cleavage compared to vehicle treatment ().
Figure 7. GSK126, EPZ-5687, and UNC1999 induced cell death in THP-1 cells. western blot analysis of apoptotic markers; PARP and caspase proteins in THP-1 cells treated with 1 µM of GSK126, UNC1999, and EPZ-5687. PARP and cleaved PARP signals were detected using anti-PARP antibody, caspase 7 and cleaved form of caspase 7 were detected suing anti caspase 7 antibody. Bands were normalized to total β-actin (loading control). N = 3 independent experiments
Suppression of EZH2 protein phosphorylation (T350) in THP-1 cells following GSK126, UNC 1999, and EPZ-5687 treatment
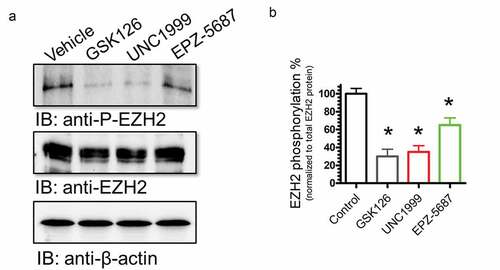
The activity of EZH2 is regulated by key phosphorylation events that have been linked to modulation of its enzyme activity. Previous studies indicated that the catalytic activity of EZH2 was regulated by phosphorylation at threonine-350 (T350) that has been linked to an increase in EZH2 activity. We therefore examined the phosphorylation status of EZH2 in THP-1 cells. Western blot analysis revealed that EZH2 is phosphorylated at T350 compared to vehicle control; however, this phosphorylation was significantly suppressed upon treatment of TPH-1 cells with EZH2i; GSK126, UNC1999, and EPZ-5687 (). These data suggest that EZH2 phosphorylation at T350 is a key regulatory mechanism of EZH2 function.
Figure 8. Phosphorylation of EZH2 was suppressed by GSK126, EPZ-5687, and UNC1999 in THP-1 cells. Western blot analysis of EZH phosphorylation levels in THP-1 cells treated with 1 µM of GSK126, UNC1999, and EPZ-5687. Phosphorylation signal was normalized to total EZH2 protein level. Bands were detected by scanning densitometry and normalized to total β-actin (loading control). N = 3 independent experiments
EZH2 inhibitors affect the key regulatory cancer signaling pathways PI3K/AKT and MAPK/ERK in a p53-independent manner
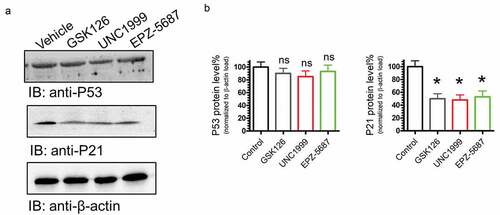
The PI3K/AKT and ERK/MAPK signaling pathways are key regulatory mechanisms of various cellular processes involved in cell growth, proliferation, differentiation, migration, and apoptosis.Citation49,Citation50 To test if treating THP-1 cells with EZH2i, GSK126, UNC1999, and EPZ-5687 would influence PI3K/AKT and MAPK/ERK pathways, inhibitor treated TPH-1 cells were tested for alterations in AKT and ERK1/2 phosphorylation. Western blot analyses showed that phosphorylation of AKT and ERK 1/2 was reduced following treatment with GSK126, UNC1999, and EPZ-5687(). THP-1 cells are characterized by a deletion mutation at codon174 of the p53 coding sequence.Citation51 We have profiled the changes of p53 and p21 proteins following the application of EZH2i to THP-1 cells (). Remarkably, our data showed that EZH2i did not alter p53 protein levels that might be attributed to mutational status of p53. However, p21 protein levels were increased after treating THP-1 cells with the EZH2i (), our data imply that the efficacy of EZH2 activity is not affected by the p53 status of the cells.
Figure 9. GSK126, EPZ-5687, and UNC1999 attenuated cell proliferation signals in THP-1 cells. Western blot analysis of cell proliferation protein signals, FLT-3, ERK 1/2, and Akt proteins. Phosphorylation levels were detected in THP-I cells treated with 1 µM of GSK126, UNC1999, and EPZ-5687. Phosphorylation signals were normalized to total protein levels. Bands were detected by scanning densitometry and normalized to total β-actin (loading control). N = 3 independent experiments
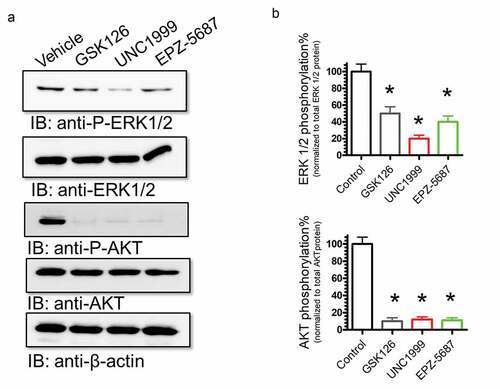
Figure 10. GSK126, EPZ-5687, and UNC1999 reduced P21 protein levels, but not P53. Western blot analysis of EZH phosphorylation levels in THP-1 cells treated with 1 µM of GSK126, UNC1999, and EPZ-5687. Protein signals for P53, P27, and P21 were detected and normalized to total β-actin (loading control)
Combination of EZH2 inhibitors and selinexor resulted in upregulation of cellular apoptotic markers
Drug combination is a highly advantageous therapeutic strategy which helps to increase cellular sensitivity to certain drugs while minimizing the generation of resistant clones by alternative pathways for survival.Citation52,Citation53 For this purpose, we studied the effects of drug combination of EZH2i with selinexor, a drug which has been known to shut down the nuclear export machinery by inhibiting the nuclear export protein XPO1.Citation54 This compound has been evaluated in multiple preclinical and clinical trials for patients with non-Hodgkin lymphoma, Citation55 bone and soft tissue sarcoma, Citation56 multiple myeloma,Citation57 and AML.Citation58 Firstly, we performed a cytotoxicity assays to evaluate the efficacy of selinexor on THP-1 cells as a single agent. Our data show that selinexor has a very potent IC50, 900 nM with minimal toxicity in control cells (hTERT) (). Then, we combined the IC50 concentration of each EZH2i with IC25 of selinexor to investigate if the combination of EZH2i and selinexor could increase the cytotoxic effects and induce apoptosis markers in treated cells. Western blot analysis revealed that combination of selinexor with either GSK126, UNC1999, or EPZ-5687 increased the cleavage signals of the pro-caspases 3,7, and 9 () which is potentially associated with elevated levels of cancer cell apoptosis.
Figure 11. Combining selinexor with GSK126, EPZ-5687, and UNC1999 increased cell apoptosis signals in THP-1 cells. (a) THP-1 cells or hTERT cells (control) were treated with increasing concentration [1 × 10−Citation4 to 100, μM] of selinexor for 96 hours. (b) Western blot analysis of caspase3, caspase7, and caspase 9 in THP-1 cells treated with 1 µM of GSK126, EPZ-5687, and UNC1999 in the absence or presence of selinexor. Levels of caspase3, 7, and 9 and their cleaved forms were detected and normalized to total β-actin protein (loading control)
![Figure 11. Combining selinexor with GSK126, EPZ-5687, and UNC1999 increased cell apoptosis signals in THP-1 cells. (a) THP-1 cells or hTERT cells (control) were treated with increasing concentration [1 × 10−Citation4 to 100, μM] of selinexor for 96 hours. (b) Western blot analysis of caspase3, caspase7, and caspase 9 in THP-1 cells treated with 1 µM of GSK126, EPZ-5687, and UNC1999 in the absence or presence of selinexor. Levels of caspase3, 7, and 9 and their cleaved forms were detected and normalized to total β-actin protein (loading control)](/cms/asset/7b38abe9-217a-40fc-bcbf-06c4f2bcbf67/kcbt_a_1902913_f0011_oc.jpg)
DISCUSSION
Molecular aberrations such as overexpression and gain-of-function mutations of EZH2 have been linked extensively to cancer. Elevated levels of EZH2 have been observed in various types of malignancies including breast cancer, bladder cancer, endometrial cancer, and melanoma.Citation59 In addition, EZH2 point mutations have been identified in non-Hodgkin lymphomas (NHL).Citation60 An increased catalytic activity of EZH2 has been shown to lead to hyper-methylation of lysine 27 on histone 3 (H3K27) which promotes global cellular modulation of gene expression and potentially assists cancer cell proliferation through regulation of essential signaling machineries such as cell cycle progression, proliferation, and apoptosis.Citation59 This histone methylation can lead to upregulation of cellular oncogenic signals and down-regulation of tumor suppressor genes.Citation61 Accumulative evidence suggests that EZH2 is a key player in the development and progression of various cancers and has been associated with poor prognosis of the disease.Citation62 This critical role(s) of EZH2 in cancer biology has intensified the development of small molecules to effectively target its enzymatic activity. In the recent past, a number of EZH2 small-molecule inhibitors have been generated and investigated for activity in different cancer models. Effectiveness of these compounds have been noted to vary depending on factors such as the EZH status (mutation and/or protein expression), type of the tumor, and the status of other disease markers.Citation63 Targeting EZH2 by small-molecule inhibitors has also been thought to be an attractive strategy in new therapeutics development for refractory pediatric tumors. In this study, we examined the molecular status of EZH2 in three distinct pediatric leukemia populations using a panel of EZH2i.
EZH2 is highly dysregulated by genetic mutations (A677) as in lymphomas, Citation64 or by protein overexpression found in prostate and ovarian cancers.Citation63 In the present study, we profiled the mutational status of A677 residue of EZH2 using DNA sequencing. Our data confirm a wild type EZH2 sequence in all of the three cell lines compared to the human database reference sequence. However, THP-1 cells showed high levels of EZH2 protein expression compared to MV4;11 and SEM cells with normalization to normal control. High levels of EZH2 protein suggest a possible role of EZH2 in THP-1 cancer cell signaling which makes EZH2 a promising druggable target in selected subtypes of AML. We have selected a panel of previously tested EZH2 small-molecule inhibitors to assess their efficacy in THP-1 cells. Our data revealed that GSK343 and EPZ-6438 are characterized by poor efficacy (IC50 > 10 µM) [] to decrease the methyltransferase activity in THP-1 cells. This might be attributed to the EZH2 status in these cells as it has been reported before that GSK343 and EPZ-6438 have pronounced activity against mutant and not wild-type EZH2 containing cells.Citation65 On the other hand, EI1, GSK503, and DZNep had anti-proliferative effects on THP-1 cells at very low micromolar ranges. However, these compounds displayed strong off-target effects on the control hTERT cells under similar conditions. Thus, these inhibitors may not be of significant potential to target in THP-1 like cells. We also noted that GSK126, UNC1999, and EPZ-5687 are very promising with very minimal effects on normal (hTERT) cells []. Our data suggest that screening of a panel of candidate compounds is important to identify highly active compounds and their desired concentrations in preclinical studies. Variable sensitivity of THP-1 cells to these compounds might be attributed to molecular status (wild type vs mutant) of EZH2 and the status of other essential components involved in tumorigenic activities of these cell lines. We have profiled the activity of EZH2 in THP-1 cells by detecting the methylation of lysine 27 on histone 3 (H3K27) following the application of our group of inhibitors. As a proof-of-concept, western blot data and immunostaining analyses showed that GSK126, UNC1999, and EPZ-5687 are selectively attenuated histone 3 methylation at K27, but not other putative lysine residues. Consistent with previous data described by Bradley and colleagues, Citation66 our data demonstrated that the kinetics of K27 demethylation by GSK126, UNC1999, and EPZ-5687 is slow and requires prolonged treatment (pronounced effects observed after 60 hours) with inhibitors to suppress EZH2 activity and to detect changes in gene expression profile.
Our investigations showed that inhibition of EZH2 was associated with a decrease in EZH2 phosphorylation at Threonine 350. It has been shown previously that the phosphorylation at this site by cyclin-dependent kinase 1 (CDK1) and cyclin-dependent kinase 2 (CDK2) is important to mediate EZH2 activity and to promote recruitment of chromatin.Citation67,Citation68 Our results indicated that phosphorylation of EZH2 at T350 was attenuated following the treatment of THP-1 cells with GSK126, UNC1999, and EPZ-5687. A possible scenario of T350 phosphorylation suppression following treatment with EZH2i might be caused by changes in the EZH2 protein conformation triggered by EZH2i that render this residue inaccessible by protein kinases. Alternatively, inhibition of EZH2 might cause alteration of CDK1 and CDK2 gene expression and consequently lessening their activity to suppress EZH2 at T350. Our results also show that the inhibition of EZH2 was associated with modulation of key signaling pathways involved in cell proliferation, survival and apoptosis. Western blot analyses revealed that EZH2-inhibited cells exhibited increased expression of apoptosis markers in addition to a remarkable reduction in AKT and ERK 1/2 phosphorylation. This indicates that the anti-cancer activity of EZH2i in THP-1 cells may be associated with a reduction in cell survival and proliferation by decreasing the activity of MAPK/ERK and PI3K/AKT pathways. P53 and p21 are tumor suppressors and they are important players in essential cellular functions such as DNA damage repair and cell cycle regulation. It has been reported previously that both genes are targets of aberrant DNA methylation which results in their inactivation in various types of cancers.Citation69–72 In this study, we profiled changes in p53 and p21 in THP-1 cells following exposure to EZH2i. Our data showed that no pronounced change in p53 protein levels was detectable following treatment with these drugs. Western blot analyses showed that p53 was insensitive to EZH2i in THP-1 cells. In agreements with a previous report that showed treating human cancer cell xenografts with DNA methylation inhibitors lead to a reduction in the growth of tumors harboring p53-mutantions not those with wild-type p53.Citation73 Previous reports also showed that the expression of p21/CDKN1A was increased following EZH2 depletion in human melanoma cells.Citation74 In this study, we found that p21 protein levels were decreased in a p53-independent manner. One major discrepancy between these two observations is that the molecular status of p53 of the cells studied; human melanoma cells have a wild type p53 status, but THP-1 cells are p53 mutant. Further analysis is required to examine the p53-dependent and the p53-independent regulatory pathways of p21 in cancer cells.
Selinexor, a first-generation selective inhibitor of nuclear export, is a promising anti-leukemic and pro-apoptotic compound which has been known to interfere with the nuclear export machinery by inhibiting the nuclear export protein XPO1.75 A previous study by Kojima and colleagues showed that an increase of XPO1 expression is significantly associated with a higher risk AML and poor overall survival (OS).Citation57 In addition, it has been shown that increased XPO1 expression in AML is highly associated with FMS-like tyrosine kinase 3 (FLT3) mutations, a key feature of cellular aberrations found in pediatric leukemias.Citation58 In this study, we evaluated the cytotoxic effects of combined treatment of EZH2i (GSK126, UNC1999, or EPZ-5687) with selinexor. We found that combining EZH2i with selinexor resulted in the upregulation of the pro-apoptotic caspases making this drug combination an attractive therapeutic approach.
Conclusion
In summary, the information presented here demonstrate the utility of examining a diverse panel of EZH2-selective small-molecule inhibitors using a cell line such as THP-1 that represents a distinct molecular subtype in pediatric leukemia. Based on these initial findings, future expanded investigations with a larger panel of leukemia cell lines with well-defined molecular aberrations will provide critical data for future targeted treatment strategies. We believe that with such studies and subsequent validation in tumor xenograft models will provide the preclinical data for the formulation of an early phase clinical trial for a challenging subgroup of pediatric leukemia patients in the future.
List of abbreviation
AML, hTERT, IC50, DMSO
Decleration
N/A
Ethics approval and consent to participate
N/A
Consent for publication
N/A
Author contributions
AAG collected the data, analyzed the data, and wrote the manuscript. AN conceived and coordinated the study. All authors reviewed the results and approved the final version of the manuscript.
Availability Of data and materials
Not applicable
Competing interests
The authors declare no potential conflict of interest
Additional information
Funding
References
- Madhusoodhan PP, Carroll WL, Bhatla T. Progress and prospects in pediatric leukemia. Curr Probl Pediatr Adolesc Health Care. 2016;46(7):229–241. doi:10.1016/j.cppeds.2016.04.003.
- Lonetti A, Pession A, Masetti R. Targeted therapies for pediatric AML: gaps and Perspective. Front Pediatr. 2019;7:463. doi:10.3389/fped.2019.00463.
- Van Eys J, Pullen J, Head D, Boyett J, Crist W, Falletta J, Humphrey GB, Jackson J, Riccardi V, Brock B, et al. The French-American-British (FAB) classification of leukemia. The pediatric oncology group experience with lymphocytic leukemia. Cancer. 1986;57(5):1046–1051. doi:10.1002/1097-0142(19860301)57:5<1046::AID-CNCR2820570529>3.0.CO;2-0.
- Tallman MS, Kim HT, Paietta E, Bennett JM, Dewald G, Cassileth PA, Wiernik PH, Rowe JM. Acute monocytic leukemia (French-American-British classification M5) does not have a worse prognosis than other subtypes of acute myeloid leukemia: a report from the Eastern cooperative oncology group. J Clin Oncol. 2004;22(7):1276–1286. doi:10.1200/JCO.2004.08.060.
- Liu LP, et al. Prognostic Stratification of Molecularly and Clinically Distinct Subgroup in Children with Acute Monocytic Leukemia. 2020;9(11):3647–3655.
- Tsuchiya S, Yamabe M, Yamaguchi Y, Kobayashi Y, Konno T, Tada K. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int J Cancer. 1980;26(2):171–176. doi:10.1002/ijc.2910260208.
- Odero MD, Zeleznik-Le NJ, Chinwalla V, Rowley JD. Cytogenetic and molecular analysis of the acute monocytic leukemia cell line THP-1 with an MLL-AF9 translocation. Genes Chromosomes Cancer. 2000;29(4):333–338. doi:10.1002/1098-2264(2000)9999:9999<::AID-GCC1040>3.0.CO;2-Z.
- Hu ZD, Wei TT, Tang QQ, Ma N, Wang LL, Qin BD, Yin JR, Zhou L, Zhong RQ. Gene expression profile of THP-1 cells treated with heat-killed Candida albicans. Ann Transl Med. 2016;4(9):170. doi:10.21037/atm.2016.05.03.
- Chanput W, Mes JJ, Wichers HJ. THP-1 cell line: an in vitro cell model for immune modulation approach. Int Immunopharmacol. 2014;23(1):37–45. doi:10.1016/j.intimp.2014.08.002.
- Suzuki S, Nakano H, Takahashi S. Epigenetic regulation of the metallothionein-1A promoter by PU.1 during differentiation of THP-1 cells. Biochem Biophys Res Commun. 2013;433(3):349–353. doi:10.1016/j.bbrc.2013.03.007.
- Davis FM, Gallagher KA. Epigenetic mechanisms in monocytes/macrophages regulate inflammation in cardiometabolic and vascular disease. Arterioscler Thromb Vasc Biol. 2019;39(4):623–634. doi:10.1161/ATVBAHA.118.312135.
- Hoeksema MA, De Winther MP. Epigenetic regulation of monocyte and macrophage function. Antioxid Redox Signal0. 2016;25(14):758–774. doi:10.1089/ars.2016.6695.
- Ciavatta DJ, et al. Epigenetic basis for aberrant upregulation of autoantigen genes in humans with ANCA vasculitis. J Clin Invest. 2010;120(9):3209–3219. doi:10.1172/JCI40034.
- Gan L, Yang Y, Li Q, Feng Y, Liu T, Guo W. Epigenetic Regulation of Cancer Progression by EZH2: From Biological Insights to Therapeutic Potential. 2018;6:10.
- Wang W, Qin JJ, Voruganti S, Nag S, Zhou J, Zhang R. Polycomb group (pcg) proteins and human cancers: multifaceted functions and therapeutic implications. Med Res Rev. 2015;35(6):1220–1267.
- Chan HL, Morey L. Emerging roles for polycomb-group proteins in stem cells and cancer. Trends Biochem Sci. 2019;44(8):688–700. doi:10.1016/j.tibs.2019.04.005.
- Pasini D, Di Croce L. Emerging roles for Polycomb proteins in cancer. Curr Opin Genet Dev. 2016;36:50–58. doi:10.1016/j.gde.2016.03.013.
- Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RGAB, Otte AP, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419(6907):624–629. doi:10.1038/nature01075.
- Raaphorst FM, Meijer CJLM, Fieret E, Blokzijl T, Mommers E, Buerger H, Packeisen J, Sewalt RAB, Ottet AP, Van Diest PJ, et al. Poorly differentiated breast carcinoma is associated with increased expression of the human polycomb group EZH2 gene. Neoplasia. 2003;5(6):481–488. doi:10.1016/S1476-5586(03)80032-5.
- Croonquist PA, Van Ness B. The polycomb group protein enhancer of zeste homolog 2 (EZH 2) is an oncogene that influences myeloma cell growth and the mutant ras phenotype. Oncogene. 2005;24(41):6269–6280. doi:10.1038/sj.onc.1208771.
- Zhang P, Xiao Z, Wang S, Zhang M, Wei Y, Hang Q, Kim J, Yao F, Rodriguez-Aguayo C, Ton BN, et al. ZRANB1 is an ezh2 deubiquitinase and a potential therapeutic target in breast cancer. Cell Rep. 2018;23(3):823–837. doi:10.1016/j.celrep.2018.03.078.
- Wang J, Cheng P, Pavlyukov MS, Yu H, Zhang Z, Kim S-H, Minata M, Mohyeldin A, Xie W, Chen D, et al. Targeting NEK2 attenuates glioblastoma growth and radioresistance by destabilizing histone methyltransferase EZH2. J Clin Invest. 2017;127(8):3075–3089. doi:10.1172/JCI89092.
- Yang PM, Hong YH, Hsu KC, Liu TP. p38α/S1P/SREBP2 activation by the SAM-competitive EZH2 inhibitor GSK343 limits its anticancer activity but creates a druggable vulnerability in hepatocellular carcinoma. Am J Cancer Res. 2019;9(10):2120–2139.
- Huang K, Sun R, Chen J, Yang Q, Wang Y, Zhang Y, Xie K, Zhang T, Li R, Zhao Q, et al. A novel EZH2 inhibitor induces synthetic lethality and apoptosis in PBRM1-deficient cancer cells. Cell Cycle. 2020;19(7):758–771. doi:10.1080/15384101.2020.1729450.
- Yomtoubian S, Lee SB, Verma A, Izzo F, Markowitz G, Choi H, Cerchietti L, Vahdat L, Brown KA, Andreopoulou E, et al. Inhibition of EZH2 catalytic activity selectively targets a metastatic subpopulation in triple-negative breast cancer. Cell Rep. 2020;30(3):755–770.e6. doi:10.1016/j.celrep.2019.12.056.
- Jin X, Yang C, Fan P, Xiao J, Zhang W, Zhan S, Liu T, Wang D, Wu H. CDK5/FBW7-dependent ubiquitination and degradation of EZH2 inhibits pancreatic cancer cell migration and invasion. J Biol Chem. 2017;292(15):6269–6280. doi:10.1074/jbc.M116.764407.
- Wang Q, Chen X, Jiang Y, Liu S, Liu H, Sun X, Zhang H, Liu Z, Tao Y, Li C, et al. Elevating H3K27me3 level sensitizes colorectal cancer to oxaliplatin. J Mol Cell Biol. 2020;12(2):125–137. doi:10.1093/jmcb/mjz032.
- Ciarapica R, Miele L, Giordano A, Locatelli F, Rota R. Enhancer of zeste homolog 2 (EZH2) in pediatric soft tissue sarcomas: first implications. BMC Med. 2011;9(1):63. doi:10.1186/1741-7015-9-63.
- Venneti S, Garimella MT, Sullivan LM, Martinez D, Huse JT, Heguy A, Santi M, Thompson CB, Judkins AR. Evaluation of histone 3 lysine 27 trimethylation (H3K27me3) and enhancer of zest 2 (EZH2) in pediatric glial and glioneuronal tumors shows decreased H3K27me3 in H3F3A K27M mutant glioblastomas. Brain Pathol. 2013;23(5):558–564. doi:10.1111/bpa.12042.
- Basheer F, Giotopoulos G. Contrasting Requirements during Disease Evolution Identify EZH2 as a Therapeutic Target in AML. J Exp Med. 2019;2164:966–981.
- Mechaal A, Menif S, Abbes S, Safra I. EZH2, new diagnosis and prognosis marker in acute myeloid leukemia patients. Adv Med Sci. 2019;64(2):395–401. doi:10.1016/j.advms.2019.07.002.
- Al-Ghabkari A, Narendran A. In vitro characterization of a potent p53-MDM2 inhibitor, RG7112 in neuroblastoma cancer cell lines. Cancer Biother Radiopharm. 2019;34(4):252–257. doi:10.1089/cbr.2018.2732.
- Al-Ghabkari A, Deng JT, McDonald PC, Dedhar S, Alshehri M, Walsh MP, MacDonald JA. A novel inhibitory effect of oxazol-5-one compounds on ROCKII signaling in human coronary artery vascular smooth muscle cells. Sci Rep. 2016;6(1):32118. doi:10.1038/srep32118.
- Huet S, Xerri L, Tesson B, Mareschal S, Taix S, Mescam-Mancini L, Sohier E, Carrère M, Lazarovici J, Casasnovas O, et al. EZH2 alterations in follicular lymphoma: biological and clinical correlations. Blood Cancer J. 2017;7(4):e555. doi:10.1038/bcj.2017.32.
- Akpa CA, Kleo K, Lenze D, Oker E, Dimitrova L, Hummel M, Bertolini F. DZNep-mediated apoptosis in B-cell lymphoma is independent of the lymphoma type, EZH2 mutation status and MYC, BCL2 or BCL6 translocations. PLoS One. 2019;14(8):e0220681. doi:10.1371/journal.pone.0220681.
- Yu T, Wang Y, Hu Q, Wu W, Wu Y, Wei W, Han D, You Y, Lin N, Liu N, et al. The EZH2 inhibitor GSK343 suppresses cancer stem-like phenotypes and reverses mesenchymal transition in glioma cells. Oncotarget. 2017;8(58):98348–98359. doi:10.18632/oncotarget.21311.
- Girard N, Bazille C, Lhuissier E, Benateau H, Llombart-Bosch A, Boumediene K, Bauge C, Alonso MM. 3-deazaneplanocin A (DZNep), an inhibitor of the histone methyltransferase EZH2, induces apoptosis and reduces cell migration in chondrosarcoma cells. PLoS One. 2014;9(5):e98176. doi:10.1371/journal.pone.0098176.
- Huang S, Wang Z, Zhou J, Huang J, Zhou L, Luo J, Wan YY, Long H, Zhu B. EZH2 inhibitor GSK126 suppresses anti-tumor immunity by driving production of myeloid-derived suppressor cells. Cancer Res. 2019;79(8):2009–2020.
- Qi W, Chan H, Teng L, Li L, Chuai S, Zhang R, Zeng J, Li M, Fan H, Lin Y, et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci U S A. 2012;109(52):21360–21365. doi:10.1073/pnas.1210371110.
- Lindsay CD, Kostiuk MA, Harris J, O’Connell DA, Seikaly H, Biron VL. Efficacy of EZH2 Inhibitory Drugs in Human Papillomavirus-positive and Human Papillomavirus-negative Oropharyngeal Squamous Cell Carcinomas. 2017;9:95.
- Konze KD, Ma A, Li F, Barsyte-Lovejoy D, Parton T, MacNevin CJ, Liu F, Gao C, Huang X-P, Kuznetsova E, et al. An orally bioavailable chemical probe of the lysine methyltransferases EZH2 and EZH1. ACS Chem Biol. 2013;8(6):1324–1334. doi:10.1021/cb400133j.
- Knutson SK, Kawano S, Minoshima Y, Warholic NM, Huang K-C, Xiao Y, Kadowaki T, Uesugi M, Kuznetsov G, Kumar N, et al. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-hodgkin lymphoma. Mol Cancer Ther. 2014;13(4):842–854. doi:10.1158/1535-7163.MCT-13-0773.
- Zingg D, Debbache J, Schaefer SM, Tuncer E, Frommel SC, Cheng P, Arenas-Ramirez N, Haeusel J, Zhang Y, Bonalli M, et al. The epigenetic modifier EZH2 controls melanoma growth and metastasis through silencing of distinct tumour suppressors. Nat Commun. 2015;6(1):6051. doi:10.1038/ncomms7051.
- Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012;13(5):343–357. doi:10.1038/nrg3173.
- Zeng D, Liu M, Pan J. Blocking EZH2 methylation transferase activity by GSK126 decreases stem cell-like myeloma cells. Oncotarget. 2017;8(2):3396–3411. doi:10.18632/oncotarget.13773.
- Adhikary G, Grun D, Balasubramanian S, Kerr C, Huang JM, Eckert RL. Survival of skin cancer stem cells requires the Ezh2 polycomb group protein. Carcinogenesis. 2015;36(7):800–810. doi:10.1093/carcin/bgv064.
- Grinshtein N, Rioseco CC, Marcellus R, Uehling D, Aman A, Lun X, Muto O, Podmore L, Lever J, Shen Y, et al. Small molecule epigenetic screen identifies novel EZH2 and HDAC inhibitors that target glioblastoma brain tumor-initiating cells. Oncotarget. 2016;7(37):59360–59376. doi:10.18632/oncotarget.10661.
- Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26(22):3279–3290. doi:10.1038/sj.onc.1210421.
- Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR signaling in cancer. Front Oncol. 2014;4:64. doi:10.3389/fonc.2014.00064.
- Sugimoto K, Toyoshima H, Sakai R, Miyagawa K, Hagiwara K, Ishikawa F, Takaku F, Yazaki Y, Hirai H. Frequent mutations in the p53 gene in human myeloid leukemia cell lines. Blood. 1992;79(9):2378–2383. doi:10.1182/blood.V79.9.2378.2378.
- Yardley DA. Drug resistance and the role of combination chemotherapy in improving patient outcomes. Int J Breast Cancer. 2013;2013:137414. doi:10.1155/2013/137414.
- Al-Ghabkari A, Perinpanayagam MA, Narendran A. Inhibition of PI3K/mTOR pathways with GDC-0980 in pediatric leukemia: impact on abnormal FLT-3 activity and cooperation with intracellular signaling targets. Curr Cancer Drug Targets. 2019;19(10):828–837. doi:10.2174/1568009619666190326120833.
- Walker CJ, Oaks JJ, Santhanam R, Neviani P, Harb JG, Ferenchak G, Ellis JJ, Landesman Y, Eisfeld A-K, Gabrail NY, et al. Preclinical and clinical efficacy of XPO1/CRM1 inhibition by the karyopherin inhibitor KPT-330 in Ph+ leukemias. Blood. 2013;122(17):3034–3044. doi:10.1182/blood-2013-04-495374.
- Kuruvilla J, Savona M, Baz R, Mau-Sorensen PM, Gabrail N, Garzon R, Stone R, Wang M, Savoie L, Martin P, et al. Selective inhibition of nuclear export with selinexor in patients with non-Hodgkin lymphoma. Blood. 2017;129(24):3175–3183. doi:10.1182/blood-2016-11-750174.
- Gounder MM, et al. Phase IB study of selinexor, a first-in-class inhibitor of nuclear export, in patients with advanced refractory bone or soft tissue sarcoma. J Clin Oncol. 2016;34(26):3166–3174. doi:10.1200/JCO.2016.67.6346.
- Bahlis NJ, Sutherland H, White D, Sebag M, Lentzsch S, Kotb R, Venner CP, Gasparetto C, Del Col A, Neri P, et al. Selinexor plus low-dose bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma. Blood. 2018;132(24):2546–2554. doi:10.1182/blood-2018-06-858852.
- Wang AY, et al. A phase I study of selinexor in combination with high-dose cytarabine and mitoxantrone for remission induction in patients with acute myeloid leukemia. J Hematol Oncol. 2018;11(1):4. doi:10.1186/s13045-017-0550-8.
- Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med. 2016;22(2):128–134. doi:10.1038/nm.4036.
- McCabe MT, et al. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc Natl Acad Sci U S A. 2012;109(8):2989–2994. doi:10.1073/pnas.1116418109.
- Christofides A, Karantanos T, Bardhan K, Boussiotis VA. Epigenetic regulation of cancer biology and anti-tumor immunity by EZH2. Oncotarget. 2016;7(51):85624–85640.
- Tabbal H, Septier A, Mathieu M, Drelon C, Rodriguez S, Djari C, Batisse-Lignier M, Tauveron I, Pointud J-C, Sahut-Barnola I, et al. EZH2 cooperates with E2F1 to stimulate expression of genes involved in adrenocortical carcinoma aggressiveness. Br J Cancer. 2019;121(5):384–394. doi:10.1038/s41416-019-0538-y.
- Papale M, Ferretti E, Battaglia G, Bellavia D, Mai A, Tafani M. EZH2, HIF-1, and their inhibitors: an overview on pediatric cancers. Front Pediatr. 2018;6:328. doi:10.3389/fped.2018.00328.
- Song X, Zhang L, Gao T, Ye T, Zhu Y, Lei Q, Feng Q, He B, Deng H, Yu L, et al. Selective inhibition of EZH2 by ZLD10A blocks H3K27 methylation and kills mutant lymphoma cells proliferation. Biomed Pharmacother. 2016;81:288–294. doi:10.1016/j.biopha.2016.04.019.
- Villanueva MT. Anticancer drugs: all roads lead to EZH2 inhibition. Nat Rev Drug Discov. 2017;16(4):239.
- Bradley WD, Arora S, Busby J, Balasubramanian S, Gehling V, Nasveschuk C, Vaswani R, Yuan -C-C, Hatton C, Zhao F, et al. EZH2 inhibitor efficacy in non-Hodgkin’s lymphoma does not require suppression of H3K27 monomethylation. Chem Biol. 2014;21(11):1463–1475. doi:10.1016/j.chembiol.2014.09.017.
- Ozes AR, Pulliam N, Ertosun MG, Yilmaz O, Tang J, Copuroglu E, Matei D, Ozes ON, Nephew KP. Protein kinase A-mediated phosphorylation regulates STAT3 activation and oncogenic EZH2 activity. Oncogene. 2018;37(26):3589–3600. doi:10.1038/s41388-018-0218-z.
- Chen S, Bohrer LR, Rai AN, Pan Y, Gan L, Zhou X, Bagchi A, Simon JA, Huang H. Cyclin-dependent kinases regulate epigenetic gene silencing through phosphorylation of EZH2. Nat Cell Biol. 2010;12(11):1108–1114. doi:10.1038/ncb2116.
- Bott SR, Arya M, Kirby RS, Williamson M. p21WAF1/CIP1 gene is inactivated in metastatic prostatic cancer cell lines by promoter methylation. Prostate Cancer Prostatic Dis. 2005;8(4):321–326. doi:10.1038/sj.pcan.4500822.
- Askari M, Sobti RC, Nikbakht M, Sharma SC. Aberrant promoter hypermethylation of p21 (WAF1/CIP1) gene and its impact on expression and role of polymorphism in the risk of breast cancer. Mol Cell Biochem. 2013;382(1–2):19–26. doi:10.1007/s11010-013-1696-5.
- Zhu J, Dou Z, Sammons MA, Levine AJ, Berger SL. Lysine methylation represses p53 activity in teratocarcinoma cancer cells. Proc Natl Acad Sci U S A. 2016;113(35):9822–9827. doi:10.1073/pnas.1610387113.
- Ito T, Nishida N, Fukuda Y, Nishimura T, Komeda T, Nakao K. Alteration of the p14(ARF) gene and p53 status in human hepatocellular carcinomas. J Gastroenterol. 2004;39(4):355–361. doi:10.1007/s00535-003-1302-9.
- Yi L, Sun Y, Levine A. Selected drugs that inhibit DNA methylation can preferentially kill p53 deficient cells. Oncotarget. 2014;5(19):8924–8936. doi:10.18632/oncotarget.2441.
- Fan T, Jiang S, Chung N, Alikhan A, Ni C, Lee CC, Hornyak TJ. EZH2-dependent suppression of a cellular senescence phenotype in melanoma cells by inhibition of p21/CDKN1A expression. Mol Cancer Res. 2011;9(4):418–429. doi:10.1158/1541-7786.MCR-10-0511.
- Talati C, Sweet KL. Nuclear transport inhibition in acute myeloid leukemia: recent advances and future perspectives. Int J Hematol Oncol. 2018;7(3):Ijh04. doi:10.2217/ijh-2018-0001.