
Abstract
High mortality among hepatocellular carcinoma (HCC) patients reflects both late diagnosis and low curability, due to pharmacoresistance. Taxol (TAX) is toxic for many human HCC-derived cell lines, yet its clinical efficacy on HCCs is poor. Combining TAX with other drugs appears a promising possibility to overcome such refractoriness. We analyzed whether combining tumor necrosis factor (TNF) with TAX would improve their toxicity. Human HCC-derived cell lines were treated with TAX or TNF, alone or combined. Apoptosis was assessed by morphology and flow-cytometry. Several pro- and anti-apoptotic molecules were evaluated by western blotting and/or enzymatic assay. After a 24 hour treatment, TNF was ineffective and TAX modestly cytotoxic, whereas HCC cells were conditionally sensitized to TNF by TAX. Indeed some relevant parameters were shifted to a prodeath setting: TNF-receptor 1 was increased, SOCS3, c-FLIP and pSTAT3 were markedly downregulated. These observations provide a significant clue to critically improve the drug susceptibility of HCC cells by combining 2 agents, TAX and TNF. The sequential application of TAX at a low dosage followed by TNF for only a short time triggered a strong apoptotic response. Of interest, prior TAX administration could also sensitize to TNF-induced apoptosis in the Yoshida AH-130 hepatoma transplanted in mice. Therefore, scrutinizing the possibility to develop similar combination drug regimens in suitable preclinical models seems highly advisable.
Abbreviations
| COL | = | colchicine |
| DAPI | = | 4,6-diamidino-2-phenylindole dihydrochloride |
| HCC | = | hepatocellular carcinoma |
| NOC | = | nocodazole |
| SOCS3 | = | suppressor of cytokine signaling 3 |
| STAT3 | = | signal transducer and activator of tanscription 3 |
| TAX | = | taxol (paclitaxel) |
| TNF | = | tumor necrosis factor-α |
| TNF-R1 | = | TNF-receptor 1 |
| TRAIL | = | tumor necrosis factor-related apoptosis-inducing ligand |
Introduction
Taxol (TAX, Paclitaxel), originally isolated from the yew tree Taxus brevifolia, is prototypic of a family of powerful anticancer agents collectively known as taxanes. These spindle poisons share a unique mechanism of action: they bind to polymerized β-tubulin, profoundly alter its dissociation constant at both microtubule ends and promote β-tubulin polymerization. The results are suppression of treadmilling and dynamic instability causing stabilization of microtubules in a straight conformation that prevents cell division.Citation1 Proliferating cells thereby undergo an arrest at the G2/M phase of the mitotic cycle,Citation2 more precisely in metaphase, followed by death, as reported for a variety of human neoplastic lines.Citation3-6 Commonly used in the treatment of solid tumors such as breast, head and neck, ovarian and non-small-cell lung carcinomas even as a front-line drug,Citation7 often their clinical efficacy is heavily hampered by chemoresistance, either primary (constitutive) or secondary (acquired as a consequence of treatment), due to a multiplicity of mechanisms.Citation8,9
HCC ranks as the sixth most frequent solid malignancy and the fifth leading cause of cancer-associated death worldwide.Citation10 Since generally diagnosed too late for surgical resection or transarterial chemoembolization to be curative, search for effective chemotherapies is a major task in this field. TAX, which easily crosses the plasma membrane, exerts a marked toxicity on HCC-derived cell lines, as shown by numerous studies,Citation3,6,Citation8,11 and significantly inhibits both tumor growth and angiogenesis in human HCCs with high metastatic potential xenografted into nude mice.Citation12 TAX compromises viability of human HCC cell lines even at quite low concentrations, just greater than 10 nM,Citation3 with IC50 values of 80 nM to 1 μM, as respectively observed in death prone and death reluctant HCC cells.Citation13 Yet most HCCs are refractory to taxanes and TAX efficacy proved negligible in phase ICitation14 and phase IICitation15 trials.
The therapeutic potential of this mitotic poison family was explored by studies aimed at unveiling the basis for the 'constitutive' drug resistance of HCCs. Function (activity/expression) of prosurvival factors was found enhanced and, by contrast, that of prodeath factors decreased in HCCs, not differently from other cancers. In particular, the imbalance between cell death and survival in HCC mainly reflects multiple modulations of antiapoptotic molecules including c-FLIP, Bcl-2/Bcl-xL Citation8 and/or signaling pathways such as RAS/ERK, PI3K/Akt, JAK/STAT, as well as defective proapoptotic mediation by p53, TGF-β, Fas, TRAIL.Citation16 Recently, an important role in the response to TAX has been advocated for the JNK activation state by Chae et al.,Citation13 who distinguished death prone and death reluctant HCC cell lines: on TAX administration, Bcl-2 was highly phosphorylated in the former, much less in the latter. Both degree of Bcl-2 phosphorylation and death of prone cells were strongly attenuated by the JNK inhibitor SP600125, whereas death reluctant cells had high inbuilt levels of phospho-JNK, JNK being much less or not any further phosphorylated upon TAX treatment. Interestingly, JNK activation was also suggested to play a role, though still debated, in HCC cell apoptosis by death receptor ligands such as TRAIL and TNF.Citation17
A different line of investigations addressed the design of combined treatments, testing a number of simultaneous or sequential associations of TAX with other antineoplastic agents. For example, TAX exhibits a synergistic lethal action on HCC cell lines when used in combination with oncolytic adenoviruses,Citation18 whereas TAX and doxorubicin exert an additive toxicity on HepG2 cells and cause a regression of the H22 tumor in mice significantly higher than that afforded by either drug individually.Citation19 Recently, the synergistic effects of IAP inhibitor LCL161 and TAX on HCC cell lines has been reported.Citation20 Along this line, a number of studies also evaluated the efficacy of combining TAX and alike drugs with death receptor ligands, particularly TRAIL and TNF (see Discussion). In this regard, the transcription factor STAT3 was reported to play a critical role in TAX sensitization to TRAIL or TNF, ascribed to decreased activity owing to its reduced degree of Tyr-phosphorylation.
The concept that sequential application of anticancer drugs can enhance neoplastic cell death by rewiring apoptotic signaling networks was worked out by Lee et al.Citation21 based on the observation that breast cancer cells were dramatically sensitized to genotoxic drugs by time-staggered EGFR inhibition, though not by simultaneous cotreatment. A distinct yet equally effective mode of rewiring that sensitizes cancer cells to apoptosis is the object of the present report, which is part of a line of investigations developed in our laboratory to dissect the mechanisms of the deadly action on HCC cells by TNF, alone or combined with other drugs.Citation17,22-24 We presently sought whether associating TAX with TNF - which individually display only moderate or no toxicity at all, respectively - would significantly enhance their deadly action on 2 different human HCC-derived cell lines, viz., Huh7 and HepG2. Both lines, but Huh7 in particular, are dramatically sensitized to TNF-induced death by simultaneous or prior exposure to TAX. Adopting various schedules of concurrent or sequential treatment disclosed a very marked toxicity when TAX and TNF were applied simultaneously or even in sequence, though not in the reverse order, at appropriate time intervals, sufficient for TAX to bring most cells to a G2/M cycle phase arrest and to reprogram relevant apoptotic pathways. These results will hopefully provide a frame of reference for future developments at a preclinical and possibly clinical level of checking.
Results
Effects of TAX on HCC cell lines
In Huh7 and HepG2 cells treated with 0.1 to 5 μM TAX for up to 24 hours the percentage of apoptotic (hypodiploid or sub-G1 or hypochromic, with <2n DNA fluorescence) cells significantly increased in a time- and dose-dependent manner (). At the lowest TAX concentration (0.1 μM), the growth curve was equally flat for both lines (), which denotes a complete growth arrest, with most cells exhibiting a rounded-up morphology and only few cells plasma membrane blebbing (). After 24 hours, an almost complete G2/M arrest () associated with a roundish morphology () were induced by TAX in both lines. Huh7 cells appeared more susceptible to TAX than HepG2 ( and ). The fraction of sub-G1 (apoptotic) cells in TAX-treated Huh7 cultures, while not differing from controls at 6 hours (about 8%), significantly increased up to 23% at 24 hours (). In TAX-treated HepG2 cultures the percentage of hypodiploid cells at 6 hours (about 5%) did not differ from controls, but at 24 hours approximated 15% ().
Figure 1. Effect of TAX on growth, morphology and survival of human HCC cells. (A) and (B) Flow-cytometric evaluation of apoptotic Huh7 and HepG2 cells after 6 and 24 hours of treatment with TAX at the concentrations indicated. Histograms report the percentage of hypodiploid (apoptotic) cells. Data are means±SD of 4 experiments. Significance of the differences vs controls: *** P < 0.001. (C) and (D) Total number of cells treated with 0.1 μM TAX for up to 24 hours. Data are means±SD of 4 experiments. Significance of the differences vs controls: ***P < 0.001. (E) and (F) Phase contrast pictures of cells untreated or treated with 0.1 μM TAX for 6 and 24 hours. Bars 30 μm. Arrows indicate apoptotic-like figures.
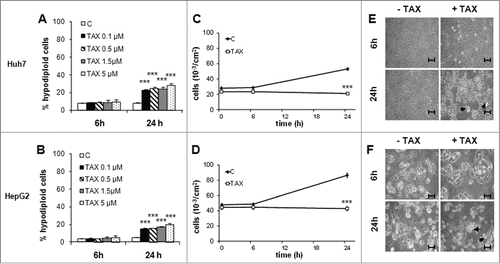
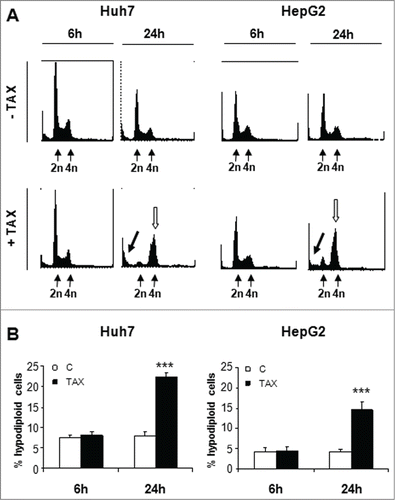
Figure 2. Effects of TAX on DNA distribution in Huh7 and HepG2 hepatoma cells, (A) and (B) DNA distribution and apoptosis quantification. Cultures exposed to 0.1 μM TAX for 6 or 24 hours. Left panels: Huh7 cells; right panels: HepG2 cells. (A) Black arrows indicate the sub-G1 population, white arrows indicate G2/M arrested cells. (B) respective fraction of apoptotic cells (hypodiploid DNA content). Data are means±SD of 4 experiments. Significance of the differences vs C: ***P < 0.001.
TAX sensitizes HCC cell lines to TNF cytotoxicity
In agreement with previous reports on HCC cell lines,Citation22,25-27 Huh7 and HepG2 cells both revealed totally resistant to TNF alone, the percentage of hypodiploid cells not differing between control and cytokine-treated cultures ()
Figure 3. TNF potentiates death induced by microtubular poisons and zVADfmk counteracts death by TAX-TNF. (A) Huh7 and HepG2 cells exposed for 24 hours to 0.1 μM TAX , 0.2 μg/mg nocodazole (NOC) or 10 μM colchicine (COL), alone or combined with 15 ng/ml TNF. Histograms indicate the percentage of hypodiploid (apoptotic) cells. Data are means±SD of 4 experiments. Significance of the differences vs untreated controls: ***P < 0.001; vs TAX: °°P < 0.01 and °°°P < 0.001; vs nocodazole: ###P < 0.001; vs colchicine §§§P < 0.001. (B) Cells exposed to 0.1 μM TAX for 12, 18, 24 and 30 hours with TNF added for the last 6 hours into the cultures to a final concentration of 15 ng/ml, or to TNF for 30 hours with TAX added for the last 6 hours, and finally to TAX alone or TAX and TNF combined for 30 hours, as indicated. Histograms indicate the percentage of hypodiploid (apoptotic) cells. Data are means±SD of 4 experiments. Significance of the differences vs untreated controls: **P < 0.01 and ***P < 0.001; vs TAX: °P < 0.05, °°P < 0.01 and °°°P < 0.001. (C) Effect of zVADfmk on apoptotic death in cells exposed to TAX or TAX+TNF for 24 hours. zVADfmk (20 μM final concentration) was added to the cultures 1 h before a 24 hour treatment with 0.1 μM TAX and TAX plus 15 ng/ml TNF. Histograms represent percentages of hypodiploid cells (apoptotic). Significance of the differences vs C: ***P < 0.001; vs TAX: °°P < 0.01 and °°°P < 0.001; vs TAX+TNF: §§§P < 0.001.
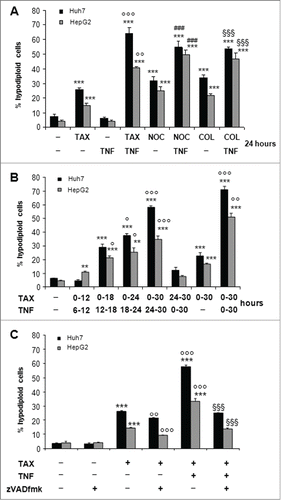
To evaluate a possible interaction between TAX and TNF, HCC cells were first exposed simultaneously to both drugs for 24 hours. Compared to TAX alone, the combined treatment resulted in a significant increase of the apoptotic population, from about 25% to 65% for Huh7 and from about 15% to 43% for HepG2 cells (). In an attempt to discriminate the respective role of the 2 drugs when used in combination, cells incubated in the presence of TAX for 12 to 30 hours were exposed also to TNF in the last 6 hours only and, reciprocally, cells treated with TNF for 30 hours were exposed to TAX in the last 6 hours. Other cultures were treated for 30 hours with TAX alone or combined with TNF. As depicted in , treatment with TAX followed by TNF significantly and time-dependently enhanced the fraction of hypodiploid cells, although the latter remained below that caused by 30 hour TAX+TNF treatment. By contrast, the percentage of apoptotic cells did not differ from controls when TAX was added for the last 6 of a 30 hour exposure to TNF.
Noteworthily, other spindle poisons such as nocodazole (NOC) or colchicine (COL), which affect microtubules through different mechanisms, had basically similar effects to those obtained with TAX on the viability of HCC cells. In fact, the percentage of apoptosing cells was comparable among cultures exposed to TAX, NOC or COL (), suggesting that sensitization of HCC cells to TNF, rather than being TAX-specific, may reflect a common effect on cells.
Caspase activation
To investigate whether caspases were involved in HCC cell death, the TAX+TNF treatment was carried out in the presence of the polycaspase inhibitor zVADfmk. As shows, 20 μM zVADfmk markedly reduced apoptosis in both cell lines, approximately down to the level observed in cells treated with TAX alone. Interestingly, while TAX-induced death proved virtually caspase–independent, in that only minimally suppressed by zVADfmk, apoptosis by TAX+TNF was largely dependent on caspases, particularly in Huh7 cells.
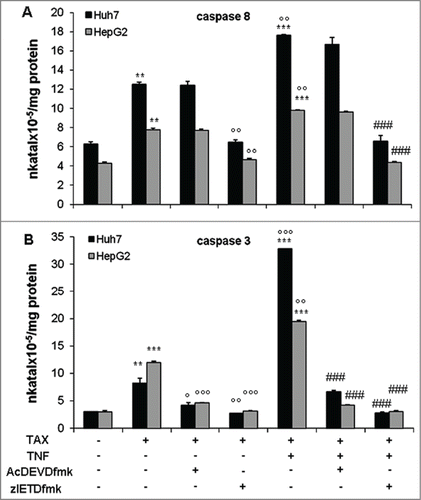
The occurrence of caspase activation was confirmed by enzymatic assays. On TAX treatment, caspase 8 activity was increased in Huh7 cells at 24 hours, while activation was already evident after 2 hours on TAX+TNF (). In HepG2 cells caspase 8 activity gradually increased from 2 to 24 hours both on TAX and, to a higher degree, on TAX+TNF (). In both cell lines, activation of caspases 3 and 9 was also observed, particularly in cultures exposed to TAX+TNF (); of interest, the increase of caspase 9 activity was detectable only after a 24 hour treatment, suggesting that its role in TAX+TNF-induced apoptosis is marginal. Moreover, the caspase 8 inhibitor zIETDfmk suppressed both caspase 8 and 3 activation, which is a plausible indication of a causal link between the 2 processes, whereas AcDEVDcmk only reduced caspase 3 (). These data are all consistent with an involvement of the extrinsic pathway in TAX+TNF-induced apoptosis.
Figure 4. Caspase 8, 9, and 3 activity in HCC cells treated with TAX or TAX+TNF. Huh7 (left panels) and HepG2 (right panels) cells exposed to 0.1 μM TAX or TAX plus 15 ng/ml TNF for 2, 6, 12 and 24 hours. Caspase activities measured as indicated in Materials and Methods, data (means±SD ) expressed as nkatal×10–5/mg protein. Panels A and B, caspase 8; panels C and D, caspase 9; panels E and F, caspase 3. Significance of the differences vs controls: *P < 0.05, **P < 0.01 and ***P < 0.001; vs TAX: °P < 0.05, °°P < 0.01 and °°°P < 0.001.
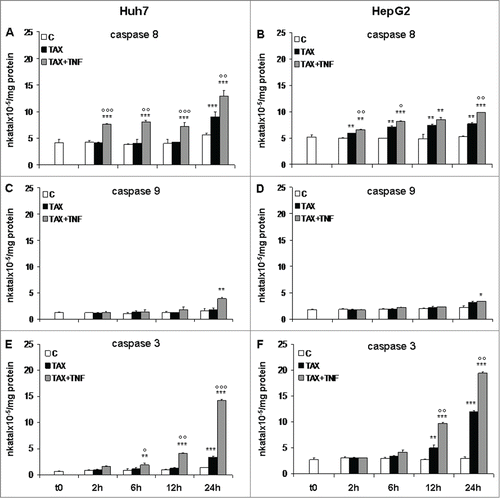
Figure 5. Effect of selective inhibitors on caspase 8 and 3 activities in HCC cells exposed to TAX or TAX+TNF for 24 hours. Caspase 8 (A) and 3 (B) activities in cells pretreated with AcDEVDcmk (20 μM) or zIETDfmk (20 μM) for 1 hour, then exposed to 0.1 μM TAX or TAX plus 15 ng/ml TNF for 24 hours. Data (means±SD ) are expressed as nkatal×10–5/mg protein. Significance of the differences vs controls: **P < 0.01 and ***P < 0.001; vs TAX: °P < 0.05, °°P < 0.01 and °°°P < 0.001; vs TAX+TNF: ###P < 0.001.
Effects of TAX on TNF-R1, SOCS3, c-FLIP and pSTAT3 levels
TNF cytotoxicity is a highly regulated process that integrates a multiplicity of positive or negative pathways and factors. A few among the latter were examined in the present work: total and cell surface expression of TNF-R1 and total levels of SOCS3 and c-FLIP, the first 2 being involved in TNF cytotoxic signal transduction, the third one in the activation of the caspase cascade that typifies the extrinsic apopotic mechanism.
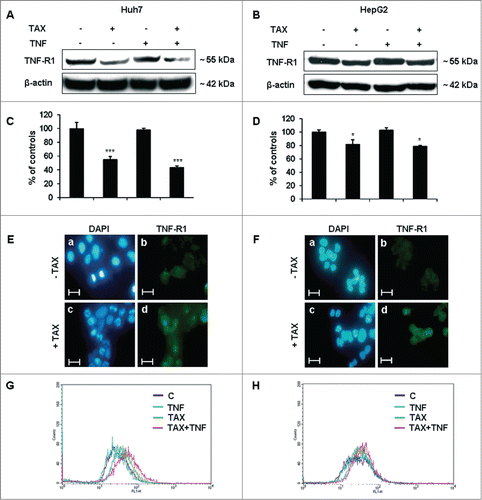
TNF-R1 levels were reduced in total extracts of both Huh7 and HepG2 cells exposed to TAX, alone or combined with TNF (). Cell-surface expression of the receptor was then evaluated on non-permeabilized cells by 2 different procedures. By immunofluorescence microscopy, TNF-R1 cell surface levels appeared higher in 24 hours TAX-treated cultures than in controls (). Although, to some extent, this finding could reflect the roundish morphology acquired by cells on TAX treatment, flow-cytometric assays indicated as well that TNF-R1 was significantly more expressed () at the surface of Huh7 cells treated with TAX and, even more, with TAX+TNF. Similar results were obtained in HepG2 cells, although TNF-R1 membrane expression did not further increase in TAX+TNF vs TAX-exposed cultures.
Figure 6. Effects of TAX and TAX+TNF (24 hours) on total and membrane TNF-R1 expression in HCC cells. Panels A and B show representative western blots of TNF-R1 in Huh7 and HepG2 cells treated with 0.1 μM TAX, 15 ng/ml TNF or TAX+TNF for 24 hours. Respective densitometric data are shown in panels C and D. Data (means±SD) are expressed as percentage of controls. Significance of the differences vs controls: *P < 0.05 and ***P < 0.001. Fluorescence microscopy of Huh7 (E) and HepG2 (F) cells after DAPI staining and immunodecoration of TNF-R1 at the cell surface. Bars 20 μm. Cell surface TNF-R1 expression measured in Huh7 (G) and HepG2 (H) cells by flow cytometry.
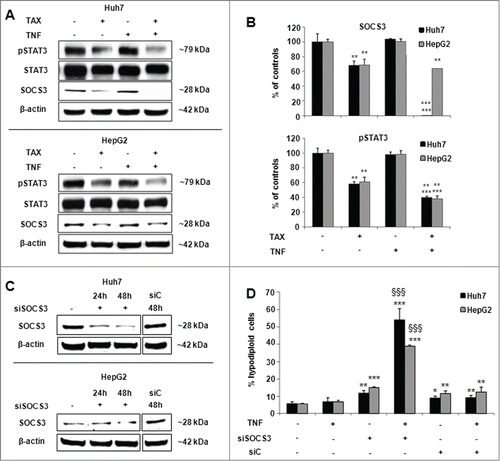
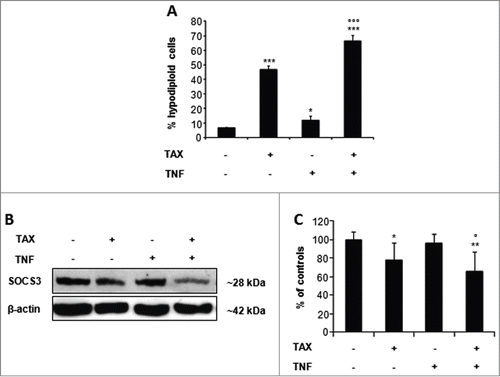
An important role in cytokine cytotoxicity is played by molecules known as SOCS, among which SOCS3 is involved in TNF action.Citation28,29 SOCS3 expression was markedly down-regulated in both Huh7 and HepG2 cells exposed to TAX and even more, though in Huh7 cells only, to TAX and TNF cotreatment (). To validate the relevance of SOCS3 down-regulation operated by TAX to TNF cytotoxicity, cells were knocked down for SOCS3 expression by the use of specific siRNA (siSOCS3). This manipulation resulted in an appreciable decrease of SOCS3 protein () and, consistently, in an increased fraction of apoptotic cells, already evident in the absence of any further treatment, but markedly enhanced on exposure of siSOCS3 cells to TNF ().
Figure 7. SOCS3 and STAT3 expression and effects of SOCS3 silencing in TAX and TAX+TNF-treated HCC cells. (A) Representative protein gel blots and (B) densitometric quantification of SOCS3, STAT3 and pSTAT3 levels in Huh7 and HepG2 cells after a 24 hour treatment with 0.1 μM TAX or 15 ng/ml TNF or TAX+TNF. Data are means±SD expressed as percentage of controls. Significance of the differences vs controls: **P < 0.01 and ***P < 0.001; vs TAX: °°°P < 0.001. (C) Representative western blot pattern of SOCS3 levels in HepG2 and Huh7 cells untreated or treated with siSOCS3 (100 pmol) for 24 and 48 hours. (D) Effect of SOCS3-siRNA or siC (100 pmol) on TNF-induced apoptosis: cells were transfected with siSOCS3 or nontargeting siRNA (siC) for 48 hours, then exposed to TNF (15 ng/ml) for further 24 hours. Histograms represent percentages of hypodiploid (apoptotic) cells in the culture. Data are means±SD of 4 experiments. Significance of the differences: vs controls **P < 0.01 and ***P < 0.001; vs TNF and siSOCS3: §§§P <0.001.
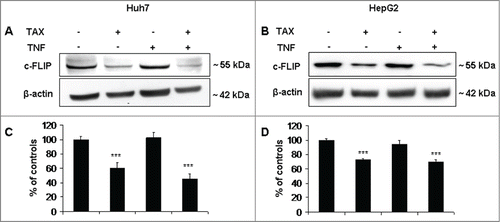
The level of c-FLIP, the physiological caspase 8 inhibitor, is known to undergo important up- or downregulations that significantly impact on the apoptotic susceptibility of cells. As shows, this level was significantly decreased in both Huh7 and, to a lesser extent, HepG2 cells by TAX or TAX+TNF treatment. Again, this change is consistent with the sensitizing action of TAX toward TNF in HCC cells.
Figure 8. c-FLIP protein levels in HCC cells treated with TAX or TAX+TNF. (A and B) show representative protein gel blots of c-FLIP in Huh7 and HepG2 cells treated with 0.1 μM TAX or 15 ng/ml TNF or TAX+TNF for 24 hours. (C and D) show the respective densitometric data. Data (means±SD ) are expressed as percentage of controls. Significance of the differences vs controls: ***P < 0.001.
Finally, Walker et al.Citation30,31 recently reported an important inhibiting effect of TAX and other microtubular drugs on STAT3. This transcription factor modulates the expression of genes among which c-FLIP and SOCS3 as a function of the degree of its Tyr-phosphorylation. This observation prompted us to evaluate the STAT3 phosphorylation status in the present experimental models. The pSTAT3 level markedly declined in both Huh7 and HepG2 cells exposed to TAX, either alone or combined with TNF, roughly paralleling SOCS3 level changes (); however, SOCS3 was almost undetectable in Huh7 cells treated with TAX+TNF.
In conclusion, whether combined or not with TNF, TAX determines changes in all the parameters herewith investigated in such a way that they should expectedly converge in enhancing the susceptibility of HCC cells to TNF. Importantly, these changes were definitely less pronounced in HepG2 than in Huh7 cells, in agreement with their dissimilar vulnerability to TAX+TNF. Whether these observations may relate to the reportsCitation32,33 of an enhanced proliferative capacity of hepatocytes in mice carrying a specific hepatocytic deletion of SOCS3 or in HCC cell lines silenced for SOCS3 expressionCitation34 is presently unclear.
Potentiation of TAX-induced cytotoxicity by TNF in a transplantable hepatoma
Preliminary experiments on a transplantable ascites hepatoma were run in order to test if the TAX-induced potentiation of TNF cytotoxicity observed in human HCC cell lines also occurs in the AH-130 Yoshida ascites hepatoma transplanted in mice. As shows, TNF or TAX alone increased the number of apoptotic (sub-G1) cells, slightly the former and markedly the latter. Simultaneous administration of the 2 drugs resulted in a further increase in the fraction of sub-G1 cells (). Qualitatively, the pattern is consistent with that reported for both Huh7 and HepG2 cells. Moreover, SOCS3 levels underwent a significant reduction in TAX-treated tumors that was further enhanced when TAX was coupled with TNF (), similarly to what observed on HCC cells.
Figure 9. TAX+TNF-induced cytotoxicity in a transplantable hepatoma. (A) Sub-G1 fraction (% of total cell population) in AH-130 hepatoma cells obtained from mice treated with TAX (4 mg/kg body weight), TNF (10 μg/mouse), or both (for treatment schedule see Materials and methods). Data are means±SD of 4 animals/group. Significance of the differences vs controls: *P < 0.05, ***P < 0.001; vs TAX °°°P < 0.001. (B) Representative western blot of SOCS3 in AH-130 hepatoma cells. (C) Respective densitometric data are means±SD expressed as percentage of untreated controls; significance of the differences vs controls: *P < 0.05, **P < 0.01; vs TAX °P < 0.05.
Discussion
TAX is poorly effective in the therapy of human HCCs,Citation14,15 despite its capability to induce apoptosis in vitro in a variety of human HCC-derived cell lines. This failure, either manifest ab initio or emerging in the course of treatment, spurred many efforts to decipher the mechanisms underlying TAX chemoresistance. Alternatively, a variety of procedures were devised to overcome such refractoriness, a preferred strategy being to combine TAX with other antineoplastic drugs or death receptor ligands such as TRAIL and TNF. Falling into the latter category, the present work was focused on the cooperation between TAX and TNF in mounting a strong toxic effect on human HCC cells and its results led to formulate the rationale for a novel antiblastic approach. In short, after a 24 hour treatment TAX caused cell cycle arrest in the M phase associated with a moderate degree of apoptosis in 2 human HCC cell lines, Huh7 and HepG2, in agreement with previous observations.Citation3,6, Citation11 By contrast, no toxic action at all on these cells was displayed by TNF per se. However, simultaneously exposing cells to TAX and TNF for up to 24/30 hours strongly enhanced apoptosis with respect to TAX alone, with Huh7 exhibiting a higher susceptibility than HepG2 cells. Noteworthily, similar results could be largely reproduced substituting TAX with spindle poisons such as COL and NOC, which compromise microtubules through a different mechanism. The data thus seem to suggest that, rather than a sensitization to TAX by TNF, the reciprocal effect came into play. To clarify the issue, Huh7 and HepG2 cells were sequentially exposed to TAX for up to 24/30 hours followed by TNF in the last 6 hours of incubation. This schedule markedly boosted apoptosis, whereas exposure to the same drugs in the reverse order (TNF followed by TAX) resulted in no significant change in the frequency of apoptotic cells with respect to TNF alone.
Some kinds of cooperation between death ligands and spindle drugs in inducing apoptosis in various tumors or neoplastic cells were previously reported. Just few examples adequately illustrate this point. Daily TNF administration over a week associated with a single quite high dose of TAX on the second day resulted in an additive or superadditive effect in mice developing tumors after inoculation of cancer cells derived from HCC or other carcinoma lines. This effect was ascribed to increased TNF-dependent antitumour efficacy of TAX;Citation35 however, the experimental design was such as to preclude an univocal interpretation about the relative contribution of each drug to anti-tumoral cytotoxicity. Apoptosis of human prostate cancer cells was conspicuously enhanced by TRAIL supplemented to cultures previously exposed to TAX,Citation36 or also to chemotherapeutic drugs such as etoposide, doxorubicin, vincristine and camptothecin.Citation37 Compared to either drug alone, combined treatment with TAX and TRAIL increased apoptotic death in human non-small-cell lung,Citation38 prostate or bladder cancer cells.Citation37 Similarly, apoptosis was enhanced in human glioblastoma multiforme and colon cancer cell lines exposed to nocodazole and TRAIL.Citation39
How a former exposure to TAX may condition cells toward TNF toxicity was herewith explored in preliminary experiments. The relevant parameters examined were found all altered by TAX in HCC cells in a way expected to favor TNF toxicity: (i) the amount of TNF-R1 exposed on the cell surface definitely increased, in spite of its decreased total levels; (ii) the content of the suppressor SOCS3 markedly decreased; (iii) c-FLIP, the physiological inhibitor of caspase 8 activation, significantly decreased as well. Notably, these changes were quite marked in Huh7 and less so in HepG2 cells, roughly paralleling their different susceptibility to TAX+TNF.
Recently, a new light was shed on the toxicity of TAX, and other microtubule inhibiting drugs as well, by the observation that these agents exert an important inhibitory action on STAT3 by decreasing its Tyr-phosphorylation.Citation30,31 STAT3 is a transcription factor involved in the control of cell proliferation, differentiation, survival, and death and c-FLIP and SOCS3 are 2 of the genes whose expression is modulated by STAT3. STAT3 activation is known to be inducible by cytokines, but also constitutive in many of the microtubule drug-responsive tumors. In both instances, it is rapidly inhibited by TAX, within 6 h of treatment, through induction of a putative negative feedback regulator.Citation31 The present data demonstrate that pSTAT3 levels markedly decrease in Huh7 cells treated with TAX or, even more, with TAX+TNF, roughly paralleling their effect on SOCS3 expression, while changes were less pronounced in HepG2 cells. The mechanism underlying the TAX effect requires further analysis, but the present observations at least support the contention that TAX somehow causes cells to reprogram TNF-triggered signaling pathways, thereby strongly promoting the susceptibility to TNF of cells otherwise not responsive at all. However, other possible mechanisms should not be ruled out as yet. Interesting, in this regard, is the recent reportCitation40 that c-FLIP downregulation by TAX depends on the action of the negative regulator MiR-512–3p, rather than on decreased mRNA levels.
The death process set in motion by TAX+TNF is caspase-mediated and zVADfmk-suppressed, largely at least, an observation in full agreement with the above changes of TNF signaling mediators elicited by TAX in HCC cells. Generally speaking, these findings also fit very well evidence in the literature suggesting that taxanes such as TAX and other antimicrotubular drugs promote cytokine-induced apoptosis, which generally occurs in association with upregulation of the cognate receptors and develops along the extrinsic pathway.Citation36,37,Citation41 Interestingly, apoptosis by TRAIL in TAX-treated PC-3 cells was found more marked than in cells exposed to the same drugs in the reverse order,Citation36 which is reminiscent of the present observations. According to several reports,Citation42 various cell lines treated with anticancer agents such as cisplatin or curcumin also increase cell surface expression and clustering of death receptors, thereby being sensitized to the cytotoxic action of death ligands. How precisely the above drugs reset the cell susceptibility to death ligands largely remains conjectural, however.
The present work shows that sequential application of 2 anticancer drugs such as TAX and TNF can enhance neoplastic cell death by some sort of rewiring of the apoptotic signaling networks. TAX strongly sensitizes to TNF cytotoxicity 2 human HCC cell lines, Huh7 and HepG2, otherwise refractory to the cytokine by itself. Exposing either cell type to TAX and TNF combined for 24/30 hours markedly enhances apoptosis, well above the low levels attained by TAX alone. A similar albeit lesser effect is achieved by applying the 2 drugs sequentially, i.e., TAX for 24/30 hours and TNF for only the last 6 hours of the total incubation time. Decreased total levels of SOCS3 or c-FLIP, to be likely ascribed to reduced Tyr-phosphorylation of STAT3, and increased TNF-R1 expression on the plasma membrane presumably concurred in TAX-engendered cell sensitization to TNF. Irrespective of the precise mechanism involved, the present findings show that a strong cytotoxic action can be achieved in cell cultures by applying the 2 drugs in an appropriate sequence. A major opportunity offered by this schedule is the possibility to employ TAX at a low concentration (0.1 μM was used in the present work) and, on the other hand, to apply TNF for only a short time (potentially profitable in view of the short half-life of serum TNF). Therefore, the present findings appear to outline a novel promising strategy to overcome drug resistance in cancer therapy: a sequential drug combination that might maximize curative effects while minimizing adverse reactions.
Materials and Methods
Cell lines
Huh7 cells originally derive from a patient affected by a well differentiated HCC, in the absence of HBV infection,Citation43 replicate continuously and are characterized by an epithelial-like morphology (Japanese Collection of Research Bioresources). HepG2 cells (ATCC), retaining features of differentiated hepatocytes and an epithelial morphology, were isolated from a hepatoblastoma. Both lines were cultured in Dulbecco's modified Eagle's medium (DMEM; Sigma) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin and 2 mM L-glutamine. Cells were grown at 37°C in a humidified atmosphere containing 5% CO2. At each experimental time point culture media were collected and centrifuged (500 × g, 10 min at 4°C). Monolayers were washed with PBS-EDTA, trypsinized and centrifuged as above. Cells obtained from medium and monolayer were resuspended in PBS, pooled and counted in the presence of trypan blue.
Drugs and treatments
Cells were exposed to 0.1 μM TAX (Sigma), 0.2 μg/ml nocodazole (NOC), or 10 μM colchicine (COL) in the presence or in the absence of 15 ng/ml TNF (Peprotech) 48 hours after plating. Cultures were supplemented with the polycaspase inhibitor zVADfmk (Alexis Laboratories) at 20 μM final concentration or with 20 μM caspase 3 or 8 selective and irreversible inhibitors AcDEVDcmk or zIETDfmk, respectively (Calbiochem) one hour before treatment with antimitotic drugs and/or TNF.
Caspase inhibitors, TAX and NOC were dissolved in DMSO (Sigma), TNF in DMEM and COL in ethanol. Control experiments ruled out any effect of DMSO or ethanol by themselves (data not shown).
DAPI staining
To detect changes in nuclear morphology characteristic of apoptosis, cells were washed with PBS, fixed with 95% ethanol for 5 min at room temperature (RT), dark-incubated for 20 min with 1 μg/ml DAPI (4,6-diamidino-2-phenylindole dihydrochloride), then washed twice with PBS to remove fluorochrome excess. Cells were observed in a fluorescence microscope (Leitz Dialux 20) and photographed (Coolpix 4500, Nikon).
Membrane expression of TNF-R1
Immunofluorescence microscopy. Cells were washed with PBS, fixed in 3.7% paraformaldehyde (Sigma-Aldrich, Milan, Italy) for 30 min, incubated at RT in 1% bovine serum albumin in PBS and overnight at 4°C with anti-TNF-R1 rabbit polyclonal antibody (1:50; Santa Cruz Biotechnology). Cells were then washed twice with PBS and incubated for 1 hour at RT with FITC-conjugated goat anti-rabbit secondary antibody (1:150; Sigma). After permeabilization with 0.05% (v/v) Triton X-100 in PBS, nuclei were stained with DAPI. Samples were observed by fluorescence microscopy and photographed.
Flow-cytometric analysis. Twenty-five×104 cells were exposed to TAX, TNF or TAX+TNF for 24 hours, washed with PBS-EDTA, trypsinized and rinsed thrice with washing buffer (WSB: PBS containing 2% fetal calf serum and 0.1% NaN3). After centrifugation at 500 × g for 1 min at RT, cells were incubated for 5 min in 10 μl of PBS containing 1% BSA and 0.1% NaN3, then in 40 μl of FITC-labeled anti-TNF-R1 monoclonal antibody (20 μg/ml; MBL) for 30 min at RT, washed with WSB, pelleted by centrifugation for 1 min at 500 × g and resuspended in 500 μl of WSB. Labeled cells were analyzed in a FACScan flow cytometer using the CellQuest software (Becton & Dickinson).
Cell cycle and DNA distribution analysis
Cell cycle progression and DNA distribution were analyzed by flow cytometry using propidium iodide staining. Cell cultures were washed with PBS-EDTA, trypsinized and centrifuged (500 × g, 10 min at 4°C). Apoptotic bodies were obtained by centrifugation of the supernatant at 1500 × g for 10 min. After washing, cells and apoptotic bodies were separately fixed in ice-cold 70% ethanol for at least 30 min at RT. Subsequently they were centrifuged as indicated above, put together in PBS containing DNase-free RNase A (0.4 mg/ml) and incubated for 30 min. Then, cells were incubated 10 min with propidium iodide (0.18 mg/ml) and analyzed by flow cytometry using the CellQuest software (Becton & Dickinson). The apoptotic population was estimated by evaluating the percentage of cells characterized by a <2n DNA fluorescence (hypodiploid or subG1 cells).Citation44
Downregulation of SOCS3 by RNA interference
One day prior to transfection, cells were seeded (104/cm2) without antibiotics in 6 cm Petri dishes in a total volume of 3 ml DMEM supplemented with 10% fetal bovine serum. Cells were then transfected using 10 μl Lipofectamine 2000 reagent (Invitrogen), 300 μl OptiMEM (Invitrogen) and 100 pmol siRNA (control and human siSOCS3 specific; MWG-Biotech AG) for 24 and 48 hours. The sequence for SOCS3-targeted siRNA (siSOCS3) was 5′-CCUGGUGGGACGAUAGCAACC-3'.Citation45 Transfected cells were then treated with TNF 48 hours after transfection. Nontargeting siRNA was used as control (siC).
Immunoblot analysis
Cells were sonicated in lysis buffer (PBS containing 1% Nonidet P40, 0.5% sodium deoxycholate, 0.1% SDS, 0.1 mM PMSF, 2 μg/ml aprotinin and 100 mM sodium orthovanadate) to obtain total extracts. Protein concentration was assayed according to a modified Bradford technique using an assay kit (Bio-Rad). Equal amounts of protein (30 μg) were heat-denaturated in sample-loading buffer (50 mM TRIS-HCl, pH 6.8, 100 mM DTT, 2% SDS, 0.1% bromophenol blue, 10% glycerol), resolved by SDS-PAGE (12% polyacrilamide, 0.1% SDS) and transferred to nitrocellulose membranes (Bio-Rad). Filters were then blocked with Tris-buffered saline (TBS) containing 0.05% (v/v) Tween and 5% non-fat dry milk, then incubated overnight with a mouse polyclonal anti-SOCS3 antibody (1:100; Santa Cruz Biotechnology) or a rabbit polyclonal anti-c-FLIP antibody (1:1000; Sigma), anti-STAT3 and anti-pSTAT3 antibodies (1:1000; Cell Signaling) and anti-TNF-R1 (1:200; Santa Cruz Biotechnology). Goat anti-rabbit and anti-mouse peroxidase-conjugated IgG (Bio-Rad) were used as secondary antibodies. Filters were then stripped by incubation in 62.5 mM Tris-HCl, pH 6.7, containing 100 mM 2-mercaptoethanol and 2% SDS for 30 min at 50°C, and reprobed with a mouse polyclonal antibody directed against β-actin (1:2000; Sigma) to normalize sample loading. The membrane-bound immune complexes were detected by enhanced chemiluminescence (Santa Cruz Biotechnology) on a photon-sensitive film (Hyperfilm ECL; Amersham Biosciences). Band quantification was performed by densitometric analysis using a specific software (TotalLab, NonLinear Dynamics).
Caspase enzymatic activity
Caspase activities were assayed on cell lysates by evaluating the cleavage of fluorogenic substrates. Cells resuspended in lysis buffer (20 mM HEPES-KOH, pH 7.5, 10 mM KCl, 1 mM Na-EDTA, 1 mM DTT, 1 mM PMSF and 10 μg/ml leupeptin), were maintained on ice for 15 min, sonicated and then centrifuged at 13,000 rpm for 15 min at 4°C (Biofuge 17 RS, Heraeus Sepatech). The supernatant was collected and protein concentration determined.
Aliquots of 30 μg protein were then incubated in reaction buffer (25 mM HEPES, pH 7.5, containing 0.1% CHAPS, 10% sucrose, 10 mM dithiothreitol, 0.1 mg/ml ovalbumin) for 60 min at 37°C in the presence of 20 μM fluorogenic substrates for caspase 3, 8 and 9 (respectively Ac-DEVD-AMC, Ac-IETD-AMC and Ac-LEHD-AMC), all purchased from Biomol (Philadelphia). Reaction was blocked with 0.01% ice-cold trichloroacetic acid and fluorescence read at 380 nm excitation and 460 nm emission in a spectrofluorometer (Perkin-Elmer). Specific activities, expressed as nkatal/mg protein, were calculated by using free AMC as working standard.
Experiments on tumor-bearing mice
The study was performed on male Balb/c mice weighing about 20 g (Charles River), cared for in compliance with the Italian Ministry of Health Guidelines (n° 86609 EEC, permit number 106/2007-B) and the Policy on Humane Care and Use of Laboratory Animals (NIH 1996). The experimental protocol was approved by the Bioethical Committee of the University of Torino. Animals were maintained on a regular dark-light cycle (light from 08:00 to 20:00), with free access to food and water during the whole experimental period. Tumor-bearing (TB) mice received an intraperitoneal (i.p.) inoculum of Yoshida AH-130 ascites hepatoma cells (5·105 cells/mouse; tumor provided years ago by Prof. U. Del Monte, University of Milano, Italy, and maintained in our lab by weekly i.p. transplantation). Despite this tumor is typically grown in rats, it also engrafts in mice. The animals were divided into 4 groups (n = 4/group), namely, untreated TB, TNF-treated TB, TAX-treated TB and TAX+TNF-treated TB. Six days after tumor transplantation both TAX and TAX+TNF-treated mice received an i.p. injection of TAX (4 mg/kg).Citation35 After 24 hours TNF and TAX+TNF-treated animals were administered TNF (10 μg/mouse, i.p.).Citation35 All the animals were sacrificed 6 hours later under isofluorane anesthesia. Tumor cells were collected from the peritoneal cavity, washed and resuspended in ice-cold 70% ethanol for FACS analysis (see above) or in lysis buffer for protein gel blotting (see above).
Data presentation and analysis
All experiments were replicated at least 3 times. Data were expressed as means±SD. Western blots were quantified by densitometry and normalized vs β-actin. Reliability of results was also checked by randomizing samples during gel loading (not shown). Significance of the differences was evaluated by one-way ANOVA; P < 0.05, P < 0.01 and P < 0.001 were considered significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank dr. C. Traboni, actually at Okairos, Pomezia, Italy, who kindly provided the Huh7 cell line.
Funding
This work was supported by 'Ministero per l'Università e la Ricerca' (MIUR, Roma; PRIN project), University of Torino (ex-60% funds), Regione Piemonte.
References
- Jordan MA, Wilson L. Microtubules and actin filaments: dynamic targets for cancer chemotherapy. Curr Opin Cell Biol 1998; 10:123-30; PMID:9484604; http://dx.doi.org/10.1016/S0955-0674(98)80095-1
- Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer 2004; 4:253-65; PMID:15057285; http://dx.doi.org/10.1038/nrc1317
- Gagandeep S, Novikoff PM, Ott M, Gupta S. Paclitaxel shows cytotoxic activity in human hepatocellular carcinoma cell lines. Cancer Lett 1999; 136:109-18; PMID:10211948; http://dx.doi.org/10.1016/S0304-3835(98)00388-7
- Yuan JH, Zhang RP, Zhang RG, Guo LX, Wang XW, Luo D, Xie Y, Xie H. Growth-inhibiting effects of taxol on human liver cancer in vitro and in nude mice. World J Gastroenterol 2000; 6:210-5; PMID:11819558
- Wan YF, Guo XQ, Wang ZH, Ying K, Yao MH. Effects of paclitaxel on proliferation and apoptosis in human acute myeloid leukemia HL-60 cells. Acta Pharmacol Sin 2004; 25:378-84; PMID:15000894
- Okano J, Nagahara T, Matsumoto K, Murawaki Y. The growth inhibition of liver cancer cells by paclitaxel and the involvement of extracellular signal-regulated kinase and apoptosis. Oncol Rep 2007; 17:1195-200; PMID:17390065
- Morris PG, Fornier MN. Microtubule active agents: beyond the taxane frontier. Clin Cancer Res 2008; 14:7167-72; PMID:19010832; http://dx.doi.org/10.1158/1078-0432.CCR-08-0169
- Chun E, Lee KY. Bcl-2 and Bcl-xL are important for the induction of paclitaxel resistance in human hepatocellular carcinoma cells. Biochem Biophys Res Commun 2004; 315:771-9; PMID:14975768; http://dx.doi.org/10.1016/j.bbrc.2004.01.118
- Le XF, Bast RC. Src family kinases and paclitaxel sensitivity. Cancer Biol Ther 2011; 12:260-9; PMID:21646863; http://dx.doi.org/10.4161/cbt.12.4.16430
- Cabrera R, Nelson DR. Review article: the management of hepatocellular carcinoma. Aliment Pharmacol Ther 2010; 31:461-76; PMID:19925500; http://dx.doi.org/10.1111/j.1365-2036.2009.04200.x
- Brenes O, Arce F, Gätjens-Boniche O, Díaz C. Characterization of cell death events induced by anti-neoplastic drugs cisplatin, paclitaxel and 5-fluorouracil on human hepatoma cell lines: Possible mechanisms of cell resistance. Biomed Pharmacother 2007; 61:347-55; PMID:17399942; http://dx.doi.org/10.1016/j.biopha.2007.02.007
- Zhang ZL, Liu ZS, Sun Q. Anti-tumor effect of thalidomide and paclitaxel on hepatocellular carcinoma in nude mice. Chin Med J (Engl) 2005; 118:1688-94; PMID:16313753
- Chae S, Kim YB, Lee JS, Cho H. Resistance to paclitaxel in hepatoma cells is related to static JNK activation and prohibition into entry of mitosis. Am J Physiol Gastrointest Liver Physiol 2012; 302:G1016-24; PMID:22323130; http://dx.doi.org/10.1152/ajpgi.00449.2011
- Strumberg D, Erhard J, Harstrick A, Klaassen U, Müller C, Eberhardt W, Wilke H, Seeber S. Phase I study of a weekly 1 h infusion of paclitaxel in patients with unresectable hepatocellular carcinoma. Eur J Cancer 1998; 34:1290-2; PMID:9849493; http://dx.doi.org/10.1016/S0959-8049(98)00054-9
- Chao Y, Chan WK, Birkhofer MJ, Hu OY, Wang SS, Huang YS, Liu M, Whang-Peng J, Chi KH, Lui WY, Lee SD. Phase II and pharmacokinetic study of paclitaxel therapy for unresectable hepatocellular carcinoma patients. Br J Cancer 1998; 78:34-9; PMID:9662247; http://dx.doi.org/10.1038/bjc.1998.438
- Fabregat I. Dysregulation of apoptosis in hepatocellular carcinoma cells. World J Gastroenterol 2009; 15:513-20
- Minero VG, Khadjavi A, Costelli P, Baccino FM, Bonelli G. JNK activation is required for TNFα-induced apoptosis in human hepatocarcinoma cells. Int Immunopharmacol 2013; 17:92-8; PMID:23751896; http://dx.doi.org/10.1016/j.intimp.2013.05.017
- Mao CY, Hua HJ, Chen P, Yu DC, Cao J, Teng LS. Combined use of chemotherapeutics and oncolytic adenovirus in treatment of AFP-expressing hepatocellular carcinoma. Hepatobiliary Pancreat Dis Int 2009; 8:282-7; PMID:19502169
- Jin C, Li H, He Y, He M, Bai L, Cao Y, Song W, Dou K. Combination chemotherapy of doxorubicin and paclitaxel for hepatocellular carcinoma in vitro and in vivo. J Cancer Res Clin Oncol 2010; 136:267-74; PMID:19693537; http://dx.doi.org/10.1007/s00432-009-0658-5
- Tian A, Wilson GS, Lie S, Wu G, Hu Z, Hebbard L, Duan W, George J, Qiao L. Synergistic effects of IAP inhibitor LCL161 and paclitaxel on hepatocellular carcinoma cells. Cancer Lett 2014; 351:232-41; PMID:24976294; http://dx.doi.org/10.1016/j.canlet.2014.06.006
- Lee MJ, Ye AS, Gardino AK, Heijink AM, Sorger PK, MacBeath G, Yaffe MB. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell 2012; 149:780-94; PMID:22579283; http://dx.doi.org/10.1016/j.cell.2012.03.031
- Autelli R, Crepaldi S, De Stefanis D, Parola M, Bonelli G, Baccino FM. Intracellular free iron and acidic pathways mediate TNF-induced death of rat hepatoma cells. Apoptosis 2005; 10:777-86; PMID:16133868; http://dx.doi.org/10.1007/s10495-005-2944-2
- Autelli R, Ullio C, Prigione E, Crepaldi S, Schiavone N, Brunk UT, Capaccioli S, Baccino FM, Bonelli G. Divergent pathways for TNF and C(2)-ceramide toxicity in HTC hepatoma cells. Biochim Biophys Acta 2009; 1793:1182-90; PMID:19328214; http://dx.doi.org/10.1016/j.bbamcr.2009.03.005
- Ullio C, Casas J, Brunk UT, Sala G, Fabriàs G, Ghidoni R, Bonelli G, Baccino FM, Autelli R. Sphingosine mediates TNFα-induced lysosomal membrane permeabilization and ensuing programmed cell death in hepatoma cells. J Lipid Res 2012; 53:1134-43; PMID:22454477; http://dx.doi.org/10.1194/jlr.M022384
- Semenkova LN, Dudich EI, Dudich IV, Shingarova LN, Korobko VG. Alpha-fetoprotein as a TNF resistance factor for the human hepatocarcinoma cell line HepG2. Tumour Biol 1997; 18:30-40; PMID:8989923; http://dx.doi.org/10.1159/000218013
- Xia L, Mo P, Huang W, Zhang L, Wang Y, Zhu H, Tian D, Liu J, Chen Z, Zhang Y, Hu H, Fan D, Nie Y, Wu K. The TNF-α/ROS/HIF-1-induced upregulation of FoxMI expression promotes HCC proliferation and resistance to apoptosis. Carcinogenesis 2012; 33:2250-9; PMID:22831955; http://dx.doi.org/10.1093/carcin/bgs249
- Fredriksson L, Herpers B, Benedetti G, Matadin Q, Puigvert JC, de Bont H, Dragovic S, Vermeulen NP, Commandeur JN, Danen E, de Graauw M, van de Water B. Diclofenac inhibits tumor necrosis factor-α-induced nuclear factor-κB activation causing synergistic hepatocyte apoptosis. Hepatology 2011; 53:2027-41; PMID:21433042; http://dx.doi.org/10.1002/hep.24314
- Yadav A, Kalita A, Dhillon S, Banerjee K. JAK/STAT3 pathway is involved in survival of neurons in response to insulin-like growth factor and negatively regulated by suppressor of cytokine signaling-3. J Biol Chem 2005; 280:31830-40; PMID:15998644; http://dx.doi.org/10.1074/jbc.M501316200
- Yoshimura A. Regulation of cytokine signaling by the SOCS and Spred family proteins. Keio J Med 2009; 58:73-83.
- Walker SR, Chaudhury M, Nelson EA, Frank DA. Microtubule-targeted chemotherapeutic agents inhibit signal transducer and activator of transcription 3 (STAT3) signaling. Mol Pharmacol 2010; 78:903-8; PMID:20693278; http://dx.doi.org/10.1124/mol.110.066316
- Walker SR, Chaudhury M, Frank DA. STAT3 Inhibition by Microtubule-Targeted Drugs: Dual Molecular Effects of Chemotherapeutic Agents. Mol Cell Pharmacol 2011; 3:13-9; PMID:21949561
- Riehle KJ, Campbell JS, McMahan RS, Johnson MM, Beyer RP, Bammler TK, Fausto N. Regulation of liver regeneration and hepatocarcinogenesis by suppressor of cytokine signaling 3. J Exp Med 2008; 205:91-103; PMID:18158318; http://dx.doi.org/10.1084/jem.20070820
- Elliott J. SOCS3 in liver regeneration and hepatocarcinoma. Mol Interv 2008; 8:19-21, 2; PMID:18332480
- Niwa Y, Kanda H, Shikauchi Y, Saiura A, Matsubara K, Kitagawa T, Yamamoto J, Kubo T, Yoshikawa H. Methylation silencing of SOCS-3 promotes cell growth and migration by enhancing JAK/STAT and FAK signalings in human hepatocellular carcinoma. Oncogene 2005; 24:6406-17; PMID:16007195
- Seong J, Milross CG, Hunter NR, Shin HC, Milas L. Potentiation of antitumor efficacy of paclitaxel by recombinant tumor necrosis factor-α. Anticancer Drugs 1997; 8:80-7; PMID:9147616; http://dx.doi.org/10.1097/00001813-199701000-00011
- Nimmanapalli R, Perkins CL, Orlando M, O'Bryan E, Nguyen D, Bhalla KN. Pretreatment with paclitaxel enhances apo-2 ligand/tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis of prostate cancer cells by inducing death receptors 4 and 5 protein levels. Cancer Res 2001; 61:759-763; PMID:11212279
- Shankar S, Chen X, Srivastava RK. Effects of sequential treatments with chemotherapeutic drugs followed by TRAIL on prostate cancer in vitro and in vivo. Prostate 2005; 62:165-186; PMID:15389801; http://dx.doi.org/10.1002/pros.20126
- Odoux C, Albers A, Amoscato AA, Lotze MT, Wong MK. TRAIL, FasL and a blocking anti-DR5 antibody augment paclitaxel-induced apoptosis in human non-small-cell lung cancer. Int J Cancer 2002; 97:458-465; PMID:11802207; http://dx.doi.org/10.1002/ijc.1640
- Kim M, Liao J, Dowling ML, Voong KR, Parker SE, Wang S, El-Deiry WS, Kao GD. TRAIL inactivates the mitotic checkpoint and potentiates death induced by microtubule-targeting agents in human cancer cells. Cancer Res 2008; 68:3440-3449; PMID:18451172; http://dx.doi.org/10.1158/0008-5472.CAN-08-0014
- Chen F, Zhu HH, Zhou LF, Wu SS, Wang J, Chen Z. Inhibition of c-FLIP expression by miR-512-3p contributes to taxol-induced apoptosis in hepatocellular carcinoma cells. Oncol Rep 2010; 23:1457-1462; PMID:20372864
- Munshi A, McDonnell TJ, Meyn RE. Chemotherapeutic agents enhance TRAIL-induced apoptosis in prostate cancer cells. Cancer Chemother Pharmacol 2002; 50:46-52; PMID:12111111; http://dx.doi.org/10.1007/s00280-002-0465-z
- Elrod HA, Sun SY. Modulation of death receptors by cancer therapeutic agents. Cancer Biol Ther 2008; 7:163-173; PMID:18059181; http://dx.doi.org/10.4161/cbt.7.2.5335
- Nakabayashi H, Taketa K, Miyano K, Yamane T, Sato J. Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res 1982; 42:3858-63; PMID:6286115
- Nicoletti I, Migliorati G, Pagliacci MC, Grignani F, Riccardi C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods 1991; 139:271-9; PMID:1710634; http://dx.doi.org/10.1016/0022-1759(91)90198-O
- Tomita S, Ishibashi K, Hashimoto K, Sugino T, Yanagida T, Kushida N, Shishido K, Aikawa K, Sato Y, Suzutani T, Yamaguchi O. Suppression of SOCS3 increases susceptibility of renal cell carcinoma to interferon-α. Cancer Sci 2011; 102:57-63; PMID:21054677; http://dx.doi.org/10.1111/j.1349-7006.2010.01751.x