
ABSTRACT
Bladder cancer (BC), a main neoplasm of urinary tract, is usually inoperable and unresponsive to chemotherapy. As a novel experimental model for muscle-invasive BC, we previously established a culture method of dog BC organoids. In the present study, the detailed in vitro and in vivo anti‐tumor effects of trametinib were investigated by using this model. In each BC organoid strain, epidermal growth factor receptor (EGFR)/ERK signaling was upregulated compared with normal bladder cells. Trametinib even at a low concentration inhibited the cell viability of BC organoids and the activation of ERK through decreasing expression of c-Myc, ELK1, SIK1, and PLA2G4A. Trametinib arrested cell cycle of BC with few apoptosis. Dual treatment of BC organoids with trametinib and YAP inhibitor, verteporfin extremely inhibited the cell viability with apoptosis induction. Moreover, trametinib induced basal to luminal differentiation of BC organoids by upregulating luminal markers and downregulating basal ones. In vivo, trametinib decreased the tumor growth of BC organoids in mice and the xenograft-derived organoids from trametinib-administered mice showed enhanced sensitivity to carboplatin due to MSH2 upregulation. Our data suggested a new strategy of trametinib-YAP inhibitor or trametinib-carboplatin combination as a promising treatment of BC.
Introduction
Bladder cancer (BC), a carcinoma of bladder, is the most common and complex neoplasm of the urinary tract and is associated with high morbidity and mortality.Citation1 The BC in human patients is mostly non-muscle invasive (NMI) type with a positive prognosis, while to a lesser degree it is muscle-invasive (MI) with a worse prognosis. The MIBC is mainly of basal type, high intricacy, stemness, epithelial-mesenchymal transition (EMT), often metastatic, and difficult to treat.Citation2 Contrarily, the NMIBC is mainly of luminal papillary type and less complex than MIBC.Citation1 Although it is the fifth most common neoplasm in the US, the precise experimental culture models that mimic the biology of the disease are few.
In dogs, BC constitutes about 2% of all naturally occurring cancers, a similar rate to that found in humans.Citation3 Besides, it mostly resembles human MIBC in histopathology and gene profiles and then could be a valuable research model for this disease.Citation4 In the previous study, we established a novel experimental organoid model of MIBC from BC diseased dogs using their urine samples.Citation4 In this model, the MIBC characteristics were successfully recapitulated. Interestingly, this model could detect the difference of drug sensitivity for each patient to the sole and/or combined anti-cancer therapies.
The mitogen-activated protein kinase (MAPK) pathway, in which mitogen-activated extracellular signal-regulated kinase (MEK) enzyme is incorporated, is constitutively activated in many tumors where it promotes cell differentiation, proliferation, angiogenesis, and survival.Citation5 The activation of MEK1/2, in turn, phosphorylates ERK1/2, which activates multiple substrates and transcription factors.Citation6 The overexpression and/or mutation of growth factor receptors such as epidermal growth factor (EGF) receptor (EGFR), erbB2 receptor, and others have also been observed in many cancers.Citation7 Therefore, MEK has emerged as a promising anti-cancer therapeutic target.Citation8
Trametinib is a type III allosteric noncompetitive and highly selective MEK1/2 inhibitor.Citation9 It suppressed RAF-dependent MEK phosphorylation and extended the inhibition of phosphorylated ERK.Citation9 Trametinib has been approved by The United States Food and Drug Administration for treatment of several cancers that harbor BRAF V600E mutation including metastatic non-small cell lung cancer,Citation10 metastatic melanoma (combined with dabrafenib),Citation11 and locally advanced or metastatic anaplastic thyroid cancer.Citation12 Nevertheless, previous reports concerning the use of trametinib in BC therapy were scarce and not well investigated. Since our established dog BC organoid model reflected the most features of human MIBC, the current study was carried out to investigate the effects of trametinib on BC organoids.
Materials and methods
Ethics approval
Collection of urine samples, generation of bladder cancer (BC) organoids, and experiments with organoids were carried out under the direction of the Institute Animal Care and Use Committee of Tokyo University of Agriculture and Technology and approved by the ethical committee (Approval number: 0016012). Written informed consent for the present study was obtained from all dog owners. In total, five strains of BC organoids were used. Dog clinical information on each dog is listed in ().
Table 1. Sample information
Anti-cancer drugs and antibodies
Anti-cancer drugs used in the present study were as follows: trametinib (Cayman Chemical, Ann Arbor, USA); carboplatin (FUJIFILM WAKO Pure Chemical Corporation, Osaka, Japan); yes-associated protein (YAP)1 inhibitor, verteporfin (VP) (Sigma-Aldrich, St. Louis, USA), mitoxantrone; vinblastine (Cayman). The antibodies used were as follows: total EGFR; phosphor-EGFR; phospho-ERK; total-ERK; total-YAP1 (Cell Signaling Technology, Inc., Danvers, MA, USA); CK5; total-VCP (GeneTex, Inc., Irvine, CA, USA); total-cyclin D1; MSH2 (Bioss, Inc., Woburn, MA, USA); total-CD44 (Bethyl Laboratories, Montgomery, TX, USA). Fluorescent secondary antibodies used were as follows: Alexa Fluor 488™ goat anti-rabbit IgG; Alexa Fluor 488™ goat anti-mouse IgG; (Thermo Fisher Scientific Inc. Waltham, MA, USA). Horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (Cayman); and HRP-conjugated anti-mouse IgG (Millipore, Temecula, CA, USA).
Organoid culture
Culture medium, supplements, conditions, handlings, and passages of the BC organoids were described before in our previous studies.Citation4 Briefly, the culture medium was composed of Advanced DMEM with 50% Wnt, Noggin, and R-spondin conditioned medium; 1% GlutaMax; 100 ug mL−1 Primocin (Thermo Fisher Scientific); 1 mM N-Acetyl-L-cysteine; 10 mM nicotinamide (Sigma-Aldrich); 50 ng mL−1 mouse EGF (PeproTech, Rocky Hill, NJ, USA); and 500 nM A83-01(Adooq Bioscience, Irvine, CA, USA).
Western blotting
To check the expression and activation level of EGF signal-related proteins, Western blot analysis was performed according to the standard procedures as described previously.Citation13 Equal amounts of protein (10 μg) were loaded and separated by SDS-PAGE (7.5%) and transferred to nitrocellulose membrane (Wako). After blocking, the membranes were incubated with primary antibody (total-EGFR; 1:500, phospho-EGFR; 1:200, total-ERK; 1:500; phospho-ERK; 1:500; total-VCP; 1:500) at 4°C overnight followed by secondary antibody (1:500 for 1 h) of goat anti-rabbit and ECL Prime (GE Healthcare, Pittsburgh, PA, USA). Images were captured with a LAS-3000 image analyzer (Fuji Film, Tokyo, Japan) and quantified by ImageJ software.
Histology (H&E staining)
Sections prepared from trametinib- or vehicle-treated BC organoids or their xenograft-derived tumors were prepared and stained with hematoxylin and eosin (H&E) as described before.Citation4 Briefly, the organoids or tumor tissues were fixed at room temperature in 4% paraformaldehyde (PFA)/phosphate-buffered saline (PBS) for 2‐3 h and then embedded in paraffin. Preparation of sections and staining procedures were performed according to the standard procedures. Images were captured by using a light microscope (BX‐52). Quantification of basal to luminal differentiation of organoids was carried out using ImageJ software (National Institutes of Health).
Cell viability assay of BC organoids
Cell viability assay of BC organoids was carried out as described before.Citation4,Citation14,Citation15 The appropriate concentrations for trametinib, VP, mitoxantrone, vinblastine, and carboplatin were determined based on their blood therapeutic level in the clinic and pharmacokinetic data.Citation9,Citation16,Citation17 Concisely, after the organoids were washed by PBS and treated with 5 mM EDTA/PBS on ice for 90 min, they were trypsinized using TrypLE (Thermo Fisher Scientific Inc.) for 5 min at 37°C. After pipetting, the organoid cells were passed through cell strainer (70 µm), neutralized by fetal bovine serum (Sigma-Aldrich), and counted. Approximately 2 × 103 cells of each BC organoid were embedded in 10 μl Matrigel with culture media in a 96-well culture plate and incubated for 24 h. Later, the cells were treated with DMSO or each drug (trametinib: 0.03, 0.1, 0.3, and 1 µM), VP: 0.3 µM, carboplatin: 0.1, 1, 10, and 100 µg ml−1, mitoxantrone: 0.1, 1, 10, and 100 ng ml−1 or vinblastine: 0.01, 0.1, 1, and 10 nM) for 72 h. Cell viability was examined using PrestoBlue kit (Thermo Fisher Scientific Inc.) by a microplate reader (TECAN, Seestrasse, Switzerland) at an emission wavelength of 585 nm. Bright-field images were captured using the light microscope (BX-52; Olympus, Japan) and the readings of the microplate reader were graphed using Sigma Plot software (Systat Software, Inc. San Jose, CA. USA).
Cell cycle analysis by flow cytometry
Cell cycle analysis was carried out to identify cell populations in various phases of the cell cycle after trametinib treatment. For this purpose, BC organoid cells (2 × 105 per well) were seeded in 24-well plates. The organoid cells were treated with DMSO or trametinib for 24 h. Thereafter, the cells were harvested, trypsinized, washed by PBS, fixed with 70% ethanol, and kept at −20°C for 20 min. Subsequently, the cells were treated with Rnase (200 μg ml−1, Sigma-Aldrich) for 30 min at room temperature and stained with propidium iodide (50 µg ml−1, Sigma-Aldrich) for 30 min at room temperature in the dark. The cells were then gently dissociated and passed through a 200 µm nylon mesh before data acquisition using flow cytometry (Guava easyCyte, Millipore) and the phase distributions were quantified using the attached software.
Quantitative real-time PCR
After extracting total RNA from control and trametinib-treated organoid cells using NucleoSpin kit (Takara Bio Inc., Shiga, Japan), the first‐strand cDNA was prepared using QuantiTect Reverse Transcription Kit (QIAGEN, Hilden, Germany) following the instructions of both manufacturers. Quantitative real‐time PCR was carried out using QuantiTect SYBR I Kit (QIAGEN) and a StepOnePlus Real-Time PCR System (Applied Biosystems, Waltham, MA, USA). The ΔΔCq method was applied for quantification and GAPDH was used as a control gene. Specific dog primers used for downstream genes of ERK signaling; c-Myc, ETS transcription factor, ELK1, SIK1, and PLA2G4A; cell cycle-related genes; cyclin D1, cyclin A2, cyclin E1, CDK4, CDK6, p21, and p16; stemness markers; CD44 and YAP1; basal cell markers, CK5 and DSG3; luminal cell markers, ERBB2 and GATA3; carboplatin sensitivity-related genes; MSH2, MLH1, and ERCC1 are listed in ().
Table 2. Primers for real-time quantitative PCR analysis
Immunofluorescence staining
Immunofluorescence staining of BC organoids and their xenograft sections were performed as described previously.Citation4,Citation14 After fixation of sections in 4% PFA for 1 h and dehydration with 30% sucrose solution at 4°C overnight, they were embedded in OCT compound. The frozen sections were made using Leica CM3050 S Research Cryostat (Leica Biosystem, Buffalo Grove, IL, USA). Sections were blocked with 1.5% normal goat serum (NGS)/PBS at room temperature for 1 h. Subsequently, they were incubated with primary antibodies (cyclin D1 1:200, CD44; 1:200, YAP1; 1:200, CK5; 1:200, MSH2; 1:200) and kept at 4°C overnight. Thereafter, the sections were washed 3 times with PBS and incubated with secondary antibody for 1 h. The images were captured by a confocal microscope (LSM 800; ZEISS, Copenhagen, Germany).
TUNEL staining
Detection of apoptosis in the frozen sections of BC organoids and their xenograft-derived tumor tissues was performed using the DeadEnd Fluorometric terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) system (Promega Co., Madison, USA) following the manufacturer’s instructions. BC organoid cells (1 × 105 cells per well) were seeded and cultured for 24 h and then treated with trametinib (1 µM) or DMSO for 72 h. Frozen sections were prepared from treated cells and kept at −80°C until apoptosis assay. The subsequent procedures for apoptosis assay were carried out following the manufacturer’s protocol. Concisely, after washing the slides with PBS for 5 min, they were fixed in 4% PFA for 15 min. Sections were then permeabilized with 2% proteinase K solution in PBS for 15 min. After PBS washing, sections were fixed again in 4% PFA for 5 min. The DNase I (0.5 U)-treated slide was prepared as a positive control. All sections were then treated with 100 ul of equilibration buffer for 10 min. After that section was treated with 100 ul of rTdT incubation buffer, covered by plastic coverslips, and incubated at 37°C inside a humidified chamber for 60 min, and the tailing reaction was then terminated by immersion of slides in 2X SCC solution for 15 min. Cell nuclei were counterstained with 3 µg mL−1 Hoechst (in PBS) for 5 min. The sections were sealed, and the fluorescein12-dUTP-labeled DNA was visualized by fluorescence imaging.
The images were captured by a confocal microscope (LSM 800; ZEISS).
Organoid xenograft studies
All experimental procedures were carried out following the recommendations of the ‘Guide for the Care and Use of Laboratory Animals’ and approved by ethics committees of Tokyo University of Agriculture and Technology (Approval number: 29–92). To study the in vivo anti-cancer effects of trametinib against xenograft BC organoids, BC organoid cells (1 × 106) were subcutaneously implanted into the back of male SCID (C.B-17/IcrHsd-Prkdcscid, Japan SLC) mice (n = 12). After forming a tumor of a certain size, the mice were randomly divided into two groups and trametinib or vehicle was intraperitoneally injected (1 mg kg−1, 6 days per week) for one month. Tumor dimensions were measured every week by caliber and their volumes (V) were estimated using V = 1/2 (L × W2), where L is the longest dimension (length) and W is the width (shortest dimension). The mean tumor volumes were recorded for each group. Six weeks post-injection of organoids, mice were anesthetized by isoflurane before cervical dislocation and tumors were isolated and weighed. The sections were prepared for H&E, TUNEL staining, and immunofluorescence staining.
Statistical analysis
Data shown are means ± SEM. Statistical evaluations were performed using a one-way analysis of variance (ANOVA) followed by Bonferroni’s test. P-values ≤0.05 were statistically significant.
Results
Effects of trametinib on cell viability and activation of cell signaling in dog BC organoids
In the previous study, we established the method of dog BC organoids and suggested that dog BC organoids become a new experimental model of human MIBC.Citation4 To clarify the molecular mechanisms in BC organoids and identify the effective molecular targeting drugs, we analyzed the activated signaling, histological characteristics, and drug sensitivity in BC organoids (). Since activation of EGF-regulated signaling was reported in several human MIBC,Citation18 we checked the expression and phosphorylation level of EGFR and ERK in dog BC organoids. As expected, expression and phosphorylation levels of EGFR and ERK in five strains of BC organoids were upregulated compared with normal bladder tissue cells ( and Figure S1). Staining of organoids with H&E showed the basal solid-like structure with squamous differentiation (), which corresponded to the typical histology of MIBC in human. We next checked the effect of a MEK inhibitor, trametinib on the cell viability of BC organoid cells. Interestingly, trametinib even at low concentration (0.03 μM) drastically inhibited the viability of organoid cells in all strains (). Thereafter, we checked the time-course effects of trametinib on activation of ERK and EGFR. As presented in (), trametinib completely inhibited phosphorylation of ERK in BC organoids even at 3 h after treatment and the effect continues to 24 h. However, there was no effect on phosphorylation of EGFR. We further checked the downstream signals of ERK cascade. Among the several transcriptional genes, trametinib significantly inhibited expression of c-Myc, ELK1, SIK1, and PLA2G4A ( and Figure S2). These findings indicate the potential role of ERK downstream signals in the growth and proliferation of BC and trametinib may become a new therapeutic agent against human MIBC.
Figure 1. Effects of trametinib on cell viability and activation of cell signaling in dog bladder cancer (BC) organoids. Experimental schema of analysis of BC organoids (a). After BC organoids were generated from urine samples of BC diseased dogs, they were used for the following experiments. Protein expression and activated level of EGF-related signals in BC organoids (b). Expression level of phosphorylation of EGFR, total EGFR, phosphorylation of ERK, and total ERK was compared between normal bladder epithelial cells (NB) and several strains of BC organoids (Or1-5) as determined by Western blotting (n = 3). Equal loading amounts of protein was confirmed by total VCP antibody. Histological analysis of BC organoids (c). Representative images of hematoxylin and eosin (H&E) staining of each BC organoid were shown. Scale bar: 100 μm. Representative phase-contrast images (scale bar: 500 μm) of the sensitivity of BC organoids to different concentrations of trametinib for 72 h using Prestoblue cell viability assay (d, n = 3–6) and its quantification (e). 100% represents cell viability of each control. Results were expressed as mean ± SEM. Effects of trametinib on activation of EGFR and ERK in BC organoids. After BC organoids were seeded into Matrigel, they were treated with trametinib for 3, 6, 12 and 24 hours. Expression level of phosphorylation of EGFR and ERK was determined by Western blotting (f, g, n = 4–5). Equal protein loading was confirmed using total actin antibody. Results were expressed as mean ± S.E.M. ﹡P < .05 vs. control. Effects of trametinib on expression of downstream signal of ERK in BC organoids. Expression of c-Myc, ETS transcription factor, ETS Like-1 protein (ELK1), Salt-Inducible Kinase 1 (SIK1), and phospholipase A2 group 4A(PLA2G4A) mRNA was determined by quantitative real-time PCR (h). Expression level of each gene was quantified based on the ration of expression level to GAPDH and shown as fold increase relative to control (n = 4). Results were expressed as mean ± S.E.M. ﹡P < .05 vs. control
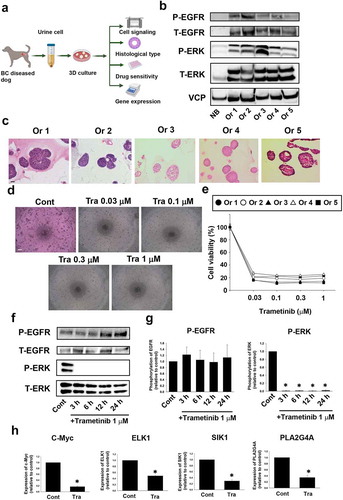
Effects of trametinib on cell cycle arrest and apoptosis in BC organoids
We next checked whether trametinib affects cell cycle in BC organoids. In the trametinib treatment of BC organoid cells, the ratio of G0/G1 phase fractions was significantly elevated, whereas the G2/M phase fractions were significantly declined (). To investigate the detailed mechanisms of cell arrest by trametinib in BC organoids, we examined the effects of trametinib on expression of cell cycle-related genes. After 24 h trametinib treatment, mRNA expression level of cell cycle-related genes was investigated. Among the several genes, cyclin D1, cyclin A2, cyclin E1, and CDK4 were significantly downregulated, whereas CDK6, p21, and p16 did not change (). Interestingly, cyclin D1 and cyclin E1 were downregulated in a time-dependent manner, whereas cyclin A2 and CDK4 were downregulated only after 24 h treatment (Figure S3). Further confirmation at protein expression level was carried out and the data revealed that cyclin D1 was significantly less expressed in the trametinib-treated organoids compared with the control (). Regarding apoptotic responses, trametinib treatment increased the ratio of TUNEL-positive cells, but the effect was minimal (). These data indicate that trametinib decreased the cell viability of BC organoids through cell arrest rather than apoptosis induction.
Figure 2. Effects of trametinib on cell cycle arrest and apoptosis in BC organoids. After BC cells were treated with trametinib (1 μM for 24 h), they were stained with propidium iodide (PI). Distribution of cell cycle phases (G0/G1, S, and G2/M) of BC organoid cells was analyzed by flow cytometry (a). The population of G0/G1, S and G2/M phases in BC organoids was expressed as mean ± S.E.M (n = 4). ﹡P < .05 vs. control. Effects of trametinib on expression of cell cycle-related genes in BC organoids (b). Expression of cyclin D1, cyclin A2, cyclin E1, CDK4, CDK6, p21, and p16 mRNA was determined by quantitative real-time PCR. Expression level of each gene was quantified based on the ration of expression level to GAPDH and shown as fold increase relative to control (n = 4). Results were expressed as mean ± S.E.M. ﹡P < .05 vs. control. Expression of cyclin D1 in trametinib-treated BC organoids. Representative photomicrographs were shown (n = 3). Scale bar: 50 μm (c). Expression level was quantified by counting the cyclin D1-positive cells (d, n = 3). Results were expressed as mean ± S.E.M. ﹡P < .05 vs. control. Effects of trametinib on apoptosis in BC organoids (e). After BC cells were treated with trametinib (1 μM for 72 h), they were stained with terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL). DNase-treated organoid cells were used as the positive control (n = 3). The green fluorescence area indicates apoptotic-positive organoids cells and the blue DAPI staining shows intact DNA. The white arrows indicate apoptotic-positive cells. Scale bar: 50 μm. Quantification of apoptosis in trametinib-treated and non-treated cells was analyzed by ImageJ software (F, n = 3). Results were expressed as mean ± S.E.M. ﹡P < .05 vs. control
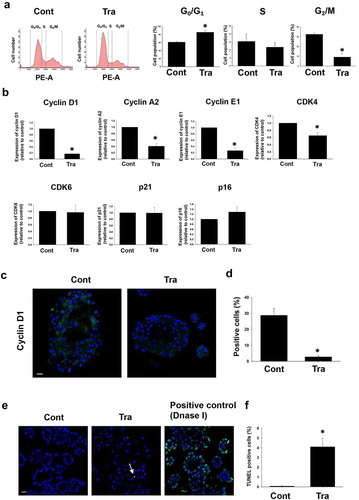
Effects of trametinib on stemness of BC organoids
Since cancer stem cell (CSC) markers are implicated in growth, invasion, angiogenesis, chemotherapy resistance, and metastasis of MIBC, the effect of trametinib on expression of two important markers, CD44 and YAP1 which were upregulated in most of MIBCCitation19 was investigated. Treatment of BC organoids with trametinib (1 µM) significantly decreased the expression level of CD44 mRNA at 12 and 24 h, while YAP1 expression was significantly upregulated in a time-dependent manner (). Further confirmation of these findings at the protein expression level was undertaken using immunofluorescence staining with CD44 and YAP1 antibodies (). Considering these data, we hypothesized that a combination treatment of trametinib and YAP inhibitor more efficiently decrease the cell viability of BC organoids. To prove this hypothesis, the synergistic effect of YAP inhibitor, verteporfin (VP) (0.3 µM) with trametinib at low concentration (0.03 µM), on the viability of BC organoids was assessed. As expected, trametinib plus VP inhibited to a greater extent the cell viability of BC organoids and the effect was significantly higher compared with the sole treatment of each drug (). Furthermore, after co-treatment with VP, the apoptotic level was assessed using TUNEL staining. Interestingly, VP plus trametinib-treated BC organoids increased apoptotic cells compared with trametinib alone or DMSO-treated (control) organoids (). These results indicate that the combinational treatment of trametinib and YAP1 inhibitor is a good therapeutic strategy for the treatment of MIBC.
Figure 3. Effects of trametinib on stemness of BC organoids. Expression levels of CD44 and YAP1 mRNA in trametinib-treated (1 μM, 24 h) BC organoids were determined by quantitative real-time PCR (a). Expression level of each gene was quantified based on the ration of expression level to GAPDH and shown as fold increase relative to control (n = 4). Results were expressed as mean ± S.E.M. ﹡P < .05 vs. control. Expression of CD44 and YAP1 protein in trametinib-treated (1 μM, 24 h) BC organoids. Representative photomicrographs were shown (b, n = 3). Scale bar: 50 μm. Combinational effects of YAP inhibitor and trametinib on cell viability of BC organoids (c). Representative phase-contrast images of the trametinib (1 μM) alone, in combination with YAP1 inhibitor, verteporfin (VP) (0.3 μM), or VP (0.3 μM) alone for 72 h on BC organoids. Scale bar: 500 μm. The enlarged images were shown below for each image. Cell viability was assessed using Prestoblue kit and 100% represents the cell viability of each control (d, n = 6). Results were expressed as mean ± S.E.M. ﹡P < .05 vs. control, #P < .05 vs. trametinib. Apoptosis level was assessed by TUNEL staining in each BC organoid (e). Scale bar: 50 μm. Quantification of apoptosis in each BC organoid was analyzed by ImageJ software (f, n = 4). Results were expressed as mean ± S.E.M. ﹡P < .05 vs. control, #P < .05 vs. trametinib
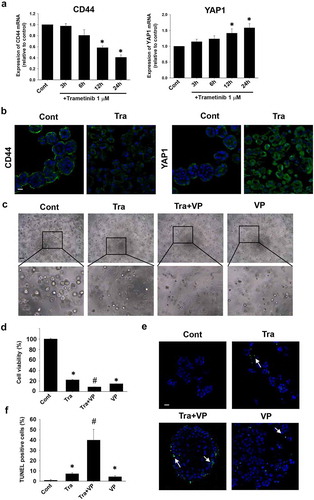
Effects of trametinib on phenotype of BC organoids
After long-term (72 h) trametinib treatment of BC organoids, we observed the morphology clearly changed. The basal solid-like structure was transformed into a luminal cystic and thin-like one (). Trametinib treatment significantly decreased the ratio of basal-like organoids, while the luminal-like ones increased compared with control (). To confirm the trametinib-induced phenotypic changes, we checked the expression of basal and luminal markers by using quantitative real-time PCR. Expression of mRNA of basal markers, CK5 and DSG3 was significantly decreased, while the expression of luminal ones, ERBB2 and GATA3 was significantly increased in the trametinib-treated BC organoids compared with control (). At the protein expression level, the immunofluorescence data revealed that trametinib significantly decreased the number of CK5-positive cells (). These results indicate that trametinib induced basal to luminal differentiation of BC organoids.
Figure 4. Effects of trametinib on morphologic/phenotypic plasticity of BC organoids. Representative bright-field images (a) and H&E staining (b) of the differentiated BC organoids compared with control ones after trametinib (1 μM) treatment for 72 h. Scale bar: 500 μm. The enlarged images were shown below for each image. The ratio of basal-like and luminal-like organoids was quantified by using ImageJ software (c, n = 4). Effects of trametinib on expression of basal and luminal marker genes in BC organoids (d). mRNA expression of basal cell markers, CK5 and DSG3 and luminal cell ones, ERBB2 and GATA3 was determined by quantitative real-time PCR. Expression level of each gene was quantified based on the ration of expression level to GAPDH (n = 4). Results were expressed as mean ± S.E.M.﹡P < .05 vs. control. Expression of CK5 in trametinib-treated BC organoids. Representative photomicrographs were shown (e). The white arrows indicate apoptotic-positive cells. Scale bar: 50 μm. Expression level was quantified by counting the CK5-positive cells (F, n = 3). Results were expressed as mean ± S.E.M. ﹡P < .05 vs. control
Effects of trametinib on tumor growth against xenografted BC organoids in immunodeficient mice
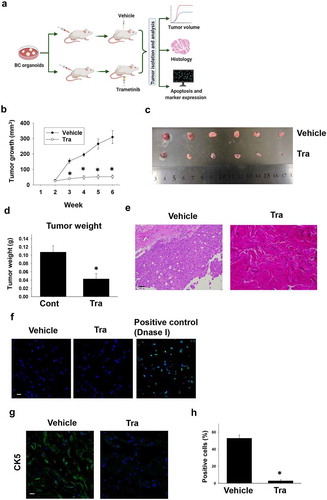
To check the effects of trametinib in vivo, we performed a xenograft experiment of BC organoids and trametinib was administrated to mice after tumor-forming (). Four weeks after administration, the tumor growth in the trametinib-administered mouse group was significantly arrested compared with the control group (). Also, the tumor weight was significantly lower in the trametinib group compared with the control group (). Histological analyses showed that trametinib induced EMT-like structural changes in the tumor tissue compared with the control tumor tissues (). TUNEL staining showed few apoptotic changes in the tumor tissues of trametinib-administered mice compared with the control ones (). We also observed that expression of CK5 in the tumor tissues of trametinib-treated mice was significantly decreased compared with the control ones (), indicating that the luminal differentiation also occurred in the tumor tissues. These results suggest the in vivo efficacy of trametinib in the treatment of MIBC.
Figure 5. Effects of trametinib on tumor growth against xenografted BC organoids in immunodeficient mice. Experimental schema of in vivo experiment (a). After BC organoid cells were subcutaneously injected into the back of SCID mice and tumors formed, trametinib and the vehicle was administered to the mice at the concentration of 1 mg kg−1 for 4 weeks. Thereafter, the tumor volume was measured by clipper every week for 6 weeks (B, n = 6). Tumor tissues of each group was isolated and used for analysis. Comparison of tumor growth curve (b), size (c), and weight (d) between trametinib- or vehicle- administered mice. Results were expressed as mean ± S.E.M. ﹡P < .05 vs. vehicle. Representative H&E staining images of the tumor tissue sections from trametinib- or vehicle- administered mice (e). Scale bar: 200 μm. Confocal microscopy fluorescence images of apoptosis as determined by TUNEL staining of frozen sections of tumor tissues from trametinib-treated or vehicle- administered mice (f, n = 3). Sections treated with DNase I were used as a positive control. Scale bar: 50 μm. Expression level of CK5 in sections from dissected tumors of trametinib- or vehicle-administered mice as determined by confocal microscopy (g, scale bar: 50 μm). Expression level was quantified by counting the CK5-positive cells (h, n = 3). Results were expressed as mean ± S.E.M. ﹡P < .05 vs. vehicle
Effects of long-term trametinib administration to mice on drug sensitivity of xenograft-derived organoids
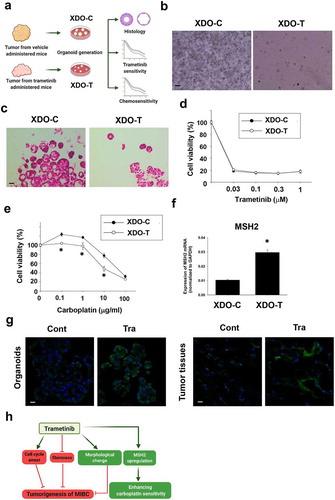
To analyze whether the long-term trametinib administration to mice affects drug sensitivity of tumor tissues, tumor tissues from trametinib-treated and control mice were dissected for developing xenograft-derived organoids and were used for experiments (). On day 3 after culture, the xenograft-derived organoids from trametinib-administered mice (XDO-T) showed a fewer number, smaller size, and lower growth speed compared with those of the control group (XDO-C) (). After passaging, their organoid-forming efficiency soon recovered. The histological difference between XDO-C and XDO-T was not observed (). Interestingly, after the long-term (four weeks) administration of trametinib, the organoid sensitivity to trametinib (), mitoxantrone, and vinblastine (Figure S4) did not change, while the sensitivity to carboplatin was enhanced (). To clarify the molecular mechanisms of the upregulation of sensitivity to carboplatin, expression level of carboplatin sensitivity-related genes, MSH2, MLH1, and ERCC1 was examined. Among these genes, MSH2 was significantly upregulated in XDO-T compared with XDO-C (). The protein expression level of MSH2 was also increased in XDO-T compared with XDO-C as well as in the corresponding tumor tissues (). These results indicate that xenograft-derived tumor tissues did not develop resistance to trametinib after 4 weeks administration period. Additionally, trametinib enhanced the sensitivity of the XDO-T to carboplatin.
Figure 6. Effects of long-term trametinib administration to mice on drug sensitivity of xenograft-derived organoids. Experimental schema of generation of xenograft-derived organoids from trametinib administered and vehicle-administered mice and the analysis (a). Bright field (b) and H&E staining (c) images of the vehicle (XDO-C) and trametinib (XDO-T) administered mice-derived tumor organoids. Scale bar: 500 μm. Sensitivity to trametinib (d, n = 3–6) and carboplatin (e, n = 3–6) was analyzed by Prestoblue kit. Results were expressed as mean ± S.E.M. 100% represents the cell viability of each control. Expression of a carboplatin sensitivity-related gene, MSH2 mRNA in each organoid was determined by quantitative real-time PCR. Expression level of gene was quantified based on the ration of expression level to GAPDH (f, n = 4). Results were expressed as mean ± S.E.M. ﹡P < .05 vs. vehicle. Expression level of MSH2 in xenografted tumor-derived organoids and original tissues (g, n = 3) from trametinib- or vehicle-administered mice. Scale bar: 50 μm. Schematic summary of the present study (h). Trametinib promoted cell cycle arrest, inhibited stemness, changed the phenotype of BC organoids, which may lead to the inhibitory effects of tumorigenesis in vivo. Besides, long-term trametinib administration enhanced the sensitivity to carboplatin through upregulation of MSH2 gene in the tumor tissues
Discussion
In the current study, we investigated the effects of trametinib on dog BC organoids. The main findings are as follows: 1) trametinib inhibited to a higher extent the viability of organoids and the activation of ERK but not EGFR () through inhibition of expression of c-Myc, ELK1, SIK1, and PLA2G4A ( and Figure S2), 2) trametinib inhibited G2/M phase fractions of the cell cycle and expression of cell cycle-related genes ( and Figure S3) with little apoptosis induction ( and ) CD44 was downregulated while YAP1 was upregulated after trametinib treatment ( and ) YAP1 inhibitor, verteporfin showed synergism with trametinib ( and ) trametinib induced basal to luminal differentiation of BC organoids with upregulation of luminal markers (ERBB2 and GATA3) and downregulation of basal ones (CK5 and DSG3) ( and ) trametinib inhibited tumor growth of engrafted BC organoids in mice with basal to luminal differentiation of tumor tissues ( and ) xenograft-derived organoids from trametinib-administered mice showed specifically enhanced sensitivity to carboplatin through upregulation of MSH2 expression (). Collectively (), our data suggest the value of our organoid model as a new tool to provide new therapeutic insights for BC.
While BC is frequent, its management is often difficult especially for the MI type which is solid and of basal originCitation2 with more stemness and EMTCitation20 and is often metastatic.Citation21 In recent years, BC overall incidence and mortality have shown a steady upward trend and have become a significant challenge to people’s health.Citation22 To improve treatment strategies, new research models are necessary, for which organoids are a promising one. Organoids are generated from auto-renovating stem cells which typically recapitulate the in vivo architecture of original tissues, functions, and genetic and molecular imprints.Citation23 It holds great promise for establishing new personalized treatments and analyzing drug resistance in the medical field.Citation13,Citation24–28 The treatment strategies for BC continue to expand and their efficacy varies depending on the type, clinical stage, and associated risk factorsCitation29 with a positive shift toward the more personalized therapy. For MIBC, the current standard of care is cisplatin-based chemotherapy followed by radical cystectomy, immunotherapy, gene therapy, or targeted therapy. Targeted therapy strategy functions the agent to identify oncogenic targets as cell surface antigen, a membrane protein molecule, or gene fragment of cancerous cells, and then induces necrosis and apoptosis of tumor cells. Recently, several oncogenic targets and signaling pathways were implicated and targeted, but mostly in BC cell lines.Citation30 For example, everolimus was used to target AKT in the mTOR pathway,Citation31 lapatinib to target HER2 (ERBB2) in the RTK/MAPK signaling pathway,Citation32 and erlotinib to target EGFR in EGF signaling pathwayCitation33. Among the several targets, ERK1/2 which is activated by MEK1/2 is important signaling pathways implicated in bladder carcinogenesis in both humansCitation18 and dogs.Citation34
In the present study, both total expression and phosphorylation level of EGFR and ERK were upregulated in the BC organoids (, Figure S1). Thus, the therapeutic targeting of an upstream protein of ERK, MEK is important to stop its growth.Citation35 Trametinib, the promising MEK1/2 inhibitor has been approved for the treatment of several cancers,Citation10 but not yet for BC due to the scarcity of data. In the present study, we investigated, in details and for the first time, the effect of trametinib on our established BC organoids of basal solid-like structure (), as a model for human MIBC.Citation4 The treatment of BC organoids with trametinib significantly inhibited phosphorylation of ERK () but not EGFR. We further checked the downstream signals of ERK cascade and found that, among the several signals, trametinib significantly inhibited the c-Myc, ETS transcription factor, ELK1, and SIK1 signals (). Interestingly, our results showed that ELK1 and SIK1 are targets of trametinib in BC. ELK1 was recently shown to be involved in BC progressionCitation36 and here for the first time in BC organoids progression. Also, in the present study and for the first time, SIK1 was shown to be involved in BC progression. Recently, SIK1 was reported to be upregulated and involved in progression of several cancers including colorectal cancer,Citation37 medulloblastoma,Citation38 and non-small cell lung cancerCitation39 and its downregulation suppressed these tumors. These data imply that trametinib inhibited BC growth by targeting the ERK downstream signals, ELK1 and SIK1.
Since many cancer stem cell (CSC) markers are implicated in growth, invasion, angiogenesis, chemotherapy resistance, and metastasis of MIBC,Citation40 the effect of trametinib on two important stem cell markers, CD44 and YAP1 was investigated in the BC organoids. Treatment of BC organoids with trametinib successfully inhibited CD44 but unexpectedly upregulated YAP1 (). CD44 is a cell surface adhesion molecule located at the basal layer of the normal urotheliumCitation41 and involved in cancer cell proliferation, migration, metastasis, angiogenesis, EMT, aggressiveness, and disease progression.Citation42 Therefore, inhibition of CD44-related stemness by trametinib decreased the viability of BC organoids in the present study. Of note, CD44 inhibition might be a consequence of inhibition of the ERK signaling pathway.Citation43 In other cancers, trametinib treatment inhibited ERK phosphorylation and decreased CD44 expression in malignant pleural mesothelioma cellsCitation44 and esophageal squamous cell carcinoma.Citation43 YAP1 is a downstream transcription coactivator and a major nuclear effector of the Hippo tumor suppressor pathway.Citation45 YAP1 is highly expressed in most BC patients, confers CSC traits,Citation46 promotes tumorigenesis,Citation47 protects BC from chemotherapy-induced DNA damage,Citation19 and contributes to the progression of it to an advanced stage and poor prognosis.Citation48 The YAP1 inhibitor, VP (a photodynamic therapy to treat macular degeneration of the retina) was used recently to target YAP1 signaling pathway in mesothelioma,Citation49 prostate cancer,Citation50 melanoma,Citation51 and BC cell lines.Citation47 In BC, VP was successfully shown to inhibit the progression of T24 and RP-B-01 BC cell lines in a dose-dependent manner via induction of apoptosis, decreasing SOX2 expression, and enhancing chemotherapy efficacy.Citation47 In the present study, VP successfully inhibited the cell viability of BC organoid cells in a dose-dependent manner (data are not shown) and showed synergetic effects on the trametinib-treated BC organoid cells through induction of apoptosis (). These results and ours highlight the oncogenic role of the YAP1 signaling pathway in BC and its pharmacological targeting by VP is important to arrest BC progression by induction of apoptosis and lowering the stemness.
BC can be grouped into basal and luminal subtypes.Citation52 Basal-subtype BC is muscle-invasive, more aggressive, metastatic, difficult to treat with poor outcomes, and higher EMT due to its enriched CSC traits than luminal one.Citation52 As noted, the tested strains of BC organoids in the current study are of basal solid-like structure with squamous differentiation ( and ) that mimics the human MIBC.Citation4,Citation52 Similarly, the basal MIBC showed more squamous featuresCitation21 and expression of the higher molecular weight keratins (CK5, CK6, CK14) and CD44 expressionCitation21 that characterizes cells in the basal layer of normal urotheliumCitation53 and MIBC with squamous differentiation.Citation21 This kind of BC revealed more aggressiveness and had shorter overall survival.Citation54 Interestingly, trametinib treatment induced morphological and molecular alterations of BC organoids (). These data indicate the efficacy of trametinib in decreasing the basal and squamous features of BC organoids and therefore might help for decreasing the aggressiveness of MIBC.
The high invasions, metastasis, and resistance of MIBC to most of the chemotherapy agents necessitate the continual search for new combinational treatment strategies to improve the survival rate of patients, especially in precision medicine. Using chemotherapy agents alone is usually toxic and intolerant for the patients. The treatment modality that combines two or more therapeutic agents with different mechanisms of action with lowering their doses, is a cornerstone of cancer therapy as it potentially reduces resistance and metastasis and increases survival rate.Citation55 Therefore, new therapeutic strategies that provide effective results and target the survival pathways at an affordable cost are being considered. In the present study, XDO-T showed a significantly enhanced sensitivity to the platinum-based adjuvant therapy, carboplatin (). Analyzing this valuable effect, we found that among the several genes (MSH2, MLH1, and ERCC1) responsible for enhancing carboplatin sensitivity, MSH2 was significantly upregulated (). Recently, high MSH2 expression was shown to mediate the death of MIBC cells by the platinum-based chemotherapyCitation56 and predicting the response to it.Citation57 The expression of ERCC1, MLH1, and MSH2 in different tumors were associated with response and outcome to platinum-based chemotherapy.Citation58 Contrarily, tumor cells that were deficient in MLH1 and MSH2 were resistant to cisplatin in vitro.Citation59,Citation60 In vivo, lower MLH1 expression levels in lung cancer patients were associated with worse prognoses.Citation61 However, patients with higher expression of ERCC1 appeared to benefit from platinum-based postoperative adjuvant chemotherapy.Citation58,Citation62 Carboplatin was tried either aloneCitation63 or with others like piroxicamCitation64 or gemcitabineCitation65 for treatment of dog BC. However, the outcomes as median survival time and progression rate were not improved.Citation64 The combination of carboplatin with trametinib was trialed recently for the treatment of locally advanced non-small-cell lung cancer,Citation66 but never for human BC. Therefore, based on our findings, using trametinib with carboplatin therapy is reasonable as a new strategic option for better treatment of both human and dog BC.
In conclusion, we for the first time investigated the effect of trametinib on BC organoids. Our results showed the efficacy of trametinib against BC organoids in vitro and in vivo (). These data suggest that trametinib has therapeutic potential for MIBC, particularly in ERK‐overexpressing one. Our data also implied a new strategy of trametinib-YAP inhibitor or trametinib-carboplatin combination as a promising treatment of BC. Further confirmation studies on the roles of ERK downstream signals, c-Myc, ELK1, SIK1, and PLA2G4A in BC organoid will help the development of future targets of MIBC.
Supplemental Material
Download MS Word (214.2 KB)Disclosure statement
The authors have no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
Additional information
Funding
References
- Kamat AM, Hahn NM, Efstathiou JA, Lerner SP, Malmström P-U, Choi W, Guo CC, Lotan Y, Kassouf W. Bladder cancer. The Lancet. 2016;388(10061):2796–2810. doi:10.1016/S0140-6736(16)30512-8.
- Rentsch CA, Muller DC, Ruiz C, Bubendorf L. Comprehensive molecular characterization of urothelial bladder carcinoma: a step closer to clinical translation? Eur Urol. 2017;72(6):960–961. doi:10.1016/j.eururo.2017.06.022.
- Lerner SP, Schoenberg M, Sternberg C. 2006. Textbook of bladder cancer. Oxon. UK: Taylor and Francis. doi:10.1201/9780367800130
- Elbadawy M, Usui T, Mori T, Tsunedomi R, Hazama S, Nabeta R, Uchide T, Fukushima R, Yoshida T, Shibutani M, et al. Establishment of a novel experimental model for muscle-invasive bladder cancer using a dog bladder cancer organoid culture. Cancer Sci. 2019;110(9):2806–2821. doi:10.1111/cas.14118.
- Sebolt-Leopold JS. Development of anticancer drugs targeting the MAP kinase pathway. Oncogene. 2000;19(56):6594–6599. doi:10.1038/sj.onc.1204083.
- Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26(22):3291–3310. doi:10.1038/sj.onc.1210422.
- Porter AC, Vaillancourt RR. Tyrosine kinase receptor-activated signal transduction pathways which lead to oncogenesis. Oncogene. 1998;17(11):1343–1352. doi:10.1038/sj.onc.1202171.
- Solit DB, Garraway LA, Pratilas CA, Sawai A, Getz G, Basso A, Ye Q, Lobo JM, She Y, Osman I, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439(7074):358–362. doi:10.1038/nature04304.
- Gilmartin AG, Bleam MR, Groy A, Moss KG, Minthorn EA, Kulkarni SG, Rominger CM, Erskine S, Fisher KE, Yang J, et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res: Off J Am Assoc Cancer Res. 2011;17(5):989–1000. doi:10.1158/1078-0432.CCR-10-2200.
- Kelly RJ. Dabrafenib and trametinib for the treatment of non-small cell lung cancer. Expert Rev Anticancer Ther. 2018;18(11):1063–1068. doi:10.1080/14737140.2018.1521272.
- Hoffner B, Benchich K. Trametinib: a targeted therapy in metastatic melanoma. J Ad Pract Oncol. 2018;9(7):741–745.
- Subbiah V, Kreitman RJ, Wainberg ZA, Cho JY, Schellens JHM, Soria JC, Wen PY, Zielinski C, Cabanillas ME, Urbanowitz G, et al. Dabrafenib and trametinib treatment in patients with locally advanced or metastatic BRAF V600–mutant anaplastic thyroid cancer. J Clin Oncol. 2018;36(1):7–13. doi:10.1200/JCO.2017.73.6785.
- Usui T, Sakurai M, Umata K, Elbadawy M, Ohama T, Yamawaki H, Hazama S, Takenouchi H, Nakajima M, Tsunedomi R, et al. Hedgehog signals mediate anti-cancer drug resistance in three-dimensional primary colorectal cancer organoid culture. Int J Mol Sci. 2018;19(4):1098. doi:10.3390/ijms19041098.
- Usui T, Sakurai M, Nishikawa S, Umata K, Nemoto Y, Haraguchi T, Itamoto K, Mizuno T, Noguchi S, Mori T, et al. Establishment of a dog primary prostate cancer organoid using the urine cancer stem cells. Cancer Sci. 2017;108(12):2383–2392. doi:10.1111/cas.13418.
- Abugomaa A, Elbadawy M, Yamanaka M, Goto Y, Hayashi K, Mori T, Uchide T, Azakami D, Fukushima R, Yoshida T, et al. Establishment of 2.5D organoid culture model using 3D bladder cancer organoid culture. Sci Rep. 2020;10(1):9393. doi:10.1038/s41598-020-66229-w.
- Dong L, Lin F, Wu W, Liu Y, Huang W. Verteporfin inhibits YAP-induced bladder cancer cell growth and invasion via Hippo signaling pathway. Int J Med Sci. 2018;15(6):645–652. doi:10.7150/ijms.23460.
- Gaver RC, George AM, Duncan GF, Morris AD, Deeb G, Faulkner HC, Farmen R. The disposition of carboplatin in the beagle dog. Cancer Chemother Pharmacol. 1988;21(3):197–202. doi:10.1007/BF00262769.
- Takeuchi A, Eto M, Shiota M, Tatsugami K, Yokomizo A, Kuroiwa K, Itsumi M, Naito S. Sunitinib enhances antitumor effects against chemotherapy-resistant bladder cancer through suppression of ERK1/2 phosphorylation. Int J Oncol. 2012;40(5):1691–1696. doi:10.3892/ijo.2012.1368.
- Ciamporcero E, Shen H, Ramakrishnan S, Ku S Y, Chintala S, Shen L, Adelaiye R, Miles KM, Ullio C, Pizzimenti S, et al. YAP activation protects urothelial cell carcinoma from treatment-induced DNA damage. Oncogene. 2016;35(12):1541–1553. doi:10.1038/onc.2015.219.
- Sjodahl G, Lauss M, Lovgren K, Chebil G, Gudjonsson S, Veerla S, Patschan O, Aine M, Fernö M, Ringnér M, et al. A molecular taxonomy for urothelial carcinoma. Clin Cancer Res: Off J Am Assoc Cancer Res. 2012;18(12):3377–3386. doi:10.1158/1078-0432.CCR-12-0077-T.
- Choi W, Porten S, Kim S, Willis D, Plimack ER, Hoffman-Censits J, Roth B, Cheng T, Tran M, Lee I-L, et al. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell. 2014;25(2):152–165. doi:10.1016/j.ccr.2014.01.009.
- Antoni S, Ferlay J, Soerjomataram I, Znaor A, Jemal A, Bray F. Bladder cancer incidence and mortality: a global overview and recent trends. Eur Urol. 2017;71(1):96–108. doi:10.1016/j.eururo.2016.06.010.
- Elbadawy M, Yamanaka M, Goto Y, Hayashi K, Tsunedomi R, Hazama S, Nagano H, Yoshida T, Shibutani M, Ichikawa R, et al. Efficacy of primary liver organoid culture from different stages of non-alcoholic steatohepatitis (NASH) mouse model. Biomaterials. 2020;237:119823. doi:10.1016/j.biomaterials.2020.119823.
- Usui T, Sakurai M, Enjoji S, Kawasaki H, Umata K, Ohama T, Fujiwara N, Yabe R, Tsuji S, Yamawaki H, et al. Establishment of a novel model for anticancer drug resistance in three-dimensional primary culture of tumor microenvironment. Stem Cells Int. 2016;2016:7053872. doi:10.1155/2016/7053872.
- Usui T, Elbadawy M, Sasaki K. Session 6: advances in anticancer therapy. J Vet Pharmacol Ther. 2018;41:28–30. doi:10.1111/jvp.12625
- Elbadawy M, Usui T, Yamawaki H, Sasaki K. Development of an experimental model for analyzing drug resistance in colorectal cancer. Cancers (Basel). 2018;10(6):164. doi:10.3390/cancers10060164.
- Abugomaa A, Elbadawy M. Patient-derived organoid analysis of drug resistance in precision medicine: is there a value? Expert Rev Precis Med Drug Dev. 2020;5(1):1–5. doi:10.1080/23808993.2020.1715794.
- Elbadawy M, Abugomaa A, Yamawaki H, Usui T, Sasaki K. Development of prostate cancer organoid culture models in basic medicine and translational research. Cancers (Basel). 2020;12(4):777. doi:10.3390/cancers12040777.
- Prasad SM, Decastro GJ, Steinberg GD. Medscape. Urothelial carcinoma of the bladder: definition, treatment and future efforts. Nat Rev Urol. 2011;8(11):631–642. doi:10.1038/nrurol.2011.144.
- Su H, Jiang H, Tao T, Kang X, Zhang X, Kang D, Li S, Li C, Wang H, Yang Z, et al. Hope and challenge: precision medicine in bladder cancer. Cancer Med. 2019;8(4):1806–1816. doi:10.1002/cam4.1979.
- Pinto-Leite R, Arantes-Rodrigues R, Sousa N, Oliveira PA, Santos L. mTOR inhibitors in urinary bladder cancer. Tumour Biol: J Int Soc Oncodev Biol Med. 2016;37(9):11541–11551. doi:10.1007/s13277-016-5083-1.
- Sakai K, Maeda S, Saeki K, Nakagawa T, Murakami M, Endo Y, Yonezawa T, Kadosawa T, Mori T, Nishimura R, et al. Anti-tumour effect of lapatinib in canine transitional cell carcinoma cell lines. Vet Comp Oncol. 2018;16(4):642–649. doi:10.1111/vco.12434.
- Tsai YC, Ho PY, Tzen KY, Tuan TF, Liu WL, Cheng AL, Pu YS, Cheng JC-H, Synergistic blockade of EGFR and HER2 by new-generation EGFR tyrosine kinase inhibitor enhances radiation effect in bladder cancer cells. Mol Cancer Ther. 2015;14(3):810–20. doi:10.1158/1535-7163.MCT-13-0951
- Hanazono K, Fukumoto S, Kawamura Y, Endo Y, Kadosawa T, Iwano H, Uchide T. Epidermal growth factor receptor expression in canine transitional cell carcinoma. J Vet Med Sci. 2015;77(1):1–6. doi:10.1292/jvms.14-0032.
- Cohen P. Protein kinases — the major drug targets of the twenty-first century? Nat Rev Drug Discov. 2002;1(4):309–315. doi:10.1038/nrd773.
- Kawahara T, Ide H, Kashiwagi E, Patterson JD, Inoue S, Shareef HK, Aljarah AK, Zheng Y, Baras AS, Miyamoto H, et al. 2015. Silodosin inhibits the growth of bladder cancer cells and enhances the cytotoxic activity of cisplatin via ELK1 inactivation. Am J Cancer Res. 5(10):2959–2968.
- Huang C, Liu J, Xu L, Hu W, Wang J, Wang M, Yao X. MicroRNA-17 promotes cell proliferation and migration in human colorectal cancer by downregulating SIK1. Cancer Manag Res. 2019;11:3521–3534. doi:10.2147/CMAR.S191087.
- Huang S, Xue P, Han X, Zhang C, Yang L, Liu L, Wang X, Li H, Fu J, Zhou Y, et al. Exosomal miR-130b-3p targets SIK1 to inhibit medulloblastoma tumorigenesis. Cell Death Dis. 2020;11(6):408. doi:10.1038/s41419-020-2621-y.
- Hollstein PE, Eichner LJ, Brun SN, Kamireddy A, Svensson RU, Vera LI, Ross DS, Rymoff TJ, Hutchins A, Galvez HM, et al. The AMPK-related kinases SIK1 and SIK3 mediate key tumor-suppressive effects of LKB1 in NSCLC. Cancer Discov. 2019;9(11):1606–1627. doi:10.1158/2159-8290.CD-18-1261.
- Abugomaa A, Elbadawy M, Yamawaki H, Usui T, Sasaki K. Emerging roles of cancer stem cells in bladder cancer progression, tumorigenesis, and resistance to chemotherapy: a potential therapeutic target for bladder cancer. Cells. 2020;9(1):235. doi:10.3390/cells9010235.
- Brandt WD, Matsui W, Rosenberg JE, He X, Ling S, Schaeffer EM, Berman DM. Urothelial carcinoma: stem cells on the edge. Cancer Metastasis Rev. 2009;28(3–4):291–304. doi:10.1007/s10555-009-9187-6.
- Naor D, Sionov RV, Ish-Shalom D. CD44: structure, function, and association with the malignant process. Adv Cancer Res. 1997;71:241–319.
- Maehara O, Suda G, Natsuizaka M, Ohnishi S, Komatsu Y, Sato F, Nakai M, Sho T, Morikawa K, Ogawa K, et al. Fibroblast growth factor-2-mediated FGFR/Erk signaling supports maintenance of cancer stem-like cells in esophageal squamous cell carcinoma. Carcinogenesis. 2017;38(11):1073–1083. doi:10.1093/carcin/bgx095.
- Cho H, Matsumoto S, Fujita Y, Kuroda A, Menju T, Sonobe M, Kondo N, Torii I, Nakano T, Lara PN, et al. Trametinib plus 4-methylumbelliferone exhibits antitumor effects by ERK blockade and CD44 downregulation and affects PD-1 and PD-L1 in malignant pleural mesothelioma. J Thorac Oncol: Off Publ Int Assoc Study Lung Cancer. 2017;12(3):477–490. doi:10.1016/j.jtho.2016.10.023.
- Beverdam A, Claxton C, Zhang X, James G, Harvey KF, Key B. Yap controls stem/progenitor cell proliferation in the mouse postnatal epidermis. J Invest Dermatol. 2013;133(6):1497–1505. doi:10.1038/jid.2012.430.
- Johnson R, Halder G. The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat Rev Drug Discov. 2014;13(1):63–79. doi:10.1038/nrd4161.
- Ooki A, Del Carmen Rodriguez Pena M, Marchionni L, Dinalankara W, Begum A, Hahn NM, VandenBussche CJ, Rasheed ZA, Mao S, Netto GJ, et al. YAP1 and COX2 coordinately regulate urothelial cancer stem-like cells. Cancer Res. 2018;78(1):168–181. doi:10.1158/0008-5472.CAN-17-0836.
- Liu JY, Li YH, Lin HX, Liao YJ, Mai SJ, Liu ZW, Zhang Z-L, Jiang L-J, Zhang J-X, Kung H-F, et al. Overexpression of YAP 1 contributes to progressive features and poor prognosis of human urothelial carcinoma of the bladder. BMC Cancer. 2013;13(1):349. doi:10.1186/1471-2407-13-349.
- Kandasamy S, Adhikary G, Rorke EA, Friedberg JS, Mickle MB, Alexander HR, Eckert RL. The YAP1 signaling inhibitors, verteporfin and CA3, suppress the mesothelioma cancer stem cell phenotype. Mol Cancer Res: MCR. 2020;18(3):343–351. doi:10.1158/1541-7786.MCR-19-0914.
- Jiang N, Hjorth-Jensen K, Hekmat O, Iglesias-Gato D, Kruse T, Wang C, Wei W, Ke B, Yan B, Niu Y, et al. In vivo quantitative phosphoproteomic profiling identifies novel regulators of castration-resistant prostate cancer growth. Oncogene. 2015;34(21):2764–2776. doi:10.1038/onc.2014.206.
- Feng X, Degese MS, Iglesias-Bartolome R, Vaque JP, Molinolo AA, Rodrigues M, Zaidi M, Ksander B, Merlino G, Sodhi A, et al. Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio-regulated rho GTPase signaling circuitry. Cancer Cell. 2014;25(6):831–845. doi:10.1016/j.ccr.2014.04.016.
- Choi W, Czerniak B, Ochoa A, Su X, Siefker-Radtke A, Dinney C, McConkey DJ. Intrinsic basal and luminal subtypes of muscle-invasive bladder cancer. Nat Rev Urol. 2014;11(7):400–410. doi:10.1038/nrurol.2014.129.
- Volkmer JP, Sahoo D, Chin RK, Ho PL, Tang C, Kurtova AV, Willingham SB, Pazhanisamy SK, Contreras-Trujillo H, Storm TA, et al. Three differentiation states risk-stratify bladder cancer into distinct subtypes. Proc Natl Acad Sci U S A. 2012;109(6):2078–2083. doi:10.1073/pnas.1120605109.
- Hurst CD, Knowles MA. Molecular subtyping of invasive bladder cancer: time to divide and rule? Cancer Cell. 2014;25(2):135–136. doi:10.1016/j.ccr.2014.01.026.
- Bayat Mokhtari R, Homayouni TS, Baluch N, Morgatskaya E, Kumar S, Das B, Yeger H. Combination therapy in combating cancer. Oncotarget. 2017;8(23):38022–38043. doi:10.18632/oncotarget.16723.
- Goodspeed A, Jean A, Costello JC. A whole-genome CRISPR screen identifies a role of MSH2 in cisplatin-mediated cell death in muscle-invasive bladder cancer. Eur Urol. 2019;75(2):242–250. doi:10.1016/j.eururo.2018.10.040.
- Mucaki EJ, Zhao JZL, Lizotte DJ, Rogan PK. Predicting responses to platin chemotherapy agents with biochemically-inspired machine learning. Signal Transduction Targeted Ther. 2019;4(1):1. doi:10.1038/s41392-018-0034-5.
- Ting S, Mairinger FD, Hager T, Welter S, Eberhardt WE, Wohlschlaeger J, Schmid KW, Christoph DC. ERCC1, MLH1, MSH2, MSH6, and betaIII-tubulin: resistance proteins associated with response and outcome to platinum-based chemotherapy in malignant pleural mesothelioma. Clin Lung Cancer. 2013;14(5):558–67 e3. doi:10.1016/j.cllc.2013.04.013.
- Gong JG, Costanzo A, Yang HQ, Melino G, Kaelin WG Jr., Levrero M, Wang JYJ. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature. 1999;399(6738):806–809. doi:10.1038/21690.
- Brown R, Hirst GL, Gallagher WM, McIlwrath AJ, Margison GP, Van Der Zee AG, Anthoney DA. hMLH1 expression and cellular responses of ovarian tumour cells to treatment with cytotoxic anticancer agents. Oncogene. 1997;15(1):45–52. doi:10.1038/sj.onc.1201167.
- Liang JY, Yang QF, Zeng YL, Liu YY, Liu YT, Gu FF, Hu Y, Zhang K, Zhong H, Liu L, et al. Clinical value of MLH1-negative circulating tumor cells in lung cancer patients. Med (Baltimore). 2019;98(25):e15721. doi:10.1097/MD.0000000000015721.
- Xie KJ, He HE, Sun AJ, Liu XB, Sun LP, Dong XJ. Expression of ERCC1, MSH2 and PARP1 in non-small cell lung cancer and prognostic value in patients treated with platinum-based chemotherapy. Asian Pac J Cancer Prev. 2014;15(6):2591–2596. doi:10.7314/APJCP.2014.15.6.2591.
- Chun R, Knapp DW, Widmer WR, DeNicola DB, Glickman NW, Kuczek T, Degortari A, Han CM. Phase II clinical trial of carboplatin in canine transitional cell carcinoma of the urinary bladder. J Vet Intern Med. 1997;11(5):279–283. doi:10.1111/j.1939-1676.1997.tb00465.x.
- Allstadt SD, Rodriguez CO Jr., Boostrom B, Rebhun RB, Skorupski KA. Randomized phase III trial of piroxicam in combination with mitoxantrone or carboplatin for first-line treatment of urogenital tract transitional cell carcinoma in dogs. J Vet Intern Med. 2015;29(1):261–267. doi:10.1111/jvim.12533.
- Hudson E, Lester JF. Gemcitabine and carboplatin in the treatment of transitional cell carcinoma of the urothelium: a single centre experience and review of the literature. Eur J Cancer Care (Engl). 2010;19(3):324–328. doi:10.1111/j.1365-2354.2008.01050.x.
- Lin SH, Mok I, Leos D, Pasia MG, Thall PF, Lin HY, Welliver MX, Villalona-Calero MA, Yao JC, Meric-Bernstam F, et al. NCI 9448: phase I study of trametinib in combination with chemoradiation for KRAS-mutant non-small cell lung cancer. J Clin Oncol. 2015;33(15_suppl): TPS7585–TPS. doi:10.1200/jco.2015.33.15_suppl.tps7585.