
ABSTRACT
Biguanide drugs (metformin and phenformin) have drawn interest for potential cancer treatments, and laboratory studies show that some cancer cells are selectively sensitive to growth-inhibitory effects of biguanides. Examining metabolic pathways affected by biguanide treatments in cancer cells that are highly sensitive to biguanides, we found that biguanide treatment depletes cellular levels of both aspartate and NAD+. Experiments to replenish these metabolites or block steps of the aspartate-malate shuttle suggest that depletion of both metabolites, rather than either aspartate of NAD+ individually, is critical for growth-inhibitory effects of biguanide exposure. Even in sensitive cancer cells, though, biguanide treatment alone over a broad range of doses only inhibits cell replication without significantly affecting cell viability. Noting that clinical observations of biguanide efficacy have used combinations of agents that typically include cisplatin, we found that biguanide treatment at a cytostatic level substantially decreases survival of lung cancer and breast cancer cells when co-treated with cisplatin at doses that alone are also non-cytotoxic. This striking enhancement of cisplatin toxicity by biguanides depends on reductions of levels of NAD+ and aspartate, since addition of either of these metabolites prevented this potentiation of cisplatin cytotoxicity. Thus, biguanide drugs can have cytotoxic effects when used in combination with other cancer drugs, such as cisplatin, and depleting cellular levels of NAD+ and aspartate is critical for enhancing the cytotoxicity of cisplatin by biguanide drugs in sensitive cancer cells.
Introduction
Findings from numerous epidemiological, clinical, and pre-clinical studies suggest that metformin and phenformin, biguanide drugs developed to treat diabetes, have anti-cancer effects.Citation1,Citation2 For example, a meta-analysis of epidemiological studies considering various types of cancers estimated a 31% reduction in overall cancer incidence or mortality in patients who are treated for diabetes with metformin rather than sulfonylureas or insulin.Citation3 Clinical evidence for metformin’s anti-cancer activity includes a significantly higher rate of complete pathological response of breast cancers to neoadjuvant therapy for diabetic patients treated with metformin in a retrospective study,Citation4 and prospective studies showing improved progression-free survival and overall survival for patients advanced lung cancer who had metformin added to standard chemotherapy.Citation5,Citation6
It is now becoming clear that biguanides have direct effects on cancer cells through inhibition of mitochondrial respiratory-chain complex I.Citation7,Citation8 How inhibition of mitochondrial complex I affects cancer cells remains an area of active investigation, with various studies proposing that important consequences of biguanide treatment are due to reductions in cellular ATP, activation of AMPK with corresponding decreases in mTOR activity,Citation9–11 or depletion of cellular levels of either aspartate or NAD+.Citation12,Citation13 Importantly, the cellular effects of biguanides have not been defined in terms that can be extropolated to improve the therapeutic use of these drugs for cancer treatment.
In this study, we investigated the cellular and metabolic effects of metformin and phenformin on highly sensitive cancer cells in vitro, and we addressed the questions of whether biguanide drugs affect cell proliferation or cell survival and thus whether effects are transient or permanent. We also examined the roles of specific metabolites that are depleted by biguanide drugs in causing these effects on cancer cells. Finally, we examined how biguanides affect cellular responses to drugs used in cancer chemotherapy, with an overall goal of understanding how biguanides might best be used in cancer therapy.
Results
Metformin and phenformin inhibit cancer cell replication without decreasing cell viability
Cell proliferation is inhibited by both metformin and phenformin, as measured by significantly decreased cell numbers in cultures of cells treated with these drugs. Previously reported data have shown that the A549 lung adenocarcinoma cell line is sensitive in vitro and in vivo to metformin.Citation14,Citation15 Supporting the use of A549 cells for investigating mechanisms of sensitivity to biguanides, we found that is particularly sensitive to growth-inhibitory effects of both metformin and phenformin relative to other cancer cell lines that we tested (), and the growth inhibition in A549 cells can be detected as early as 24 hours after treatment (). Remarkably, there is a plateau in the dose-dependent inhibition of proliferation by these drugs, with similar levels of inhibition seen over a 100-fold range of drug dose (). Moreover, this inhibition of proliferation is reversible, and no significant loss in cell viability is noted when cells treated for up to three days with 10 µM phenformin are subsequently incubated with drug-free media ().
Figure 1. Reversible inhibition of cell proliferation in cancer cells by biguanide drugs. A) Cell proliferation is inhibited by both 1 mM metformin (light columns) and 10 µM phenformin (dark columns), with variable responses among cancer cell lines. Data shown represents change in cell numbers relative to control, with mean and standard deviation for three measurements. B) Growth inhibition in A549 cells can be detected as early as 24 hours after treatment. Cell numbers were enumerated at 24, 48, and 72 hours in cultures treated in triplicate. C) Growth in A549 cultures treated with variable levels of phenformin for 3 days. All cultures (including control) were plated at clonal density and grown for 7 days total (3 with treatment, 4 without). Note that dose-dependent inhibition of proliferation by phenformin is similar over range of 10 to 100 µM. D) Inhibition of proliferation by phenformin is reversible. When clonal density cultures treated with 10 µM phenformin for 3 days are subsequently incubated with drug-free media, the numbers and sizes of colonies are similar to cultures incubated only with drug-free media for 7 days
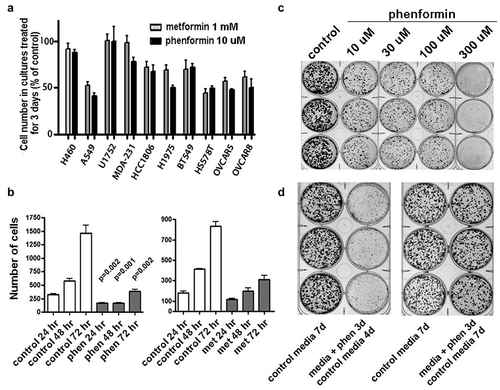
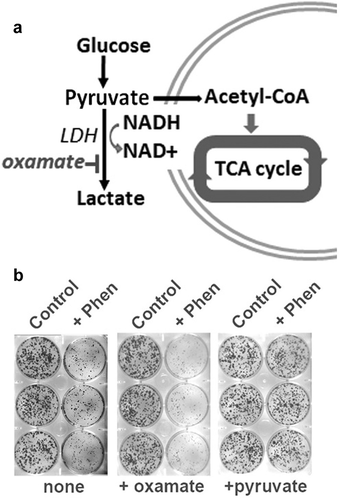
Investigating possible mechanisms to explain how biguanides inhibit proliferation in A549 cells, we focused on the known effect of biguanides of blocking mitochondrial electron transport,Citation7,Citation16 It is accepted that even in the setting of deficient mitochondrial electron transport, glycolysis is sufficient to generate ATP if NAD+ is regenerated by transfer of electrons from pyruvate to lactate by lactate dehydrogenase (LDH) (). We tested the importance of lactate dehydrogenase for maintaining cell proliferation when electron transport is blocked by co-incubating biguanide-treated cultures with oxamate, an inhibitor of LDH. Oxamate and other inhibitors of LDH have been shown previously to inhibit growth of cancer cells,Citation17,Citation18 and we found that adding sodium oxamate (1 mM) to media, even at levels that alone have no effects on cell growth, enhances the inhibition of cell proliferation by metformin or phenformin (). By contrast, studies conducted several decades ago showed that cells devoid of mtDNA and lacking a functional mitochondrial electron transport chain (ETC) can proliferate when cultured with supra-physiological levels of pyruvate.Citation19 Consistent with expectations from these and other previously reported findings,Citation13 the proliferative ability of phenformin-treated A549 cells was completely restored by co-treatment of cultures with 1 mM sodium pyruvate ().
Figure 2. Glycolysis and generation of NAD+ in activity of biguanide drugs. A) Diagram reviewing how NAD+ is regenerated by transfer of electrons from pyruvate to lactate by lactate dehydrogenase (LDH). B) Inhibition of cell proliferation in cultures treated with phenformin (10 µM) is enhanced by co-treatment with oxamate, which blocks LDH, but offset by co-treatment with pyruvate, which is a substrate for LDH
Depletion of NAD+ and aspartate are both linked to anti-proliferative activity of biguanides
The importance of LDH activity in cell growth and the ability of pyruvate to restore cell proliferation in biguanide-treated cultures suggest that depleting levels of NAD+ has a central role in the inhibitory effects of biguanides on A549 cells. Indeed, we found that treatments of A549 cells with levels of metformin or phenformin that inhibit proliferation result in substantial decreases in cellular NAD+/NADH ratios within 4 hours of treatment (). Previous studies have also suggested that depletion of aspartate is responsible for the effects of biguanides on cell proliferation.Citation12,Citation13 We measured aspartate in phenformin-treated A549 lung cancer cells using magnetic resonance spectroscopy (MRS), and consistent with previous reports, we found depletion of aspartate after 16 hours treatment with 10 µM phenformin (). Interestingly, a recent study reported that asparagine bioavailability strongly influences metastatic potential of breast cancer cells,Citation20 and our MRS measurements also found that phenformin treatment causes depletion of cellular asparagine levels in parallel with depletion of aspartate.
Figure 3. Depletion of NAD+ and aspartate in cancer cells by biguanide drugs. A) Treatments of A549 cells with levels of metformin or phenformin that inhibit proliferation result in substantial decreases in cellular NAD+/ NADH ratios within 4 hours of treatment. B, C) Aspartate and asparagine (measured using magnetic resonance spectroscopy) are also depleted in phenformin-treated A549 lung cancer cells. By contrast, levels of methionine (a control amino acid) are stable. Columns and error bars in Panel C represent means and standard deviations for triplicate measurements. D) Addition of exogenous NAD+ (free acid) or aspartate to culture media attenuated the inhibition of cell proliferation by phenformin, demonstrating roles for both NAD+ and aspartate in maintaining the ability of A549 cells to proliferate when exposed to biguanide. E) Addition of aspartate (10 mM) to media of cultures does not prevent depletion of NAD+ by phenformin, and addition of NAD+ (1 mM) to culture media does not prevent depletion of aspartate by phenformin. Columns and error bars represent means and standard deviations for triplicate measurements
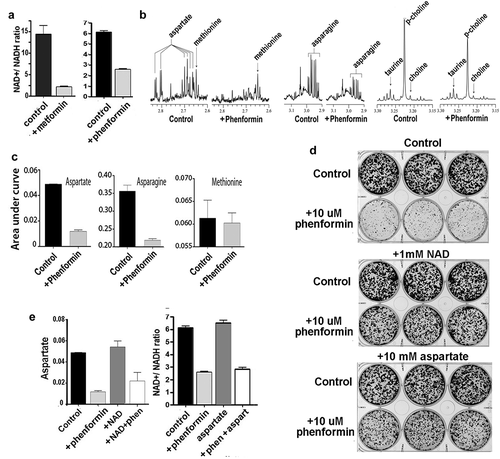
Roles for both NAD+ and aspartate in maintaining the ability of A549 cells to proliferate was directly demonstrated by experiments showing that addition of supra-physiological levels of either NAD+ (free acid) or aspartate to culture media prevented inhibition of cell proliferation by phenformin (). Although aspartate is potentially capable of increasing cytosolic levels of NAD+ in cells through the aspartate-malate shuttle, our experiments found that addition of aspartate to culture media did not restore NAD+ levels in phenformin-treated cultures (). Moreover, consistent with previous reports,Citation14 addition of exogenous NAD to culture media did not restore aspartate levels in phenformin-treated cultures, even though this level of NAD restored the proliferative potential of these cultures with phenformin treatment ().
The aspartate-malate shuttle can maintain cellular NAD+ and aspartate
In further efforts to clarify whether depletion of aspartate or depletion of NAD+ is responsible for mediating the growth inhibitory effects of biguanides on A549 cancer cells, we examined how aspartate and NAD+ interact in the aspartate-malate shuttle. The aspartate-malate shuttle is mechanism for translocating electrons produced during glycolysis across the inner mitochondrial membrane for oxidative phosphorylation, and as diagramed in , the aspartate-malate shuttle potentially allows abundant cytosolic aspartate to be a source of NAD+, and conversely, abundant cytosolic NAD+ can contribute to restoration of depleted aspartate.
Figure 4. Role of the aspartate-malate shuttle in biguanide activity. A) Diagram summarizing how the aspartate-malate shuttle potentially allows abundant cytosolic aspartate to be a source of NAD+, and conversely, how abundant cytosolic NAD+ can contribute to restoration of depleted aspartate. B) Cell proliferation in phenformin-exposed A549 cells is maintained by co-treatment with oxaloacetate, an intermediate metabolite of the aspartate-malate shuttle. C) Cell proliferation as measured by counts of A549 cells with CRISPR/ Cas9-mediated knockout of GOT1 and MDH1 after treatments for three days with phenformin (10 µM) and NAD+ (1 mM), alone or in combination as indicated, and compared to control cultures. Columns and error bars represent means and standard deviations for triplicate measurements. In all situations, co-treatment with exogenous NAD+ increases cell proliferation in phenformin-treated cultures compared to phenformin treatment alone (p < .02). D) Growth of A549 cells with knock-out of GOT1 or knock-out of MDH1, plated at clonal density, and treated for 3 days with phenformin followed by incubation for 4 days in standard media. Co-treatment with pyruvate, aspartate, oxaloacetate, NAD+, or α-ketoglutarate results in increased proliferation, to variable extents
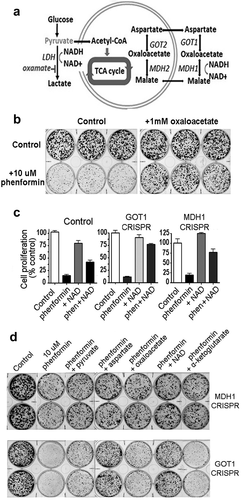
Other intermediate metabolites can also contribute to maintaining cellular levels of NAD+ and aspartate via the aspartate-malate shuttle, and as seen in , growth-inhibitory effects of phenformin were substantially abrogated by supplementing the culture media with oxaloacetate, which has the ability to restore levels of both NAD+ and aspartate, depending on the activity of the glutamate-oxaloacetate transferase (GOT1) or the malate dehydrogenase (MDH1) enzymes. To distinguish the relative importance of replenishing aspartate and NAD by oxaloacetate, we used CRISPR/Cas9 editing to knock out the genes for GOT1 and MDH1 enzymes in A549 cells and then re-tested the ability of various metabolites to overcome growth inhibitory effects of phenformin. Cultures with knockout of GOT1 showed greater sensitivity to growth-inhibitory effects of phenformin than cultures with knockout of MDH1 (), consistent with the previous findings of a CRISPR-based negative selection screen in human Jurkat leukemic T cells, where anti-proliferative effects of phenformin were substantially potentiated by loss of GOT1.Citation12 In both GOT1-KO cells and MDH1-KO cells, growth-inhibitory effects of phenformin were partially (and variably) overcome by adding pyruvate, oxaloacetate, NAD, or aspartate to culture media (). Notably, in GOT1-KO cells, addition of NAD to culture media was as effective in restoring cell proliferation as was addition of aspartate. These experiments again point to important roles for both GOT1 and MDH1 in protecting cells from growth inhibitory effects of biguanides but do not implicate either NAD or aspartate individually as a primary metabolite that is critically depleted by biguanide exposure.
Phenformin sensitizes lung cancer cells to cell killing by cisplatin
Clinical evidence for metformin’s anti-cancer activity comes from both retrospective studies and prospective clinical trials where metformin was added to platinum-based chemotherapy.Citation4–6 Since these clinical observations are contrary to the concept that active cell replication enhances the cytotoxic effects of platinum agents,Citation21,Citation22 we then tested how inhibiting cell replication with biguanide treatment would affect survival of cancer cells when also treated with cisplatin.
For these experiments, we focused on responses of two cell lines that are relatively sensitive to growth-inhibitory effects of biguanides: A549 lung cancer cells and Hs578T breast cancer cells. We enumerated live and dead cells after trypan blue staining in cultures that were treated with cisplatin, metformin, phenformin, or combinations of cisplatin plus either metformin or phenformin. Consistent with our results shown above, these assays showed significantly (p < .02) reduced live cell numbers in cultures of the sensitive A549 and Hs578T cells after 72 hour treatments with either metformin (1 mM) or phenformin (10 µM) () compared to control cultures. We also tested HCC1806 breast cancer cells, which are relatively insensitive to growth-inhibitory effects of biguanides and found no changes in cell numbers after treatment with drugs at these doses. Treating cultures with 0.2 µM cisplatin alone did not significantly decrease the number of live cells after 72 hr treatment, but A549 and Hs578T cultures treated with combinations of cisplatin plus either metformin or phenformin showed significantly few live cells than control cultures or cultures treated with biguanide alone ().
Figure 5. Phenformin and metformin sensitize cancer cells to cisplatin-induced cytotoxicity and loss of replicative potential. A) Live-dead cell assays show significantly (p < .02) reduced cell numbers in cultures of the sensitive A549 and Hs578T cells after 72 hour treatments with either metformin (1 mM) or phenformin (10 µM), compared to control cultures. This reduction in cell number is not seen with HCC1806 cells. Treating all cultures with 0.2 µM cisplatin did not significantly result in decreased cell numbers after 72 hr treatment, compared to control cultures. Treatments with metformin, phenformin, or cisplatin did result in increased numbers of dead cells in A549 cultures, but not in Hs578T or HCC1806 cultures. Notably, the combination of cisplatin with either metformin or phenformin resulted in markedly increased numbers of dead cells in the A549 and Hs578T cultures compared to control or to cultures treated with any single agent (p < .001). Increases in dead cells with a combination of cisplatin with either metformin or phenformin resulted in markedly increased numbers of dead cells in HCC1806 cultures, but to a lesser extent than what occurred in A549 or Hs578T cultures. B) In clonogenic assays where cultures plated at clonal density and allowed to grow for seven days, A549 or Hs578T cultures treated with 10 µM phenformin for the first three days show severely stunted growth. Cisplatin treatment alone (200 nM for Hs578T and 500 nM for A549) for the first three days shows only modest changes in growth, but combined phenformin plus cisplatin also shows markedly decreased growth. C) To determine whether various treatments for three days affects replicative ability survival, treated cultures were allowed to fully recover by culturing cells in standard media for seven days after completion of treatments. After this full recovery, cultures that had been treated with phenformin for three days show the ability to form colonies, as do cultures treated with cisplatin, indicating that these treatments individually are not cytotoxic. However, cultures treated with both phenformin and cisplatin do not recover, showing substantially decreased ability to form colonies even after culture with seven additional days of standard media. D) Reduced survival with combined cisplatin and biguanide treatment in A549 cells is prevented by addition of aspartate or NAD+ to culture media
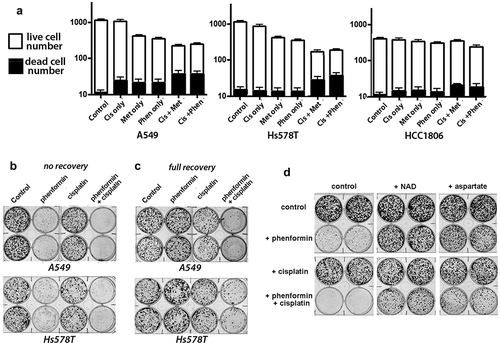
Treatments with metformin, phenformin, or cisplatin individually did result in modestly increased numbers of dead cells in A549 cultures, but not in Hs578T or HCC1806 cultures. Most notably, combining cisplatin with either metformin or phenformin resulted in markedly increased numbers of dead cells in the A549 and Hs578T cultures compared to control or any single agent (p < .001 for all comparisons) (). Combinations of cisplatin with either metformin or phenformin also resulted in increased numbers of dead cells in HCC1806 cultures, but to a lesser extent than what occurred in A549 or Hs578T cultures.
Assays to measure cells that exclude vital dyes or apoptotic cells do not fully reflect loss of cell viability, since necrotic and apoptotic cells invariably disintegrate over time, and because damaged cells that no longer have the ability to replicate can still appear live in vital dye exclusion assays. Accordingly, to measure the effect of these biguanide and cisplatin treatments on the ability of cells to replicate, we performed colony growth assays. Using the same strategy described above in , we compared colony growth of cells after three-day treatments with cisplatin or phenformin – alone or in combination – to control cultures, with all cultures after seven days total, including the three days of treatment, or stained after seven days in standard media without drugs. Again consistent with previous results, phenformin treatment alone (10 µM) resulted in stunted colony growth, while treating cells with cisplatin alone (500 nM for A549 and 200 nM for Hs578T) showed no effects or modest effects on colony number and size, () that could not be readily differentiated from the effect of cisplatin plus phenformin in this assay. To more accurately reflect cell viability, we conducted an assay where drug-treated cultures were allowed to “recover” and grow for seven full days after the completion of drug treatments. After allowing this full recovery, we showed that although the dose of phenformin tested (10 µM) inhibits cell proliferation, it does not decrease cell viability. However, this inhibitory level of phenformin substantially reduced survival of both A549 and Hs578T cells co-treated with cisplatin at doses that do not alone affect cell proliferation or survival ().
Additional experiments showed that addition of either exogenous NAD or exogenous aspartate (10 mM) markedly protects cancer cells from these cytotoxic effects of combined phenformin and cisplatin (), indicating that metabolite depletion (NAD+ and aspartate) by biguanide treatment is responsible for potentiating the cell lethality of cisplatin. These findings of enhanced cell killing by co-treatment with doses of phenformin that inhibit cell replication (but not reduce viability) and doses of cisplatin that alone show no significant effect on cell survival or replication suggest that biguanides might be more effective for enhancing the efficacy of agents such as cisplatin than as a single agent in cancer treatment.
Discussion
Ongoing interest in the potential use of metformin in cancer therapy has led to numerous laboratory studies as well as several clinical trials. Understanding mechanisms for the anti-cancer effects of metformin remain a research priority, with an expectation that understanding these mechanisms could help improve the use of these drugs in cancer treatment. Early studies, focusing on biguanides as anti-diabetic drugs, led to recognition that biguanides inhibit complex 1 in mitochondria. More recent studies focusing on effects of biguanides on cancer have led to recognition that the primary effects of biguanides against cancer are due to direct effects on cancer cells, rather than due to systemic effects, such as lowering insulin levels. Moreover, studies over recent years have shown that biguanides lead to depletion of metabolic intermediates, including ATP, NAD+, and aspartate.
Identifying a specific metabolic intermediate that is critical for the effects on cancer cells remains elusive. Our results point to depletion of both aspartate and NAD+, rather than one metabolite individually, as being key for the growth inhibitory effects of biguanides. This conclusion is supported by our findings that addition of either aspartate or NAD+ to media of biguanide-treated cancer cells can ameliorate the growth-inhibitory effects of the drug, even without increasing cellular concentrations of the other metabolite. In other experiments, we sought to distinguish the importance of replenishing aspartate from that of replenishing NAD by using CRISPR/Cas9 editing to knock out the genes for GOT1 and MDH1 enzymes in A549 cells; thus, specifically targeting the ability of the aspartate-malate shuttle to replenish aspartate and NAD. Most notably, in cultures with knock-out of GOT1, the protective effects of NAD and aspartate were similar, suggesting that neither NAD nor aspartate alone is a principal metabolite that critically depleted by biguanide exposure.
Previous studies have pointed to aspartate biosynthesis as the critical function of cellular respiration for maintaining cellular proliferation,Citation12,Citation13,Citation23,Citation24 while other studies provide evidence to indicate that metformin’s anti-proliferative effect is due to both decreases in NAD+ and inhibition of aspartate biosynthesis,Citation14,Citation25 In our studies, phenformin treatment resulted in decreases in both the NAD+/NADH ratio and cellular levels of aspartate, and growth inhibitory effects of phenformin could be overcome by replenishing NAD without increasing cellular aspartate levels, or by replenishing aspartate without increasing the NAD+/NADH ratio. While the available data overall seem to be unable to implicate either aspartate or NAD+ as a single metabolite critical for mediating the growth inhibitory effects of biguanides on cancer cells, it appears likely that depletion of both NAD+ and aspartate are necessary for growth inhibition by biguanide drugs.
More important for clinical application of biguanides in cancer therapy, our experiments demonstrate that over a 10-fold dose range, the effect of biguanide drugs on cancer cells as a single agent is limited to reversibly inhibiting cell proliferation. Thus, as single agents, these drugs might have limited efficacy in treatment of established tumors. However, substantial killing of tumor cells is seen when cells are treated with a combination of a biguanide (either metformin or phenformin) and cisplatin, even at doses of the drugs that individually do not appreciably affect cell growth or survival. These laboratory findings correspond to the markedly reduced tumor size seen in mice with A549 xenografts that were treated with a combination of metformin and cisplatin compared to either drug alone,Citation15 as well as the improved clinical responses of patients who are treated simultaneously with metformin and platin-based chemotherapy.
Materials and methods
Cell culture, reagents, and treatments: All cell lines (A548, BT549, H460, H1975, HCC1805, Hs578T, MDA-MB-231, OVCAR-5, OVCAR-8, and U1752) were obtained from the ATCC, maintained in recommended media, and cultured at 37°C/5% CO2. Cell cultures in the laboratory were tested for mycoplasma contamination and identify verified by short tandem repeat profiling. Phenformin hydro-chloride, sodium oxamate, L-Aspartic acid potassium salt, Cisplatin, and B-Nicotinamide adenine dinucleotide hydrate were purchased from Sigma Aldrich, and metformin was purchased from Millipore Sigma.
Cell survival and growth assays: To measure numbers of cultured cells after various treatments, cells were plated in triplicate in 6-well culture plates at a density of 1,000 cells per well. The next day, various agents were added to wells at indicated concentrations and cells were incubated for 72 hours before replacing treatment media with standard media and incubating for an additional 4 days (to detect effects of treatment on cell proliferation), or an additional 7 days (to detect effects of treatment on proliferation potential). The cell plates were then fixed in 100% ethanol and stained with crystal violet. Effects of treatments on cell survival (live/dead cell assay) were also quantitatively determined by detaching cells from culture plate wells after 72 hours of treatment, preparing a 1:1 dilution of cell suspension using a 0.4% Trypan Blue solution, and counting cells with a hemocytometer. All cell growth assays were independently repeated at least one time.
CRISPR/Cas-9 knock-out of GOT1 and MDH1: For knockout of GOT1 and MDH1, LentiCRISPRv2 plasmid was annealed to oligonucleotides specific for target genes as follows: GOT1 (Forward 1: 5ʹ- CACCGCAAGCTCACTGCCGACTTCA-3′, Reverse 1: 5ʹ- AAACTGAAGTCGGCAGTGAGCTTGC-3′, or Forward 2:5′- CACCGTCACGAGTATCTGCCAATCC-3′, Reverse 2:5′-AAACGGATTGGCAGATACTCGTGAC-3′); MDH1 (Forward 1: 5ʹ- CACCGCCAATCAGAGTCCTTGTGAC-3′, Reverse 1:5ʹ AAACGTCACAAGGACTCTGATTGGC-3′, or Forward 2:5′- CACCGTCCAGATGTCAACCATGCCA-3′, Reverse 2:5′- AAACTGGCATGGTTGACATCTGGAC-3′. Plasmid constructs were sequence verified and transfected into A549 cells using methods described previously.Citation26
Measurements of NAD+/NADH
NAD+/NADH was measured using the NAD/NADH Glo Assay from Promega. In brief, cells were plated in six well dishes at a density of 20,000 cells/well. The next day, cells were treated for 6 hours with specified agents, then washed 3X in ice-cold PBS, extracted in 100ul extraction buffer, (0.2 N NaOH +1% DTAB diluted 1:1 in PBS), and frozen at −80 C. For measuring NAD+, 0.4 N HCL was added to aliquots at recommended levels before incubating at 60°C for 15 minutes, followed by neutralizing samples with recommended levels of 0.5 M Trizma base. For measuring NADH, samples were heated for 30 min at 75°C, and after cooling to room temperature, samples were neutralized by 40 µl of HCL/Trizma to each tube. The luciferin Glo detection reagent was added as recommended and luminescence was measured using a luminometer, and all measurements were repeated in independent experiments.
Magnetic resonance spectroscopy
Approximately 108 cells in each treatment group were harvested by trypsinization, rinsed three times with PBS, counted, and stored at −80°C until assayed. Stored samples were then thawed on ice, mixed with 650 µL of deuterated PBS and sonicated for 3 min under ice-cooled condition to make homogenized cell lysate, and centrifuged at 12 K rpm. High-resolution 1 H MR spectra were then acquired on supernatants using a Bruker Biospin Avance-III 750 MHz NMR spectrometer with a 5-mm broad band inverse (BBI) probe. Proton (1 H) MR Spectra were also acquired with a one-dimensional CPMG pulse sequence with water suppression. Spectral processing, metabolic identification and quantification are described in previously reported NMR methods.Citation27
Acknowledgments
The Lungevity Foundation is acknowledged for support to initiate these studies. Dr. Anne Le is acknowledged for providing helpful critques and suggestions in our studies.
Disclosure statement
All authors contributed to the conception, design, and interpretation of data, and to the composition of the manuscript. ET, SB, and JW contributed to the acquisition of data. The authors report no potential financial conflicts of interest.
Additional information
Funding
References
- Bowker SL, Majumdar SR, Veugelers P, Johnson JA. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin: response to farooki and schneider. Diabetes Care. 2006;29(8):1990–1991. doi:https://doi.org/10.2337/dc06-0997.
- Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JM. New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care. 2009;32(9):1620–1625. doi:https://doi.org/10.2337/dc08-2175.
- Decensi A, Puntoni M, Goodwin P, Cazzaniga M, Gennari A, Bonanni B, Gandini S. Metformin and cancer risk in diabetic patients: a systematic review and meta-analysis. Cancer Prev Res (Phila). 2010;3(11):1451–1461. doi:https://doi.org/10.1158/1940-6207.CAPR-10-0157.
- Jiralerspong S, Palla SL, Giordano SH, Meric-Bernstam F, Liedtke C, Barnett CM, Hsu L, Hung M-C, Hortobagyi GN, Gonzalez-Angulo AM, et al. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J Clin Oncol. 2009;27(20):3297–3302. doi:https://doi.org/10.1200/JCO.2009.19.6410.
- Marrone KA, Zhou X, Forde PM, Purtell M, Brahmer JR, Hann CL, Kelly RJ, Coleman B, Gabrielson E, Rosner GL, et al. A randomized phase ii study of metformin plus paclitaxel/carboplatin/bevacizumab in patients with chemotherapy-naive advanced or metastatic nonsquamous non-small cell lung cancer. Oncologist. 2018;23(7):859–865. doi:https://doi.org/10.1634/theoncologist.2017-0465.
- Tan BX, Yao WX, Ge J, Peng XC, Du XB, Zhang R, Yao B, Xie K, Li L-H, Dong H, et al. Prognostic influence of metformin as first-line chemotherapy for advanced nonsmall cell lung cancer in patients with type 2 diabetes. Cancer. 2011;117(22):5103–5111. doi:https://doi.org/10.1002/cncr.26151.
- Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000;348(3):607–614. doi:https://doi.org/10.1042/bj3480607.
- El-Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. 2000;275(1):223–228. doi:https://doi.org/10.1074/jbc.275.1.223.
- Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007;67(22):10804–10812. doi:https://doi.org/10.1158/0008-5472.CAN-07-2310.
- Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9(8):563–575. doi:https://doi.org/10.1038/nrc2676.
- Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006;66(21):10269–10273. doi:https://doi.org/10.1158/0008-5472.CAN-06-1500.
- Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, Sabatini DM. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell. 2015;162(3):540–551. doi:https://doi.org/10.1016/j.cell.2015.07.016.
- Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, Vander Heiden MG. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell. 2015;162(3):552–563. doi:https://doi.org/10.1016/j.cell.2015.07.017.
- Gui DY, Sullivan LB, Luengo A, Hosios AM, Bush LN, Gitego N, Davidson S, Freinkman E, Thomas C, Vander Heiden M, et al. Environment Dictates Dependence on Mitochondrial Complex I for NAD+ and Aspartate Production and Determines Cancer Cell Sensitivity to Metformin. Cell Metab. 2016;24(5):716–727. doi:https://doi.org/10.1016/j.cmet.2016.09.006.
- Wang J, Gao Q, Wang D, Wang Z, Hu C. Metformin inhibits growth of lung adenocarcinoma cells by inducing apoptosis via the mitochondria-mediated pathway. Oncol Lett. 2015;10(3):1343–1349. doi:https://doi.org/10.3892/ol.2015.3450.
- Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, Glasauer A, Dufour E, Mutlu GM, Budigner GS, et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife. 2014;3:e02242. doi:https://doi.org/10.7554/eLife.02242.
- Miskimins WK, Ahn HJ, Kim JY, Ryu S, Jung YS, Choi JY, Moreno-Sanchez R. Synergistic anti-cancer effect of phenformin and oxamate. PLoS One. 2014;9(1):e85576. doi:https://doi.org/10.1371/journal.pone.0085576.
- Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV, et al. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A. 2010;107(5):2037–2042. doi:https://doi.org/10.1073/pnas.0914433107.
- King MP, Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science. 1989;246(4929):500–503. doi:https://doi.org/10.1126/science.2814477.
- Knott SRV, Wagenblast E, Khan S, Kim SY, Soto M, Wagner M, Turgeon M-O, Fish L, Erard N, Gable AL, et al. Asparagine bioavailability governs metastasis in a model of breast cancer. Nature. 2018;554(7692):378–381. doi:https://doi.org/10.1038/nature25465.
- Trimmer EE, Essigmann JM, Ballou DP. Cisplatin. Essays Biochem. 1999;34:191–211. doi:https://doi.org/10.1042/bse0340191.
- Florea AM, Busselberg D. Cisplatin as an anti-tumor drug: cellular mechanisms of activity, drug resistance and induced side effects. Cancers (Basel). 2011;3(1):1351–1371. doi:https://doi.org/10.3390/cancers3011351.
- Sullivan LB, Luengo A, Danai LV, Bush LN, Diehl FF, Hosios AM, Lau AN, Elmiligy S, Malstrom S, Lewis CA, et al. Aspartate is an endogenous metabolic limitation for tumour growth. Nat Cell Biol. 2018;20(7):782–788. doi:https://doi.org/10.1038/s41556-018-0125-0.
- Garcia-Bermudez J, Baudrier L, La K, Zhu XG, Fidelin J, Sviderskiy VO, Papagiannakopoulos T, Molina H, Snuderl M, Lewis CA, et al. Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat Cell Biol. 2018;20(7):775–781. doi:https://doi.org/10.1038/s41556-018-0118-z.
- Cameron AR, Logie L, Patel K, Erhardt S, Bacon S, Middleton P, Harthill J, Forteath T, Coats J, Kerr C, et al. Metformin selectively targets redox control of complex I energy transduction. Redox Biol. 2018;14:187–197. doi:https://doi.org/10.1016/j.redox.2017.08.018.
- Walker A, Singh A, Tully E, Woo J, Le A, Nguyen T, Biswal S, Sharma D, Gabrielson E. Nrf2 signaling and autophagy are complementary in protecting breast cancer cells during glucose deprivation. Free Radic Biol Med. 2018;120:716–727. doi:https://doi.org/10.1016/j.freeradbiomed.2018.04.009.
- Bharti SK, Wildes F, Hung CF, Wu TC, Bhujwalla ZM, Penet MF. Metabolomic characterization of experimental ovarian cancer ascitic fluid. Metabolomics. 2017; 13:113. doi:https://doi.org/10.1007/s11306-017-1254-3.