
Abstract
Interstrand crosslinks induce DNA replication fork stalling that in turn activates the ATR-dependent checkpoint and DNA repair on nuclear chromatin. Mitomycin C (MC) and Decarbamoyl Mitomycin C (DMC) induce different types of DNA crosslinks with DMC being a more cytotoxic agent. We previously reported that the novel DMC induced β–interstrand DNA crosslinks induce a p53-independent form of cell death. The p53-independent DMC cytotoxicity associates with the activation, and subsequent depletion, of Chk1. In this study we further dissect the novel DMC signal transduction pathway and asked how it influences chromatin-associated proteins. We found that treatment with DMC, but not MC, stimulated the disassociation of ATR from chromatin and re-localization of ATR to the cytoplasm. The chromatin eviction of ATR was coupled with the formation of nuclear Rad51 foci and the phosphorylation of Chk1. Furthermore, DMC but not MC, activated expression of gadd45α mRNA. Importantly, knocking down p53 via shRNA did not inhibit the DMC-induced disassociation of ATR from chromatin or reduce the activation of transcription of gadd45α. Our results suggest that DMC induces a p53-independent disassociation of ATR from chromatin that facilitates Chk1 checkpoint activation and Rad51 chromatin recruitment. Our findings provide evidence that ATR chromatin eviction in breast cancer cells is an area of study that should be focused on for inducing p53-independent cell death.
Abbreviations
| DMC | = | Decarbamoyl Mitomycin C |
| ATR | = | Ataxia Telangiectasia and Rad3 Related |
| MC | = | Mitomycin C |
| ICLs | = | interstrand cross-links |
| RPA | = | replication protein A |
| Chk1 | = | checkpoint serine/threonine protein kinase 1 |
| TNBCs | = | triple negative breast cancers |
| HR | = | homologous recombination |
| NER | = | nuclear excision repair |
| PCNA | = | Proliferating Cell Nuclear Antigen |
| DDR | = | DNA damage response |
| DOX | = | doxycycline |
| ChIP | = | chromatin immunoprecipitation |
| Cdc25 | = | cell division cycle 25. |
Introduction
The alkylating agent Mitomycin C (MC) has been actively used in the treatment of many tumors, including stomach, pancreas, anal, bladder, cervical, colorectal, head and neck, non-small cell lung cancer, and breast cancer, with favorable response rates of about 15–20% (reviewed inCitation1,2). It has been shown that MC generates different types of DNA lesions, including monoadducts, intrastrand crosslinks and interstrand crosslinks.Citation3 DNA interstrand cross-links (ICLs) covalently crosslink both strands of DNA and block DNA metabolism. DNA replication and transcription are blocked by ICLs making them highly toxic to dividing cells. It has been estimated that the presence of approximately 40 unrepaired ICLs can kill mammalian cells.Citation4 The MC derivative, 10-decarbamoyl mitomycin C (DMC) shows a stereochemical preference of linkage to the guanine-2-amino group opposite from that of MC induced ICL adducts.Citation3 The predominant stereochemistry of the 1′′-mitosene linkage of the DMC induced interstrand crosslink to DNA is β, this is opposite to the predominant α stereochemistry of the MC interstrand crosslink.Citation5 The striking difference between the DNA interstrand crosslinks of MC and DMC in the breast cancer cell line MCF7 has been revealed to induce higher p53-independent cytotoxicity.Citation5 This higher cytotoxicity involves activation, and then subsequent depletion of cell cycle checkpoint serine/threonine protein kinase (Chk1).Citation5 Importantly, DMC induces cell death more rapidly than MC in the presence and absence of wild-type p53.Citation6 7 This suggests that further elucidating the mechanism of action of DMC induced cytotoxicity will provide information toward the development of chemotherapeutics for multiple forms of cancer with or without p53 mutations.
The p53 gene is mutated in over half of human cancers.Citation8 The mutation of p53 is present in 80% of triple-negative breast cancers (TNBCs).Citation9 The TNBCs are very aggressive and currently lack effective targeted therapies, however their mutated p53 (and thus wild-type p53 deficient state) may provide a new targeting approach. It has been found that wild-type p53 deficient cells are sensitive to ATR inhibition.Citation10 In addition, ATR is preferentially required for proliferation of p53-deficient human cancer cells.Citation11 In light of the fact that the p53 dependent cell death pathway is dysfunctional in 80% of TNBC, inducing a p53-indepedent cell death could be an approach for TNBC cancer therapy.
The replication protein A (RPA)–Ataxia Telangiectasia and Rad3‑related (ATR)–cell cycle checkpoint serine/threonine protein kinase (Chk1) signaling cascade participates in ICL repair.Citation12 ATR is a protein kinase that regulates cell cycle checkpoints and DNA repair. Defects of ATR such as splicing mutations in human ATR cause the autosomal-recessive disease Seckel syndrome.Citation13 It has been shown that complete loss of ATR in mice leads to embryonic lethality.Citation14 Chk1 is a direct downstream target of ATR and Chk1-mediated phosphorylation of Cdc25A in response to DNA damage induces G2/M cell-cycle arrest.Citation12 Additionally, ATR directly phosphorylates BRCA1 in response to damaged DNA.Citation15 Importantly, the BRCA1/BRCA2 and Rad51complexes initiate and regulate DNA repair via homologous recombination (HR).Citation12 Recently, it was reported that cells lacking ATR have decreased density with abnormal morphology, a decreased frequency of HR and an increased level of chromosomal damage in vivo.Citation16
ATR mediates p38 MAPK-dependent activation of MK2, and MK2 is required in p53-deficient cells to arrest the G1/S, intra-S phase and G2/M transition after cisplatin and doxorubicin treatment.Citation17 Significantly, MK2 activates a cytoplasmic cell cycle checkpoint network.Citation18 MK2 directly phosphorylates Cdc25 family members, resulting in a G1/S and G2/M arrest.Citation17 In addition, p38/MK2 dependent phosphorylation of hnRNPA0, TIAR and PARN stabilizes the gadd45 transcript through its 3′UTR.Citation18 The Gadd45 protein participates in nuclear excision repair (NER) through interaction with the Proliferating Cell Nuclear Antigen (PCNA).Citation19 Importantly, after DNA damage cells lacking a functional p53 pathway rely on cytoplasmic p38/MK2 and nuclear Chk1 activation to arrest the cell cycle.Citation17
Chk1 is activated and then depleted following DMC treatment and knocking down of Chk1 increases the cytotoxicity of MC.Citation5 The detailed mechanism of action of the β- interstrand cross-linking agent DMC induced cell death pathway is not fully defined. In this study, we investigated how nuclear β-interstrand crosslinks in breast cancer cells influenced nuclear and cytoplasmic responses. We compared MC and DMC signal transduction pathways. We found that DMC, but not MC, induced ATR disassociation from chromatin and activation of the Chk1 kinase pathway. In addition, DMC, but not MC, transcriptionally stimulated gadd45 induction. Furthermore we utilized an inducible p53 shRNA stable cell line to knock down wild-type p53 in order to confirm the p53-independent mechanism of action of DMC in the DNA damage response (DDR).
Results
DMC induces Chk1 phosphorylation at ser 345, p38MAPK phosphorylation, ATR chromatin eviction and recruitment of homologous recombination protein Rad51 to DNA damage foci
ATR activation occurs in response to DNA replication fork stalling, and ATR kinase activity can be examined by the level of phosphorylation of its downstream target Chk1.Citation12 ATR phosphorylates Chk1 at ser-317 and ser-345 resulting in increased Chk1 kinase activity in response to DNA damage.Citation20 We previously reported that DMC has higher cytotoxicity compared to MC in the presence or absence of wild-type p53.Citation6 DMC first activates Chk1 but following 12 hours of drug treatment Chk1 is depleted.Citation6 DMC induced Chk1 depletion is caused by increased Chk1 ubiquitination and subsequent degradation of the kinase by the proteasome.Citation5 To study how β interstrand crosslinks induced by DMC initiate the DNA damage checkpoint activation, we examined an earlier time point of 4 hours of drug treatments. Ten μM of DMC, but not 10 μM of MC treatment strongly induced an 8.6-fold induction of phosphorylation of nuclear Chk1 at ser-345 ( compare lanes 4, 5 and 6). The total Chk1 protein levels did not change significantly after either short term treatment with MC or DMC. In addition, at the 4 hours treatment time point we examined the phosphorylation of p38 MAPK. Higher levels of phosphorylation of cytoplasmic p38 MAPK at Thr180/Tyr182 were detected ( compare lanes 1, 2 and 3). These results demonstrate that DMC, as compared to MC, induced a more rapid ATR kinase activation compared to MC.
Figure 1. DMC induces Chk1 phosphorylation at ser 345, p38MAPK phosphorylation, ATR chromatin eviction and chromatin recruitment homologous recombination protein Rad51. (A) Cytoplasmic and nuclear proteins were extracted from MCF7 cells, untreated (Con) or treated with 10 μM of MC or DMC for 4 hours. 50 μg of cytoplasmic or nuclear extracts were loaded on a 4–12% SDS-PAGE gel, immunoblotted with p-ser345 Chk1, Chk1, p38MAPK, pT-180 pY182 p38 MAPK and Actin antibodies. Densitometry analysis of p-ser345 Chk1 or pT-180 pY182 p38 MAPK was performed using NIH ImageJ, and values were normalized to total Chk1 or p38 MAPK. Number represents average of 2 experiments. (B) MCF7 cells, untreated or treated with 10 μM MC or DMC for 4 hours were fixed and double stained with ATR (red) and Lamin (green) antibodies. Nuclear DNA was stained with DAPI (blue). ATR staining is representation of 3 experiments. (C) MCF7 cells, untreated or treated with 10 μM MC or DMC for 4 hours were fractionated into cytosol, nuclear soluble and chromatin bound fractions. 50 μg of cytosolic or chromatin-bound fractions were resolved by SDS/PAGE and immunobloted with RPA, ATR, Chk1, p53 and Histone 3 antibodies. (D) Immunofluoresence analysis of Rad51 foci formation after 10 μM MC or DMC for 4 hours treatment of MCF7 cells. Rad51 signals are shown in green and DNA was counter-stained with DAPI (blue). Rad51 staining is representation of 2 experiments. The RAD51 foci are quantified in 4 hours 10 μM of MC or DMC treated MCF7 cells.
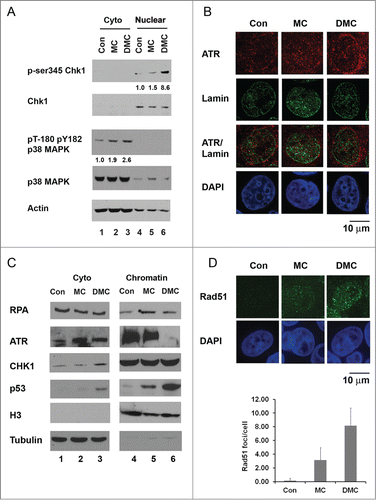
In order to further address the mechanism of action of DMC we considered the prevailing model for how ATR is activated to regulate the phosphorylation of Chk1. This involves an ATR chromatin recruitment paradigm.Citation21,22 However it is not clear if ATR must remain associated with chromatin in order for Chk1 to be phosphorylated. DNA damage by cisplatin (in the absence of hMSH2) can induce Chk1 phosphorylation without clear ATR recruitment.Citation22 Increasing evidence points to the eviction of certain proteins from chromatin during certain catastrophic DNA damage scenarios. Therefore we also considered the possibility that ATR might be evicted from the chromatin. We carried out confocal microscopy immunofluorescence analysis to compare the subcellular MC and DMC influences on ATR localization (). In response to DMC, but not MC treatment, we observed translocation of ATR from the nucleus to the cytoplasm (Lamin showed the nuclear envelope). These data suggest that translocation of ATR from the nucleus to the cytoplasm performs a functionally important step in DMC-induced DDR catastrophe. Importantly, ATR prohibits replication catastrophe mediated cell death by preventing global exhaustion of RPACitation23 and we see strong DMC-induced cell death. Dynamic association and release of DDR proteins at active and damaged replication folks has been reported.Citation24 When chromatin integrity is damaged by genotoxic agents the cellular DDR proteins are recruited to sites of DNA damage and the kinase activity of ATR is stimulated.Citation25 However, the detailed mechanistic insight after the ATR recruitment for what signals cause growth arrest versus those that cause cell death are lacking.
The observed ATR nuclear eviction supported current research that suggests that chromatin eviction is a central player in cell death signal transduction.Citation26 We investigated ATR proteins binding verses disassociation with chromatin by using cell fractionation and immunobloting analysis to gain more knowledge of molecular signaling of DNA damage caused by MC vs. the DMC. MCF7 cells with 4 hours of drug treatment were lysed and the cytosolic and chromatin fractions were isolated. We analyzed the RPA, ATR, Chk1 and p53 proteins associated with the chromatin in . In untreated cells ( lane 4), these proteins were detected in the chromatin fraction with only low levels of p53 protein detected supporting the well-established central functions of these proteins in DNA damage signaling and genome maintenance.Citation25 Upon MC or DMC treatment, as expected, increased levels of p53 were found on chromatin ( compare lane 4 through 6). Interestingly, after DMC treatment, a reproducibly distinct absence of ATR association on damaged chromatin was observed ( compare lane 6 to 4). A slight increase in cytosolic ATR was detected after DMC treatment ( compare lane 3 to 1). The total chromatin associated Chk1 protein levels remained unchanged in the presence or absence of DNA damage ( lane 4–6). We further carried out variable durations of treatment (1, 2, 3 and 4 hours) to monitor the association of ATR on the chromatin after MC or DMC treatment. Chromatin bound ATR was dissociated after only 4 hours of DMC treatment (data not shown).
It has been reported that cells demonstrate an accumulation of Rad51 into foci after various forms of DNA damage caused by single-strand DNA breaks, marking the sites of lesion.Citation27 To determine whether the DMC treatment caused rapid formation of Rad51 foci in MCF7 cells, we performed immunofluorescence to detect the sub-cellular localization of Rad51 (). After treatment with 10 μM DMC, but not 10 μM MC, for 4 hours we observed an increase in Rad51 foci. These results suggest that DMC treatment also induced activation of homologous recombination DNA damage repair.
DMC stimulates genome wide nuclear proteins dissociation from DNA
The chromatin eviction of ATR suggested that DMC treatment might signal for other proteins to no longer maintain DNA binding activity. To investigate the DNA binding activity of nuclear proteins upon DMC treatment, we carried out an electrophoretic mobility shift assay (EMSA) with nuclear proteins isolated from MCF7 cells treated with either MC or DMC and micrococcal nuclease prepared mono-nucleosomal size (150 bp) naked genomic DNA from untreated MCF7 cells. This allowed us to examine general DNA binding activity to the entire genome sequence. Interestingly decreased DNA binding activity was detected for proteins isolated from DMC treated samples, as the shifted DNA was 60% less compared by phosphor imaging quantification analysis to untreated samples (compare lanes 2 and 4). Nuclear proteins isolated from MC treated cells displayed slightly increased DNA binding activity (compare lanes 2 and 3). The global release may mean that many signaling molecules are released during DMC treatment. However a great deal of p53 was chromatin associated as shown in . The possibility exists that the ATR release and p38 MAPK phosphorylation may be a small part of a bigger general housekeeping protein picture. p53 can activate apoptosis by interacting directly with the mitochondria so it interacts with Bcl-2 member family proteins to promote cytochrome c release.Citation28 Therefore if some of the p53 were released from the chromatin by DMC treatment it might help facilitate transcription-independent cell death.
Figure 2. DMC treatment inhibits the interaction of proteins with the DNA. (A) Nuclei were isolated from MCF7 cells and treated with MNase to obtain mononucleosome fragmented DNA (150 bp). Mononucleosomal DNA was extracted and labeled with [32P]. DNA-binding activity of nuclear proteins of untreated, 10 μM MC or DMC treated for 4 hours was analyzed by EMSA. (B) The cell cycle profile was checked by Fluorescence activated cell sorting (FACS). MCF7 cells left untreated, or treated with 10 μM MC or DMC for 4 hours and fixed in 30% ethanol and cellular DNA were stained with propidium iodide. (C) Chromatin immunoprecipitation (ChIP) was carried out in untreated, 10 μM MC or DMC treated for 4 hours and UV (50J/m2) irradiated MCF7 cells. ChIP was performed in 400 μg of cross-linked and sonicated cell lysates and antibody against ATR. Immunoprecipitated DNA was amplified by real-time quantitative PCR with primers for p53 intron 7 gene. Non-specific IgG was used to subtract the background. Values were normalized to IgG and inputs, followed by normalization to the untreated samples.
![Figure 2. DMC treatment inhibits the interaction of proteins with the DNA. (A) Nuclei were isolated from MCF7 cells and treated with MNase to obtain mononucleosome fragmented DNA (150 bp). Mononucleosomal DNA was extracted and labeled with [32P]. DNA-binding activity of nuclear proteins of untreated, 10 μM MC or DMC treated for 4 hours was analyzed by EMSA. (B) The cell cycle profile was checked by Fluorescence activated cell sorting (FACS). MCF7 cells left untreated, or treated with 10 μM MC or DMC for 4 hours and fixed in 30% ethanol and cellular DNA were stained with propidium iodide. (C) Chromatin immunoprecipitation (ChIP) was carried out in untreated, 10 μM MC or DMC treated for 4 hours and UV (50J/m2) irradiated MCF7 cells. ChIP was performed in 400 μg of cross-linked and sonicated cell lysates and antibody against ATR. Immunoprecipitated DNA was amplified by real-time quantitative PCR with primers for p53 intron 7 gene. Non-specific IgG was used to subtract the background. Values were normalized to IgG and inputs, followed by normalization to the untreated samples.](/cms/asset/b05af364-6757-4fcd-a47a-770a147a945b/kccy_a_997517_f0002_b.gif)
It has been reported that recruitment of ATR to chromatin occurs during S-phase and that ATR exhibits a decreased affinity for chromatin in quiescent cells.Citation29 To make sure that the chromatin eviction of ATR by DMC was not cell cycle phase dependent we carried out FACS for cell cycle analysis. We treated MCF7 cells with 10 μM MC or DMC for 4 hours and the cell cycle profiles (sub G1, G1, S and G2/M) were examined (). No significant cell cycle changes were detected in S phase when compared with the untreated control cells to MC or DMC treated cells (14.1% in untreated cells, 14.8% in MC and 15.61% in DMC treated cells, respectively). The FACS analysis indicated that disassociation of ATR from chromatin after DMC treatment was not due to a shift in cell cycle stages.
We further investigated the DNA binding of ATR to a specific region of chromatin using quantitative chromatin immunoprecipitation (ChIP) experiments to the p53 intron 7 region, which has been extensively studied as an ATR recruitment site.Citation30 Recruitment of ATR to UV mediated base lesions in this region are found after UV damage.Citation30 Interestingly, in our ChIP assay at the p53 intron 7 region, after both MC and DMC treatment a reduction in chromatin associated ATR was found though the recruitment of ATR was detected after UV treatment (). Taken together, our results strongly demonstrated the DMC-induced dissociation of ATR from nuclear chromatin at both genome-wide and site specific DNA regions.
ATR induces cytoplasmic DDR by activation of gadd45α
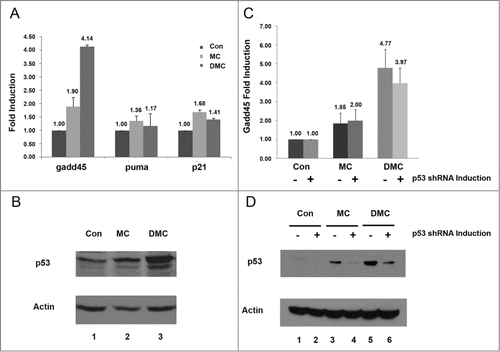
As the nuclear export of ATR was observed following DMC, but not MC, treatment we investigated the possibility that DMC induced the cytoplasmic DNA damage response of ATR when the replication forks were stalled. ATR dependent phosphorylation of p38 MAPK performs the signal transmission between the nucleus and cytoplasm by regulating gadd45α mRNAs stability after DNA damage.Citation18 We analyzed mRNA levels of gadd45α, puma and p21 in the MCF7 cells treated with 10 μM MC or DMC for 4 hours (). In DMC, but not MC, treated cells we detected a significant increase in gadd45α mRNA ( gadd45), while MC and DMC treatment had a similar moderate impact on the transcript level of puma and p21. With DMC treatment we detected a more profound increase in p53 protein levels (, lane 3). As our results showed that DMC resulted in higher gadd45α transcript levels, and gadd45α is a p53 target, we further investigated whether this induction of gadd45α was p53-dependent. We utilized MCF7 isogenic lines that were generated in our lab for inducible knockdown of p53 by mir30-based shRNA to a vector control cell line in the presence of doxycycline.Citation31 MC and DMC treatment increased the level of p53 and p53.shRNA expression resulted in decreased p53 expression (). Importantly, the gadd45α increased transcript level was not significantly reduced by the reduction in p53 protein (). The DMC induced gadd45α mRNA was 3.97 fold higher compared to untreated samples when p53 was knocked down while the DMC induced gadd45 mRNA was 4.77 fold compared to untreated cells with no knockdown (no significant statistical difference was found when tested by the Student t-test). At the same time, we found a 3-fold reduction of p53 protein levels after p53 shRNA induction in the DMC treated samples by ImageJ analysis ( lane 5 and lane 6). Taken together, our results indicate that the DMC can activate p53-independent ATR induced gadd45α mRNA stabilization.
Figure 3. p53-independent activation of gadd45 by DMC. (A and B) MCF7 cells, untreated or treated with 10 μM MC or DMC for 4 hours and RNA (A) or protein (B) were extracted. The DNA damage response genes, gadd45α, puma and p21 mRNA, were analyzed by quantitative real-time reverse transcriptase-polymerase chain reaction PCR (qRT-PCR). Results were normalized to untreated samples and gapdh values. Graphs show means and standard errors of 2 independent experiments. Western blot analysis of p53 and Actin protein levels from 50 μg of whole cell lysates in response to MC or DMC treatment. (C and D) The MCF7 cell line with inducible p53.shRNA was either treated with 2 μg/ml doxycycline (DOX) for 7 d to induce shRNA expression or left without DOX treatment. Cells were then treated with 10 μM of MC or DMC for 4 hours. Gadd45α mRNA levels were analyzed by qRT-PCR. Results were normalized to untreated samples and gapdh values. Graphs show means and standard errors of 2 independent experiments. Western blot analysis shows p53 and Actin protein levels from 50 μg of whole cell lysates in response to MC or DMC treatment.
DMC induced p53-independent higher cytotoxicity than MC
We have previously documented that DMC is more cytotoxic than MC in human cancer cell lines.Citation6 Recently, it was reported that RPA exhaustion can cause replication catastrophe and ATR signaling prevents RPA exhaustion.Citation23 To further understand the cytotoxicity caused by DMC, we treated MCF7 cells with 1, 5, 10, 50 and 100 μM MC or DMC for 48 hours. We observed that MCF7 cells were remarkably more sensitive to DMC than MC (). Reduction in cell viability was found in a dose-dependent manner after exposure to MC or DMC, but DMC showed significantly more cytotoxicity than MC ().
Figure 4. DMC induced p53-independent cell death in MCF7 cells. MCF7 cells were left either untreated or treated with 1, 5, 10, 50 and 100 μM MC or DMC for 48 hours. (A) Phase contrast microscopy image of MCF7 cells treated with MC and DMC as indicated concentrations at ×20 magnification. (B) Quantification of cell viability to MC or DMC treatment was carried out by Guava ViaCount. (C) The MCF7 cell line with inducible p53.shRNA was either treated with 2 μg/ml doxycycline (DOX) for 5 d to induce shRNA expression or left without DOX treatment, followed by treatment with 1, 5, 10, 50 and 100 μM DMC for 48 hours. (D) Isogenic MCF7 cell lines with p53.shRNA or control vector were treated with 2 μg/ml doxycycline (DOX) for 7 d to induce shRNA expression. 5000 cells were plated on the E-plate for real-time growth assay. After 6 hours the cells were treated or untreated with 10 μM DMC. The growth of untreated or 10 μM DMC treated MCF7 cells with either p53 shRNA or control vector were monitored for a total of 26 hours by xCELLigence System. Arrow indicates the 4 hours DMC treatment.
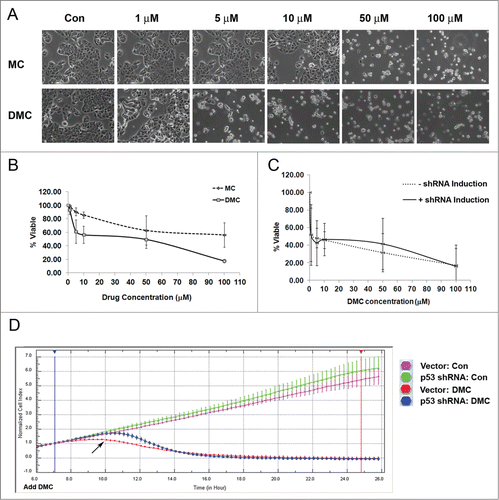
As our results showed that DMC induced higher cell death, we further investigated whether p53 played a role in DMC induced cell cytotoxicity. We compared the cytotoxicity in MCF7 isogenic lines (with and without p53 expression) by Viacount cell assay and a real-time impedance based assay (). To induce shRNA expression, cells were treated with 2 μg/ml doxycycline (DOX) for 5 d then followed with 48 hours of DMC treatment at concentrations of 1, 5, 10, 50 and 100 μM. Knocking down wt-p53 did not influence cell viability (shRNA induction vs. –shRNA induction ). The real-time growth profile in response to DMC treatment and knocking down wt-p53 of these isogenic lines was obtained with the xCELLigence System (). The growth of untreated or 10 μM DMC treated MCF7 cells with either p53.shRNA or control vector was monitored. At 4 hours of DMC treatment the cells expressing wild-type p53 had reduced proliferation while the p53 knockdown cells continued to proliferate (), while following DMC treatment for 10 hours the proliferation was drastically reduced in both populations.
DMC induced ATR disassociation from chromatin is not influenced by p53
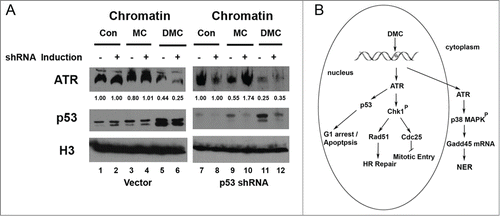
We investigated if the release of ATR from chromatin in response to DMC required p53 to work as a part of the signaling. To determine the localization of protein before and after treatment a cell fractionation protocol was carried out for the MCF7 isogenic lines: p53.shRNA and control vector (). Chromatin bound p53 was greatly induced after DMC treatment in the control vector cell line and the MCF7.shp53 inducible cell line ( compare lanes 5 and 6 to lanes 1 and 2; compare lane 11 and 12 to lanes 7 and 8). Following induction of the p53. shRNA we clearly observed the reduction of p53 ( compare lane 11 to 12). Importantly, ATR chromatin eviction was detected after DMC treatment in both cell lines ( lanes 5, 6, 11 and 12). Therefore, the reduction of p53 had no significant effect on the DMC mediated ATR chromatin eviction ( compare lane 11 to 12). These data suggest that DMC activates a p53-independent ATR-mediated DDR signal cascade that involves a chromatin eviction mechanism.
Figure 5. DMC-mediated ATR dissociation from chromatin is p53-independent. (A) MCF7 cell lines with either p53.shRNA or control vector were either treated with 2 μg/ml doxycycline (DOX) for 7 d or left DOX untreated. Cells were then treated with 10 μM of MC or DMC for 4 hours. The cell fractionation protocol was carried out to separate cellular proteins. 50 μg of chromatin bound proteins were resolved by SDS/PAGE and immunoblotted with ATR, p53 and Histone H3 antibodies. Densitometry analysis of chromatin bound ATR was performed using NIH ImageJ, and values were normalized to untreated samples (Con). (B) A simplified scheme is shown to model the signaling pathways for DMC-activated cell death through eviction of ATR from chromatin and activation of both Chk1 and gadd45 pathways.
Discussion
Dynamic activation of DDR proteins on chromatin in response to DMC induced β-interstrand crosslink
In this manuscript, we investigated the molecular mechanism of the higher cytotoxicity induced by the β−interstrand crosslink agent DMC. By cell fractionation, immunofluorescence and ChIP we identified that DMC, but not MC, treatment reduced the association of ATR with the chromatin and activated Chk1 phosphorylation and Rad51 foci formation. Moreover, using gel-shift experiments we observed that DMC treatment caused a general reduction of DNA binding activity of many nuclear proteins.
The topoisomerase II (TopoII) inhibitor Doxorubicin (Doxo) induces H2A/2B and H3/H4 chromatin eviction.Citation32 Furthermore, Doxo induces H2AX chromatin eviction and impaired DDR signaling but also can stimulate epigenetic alterations and apoptosis induction.Citation32 Our chromatin fractionation data support a model for a rapid activation of the DDR induced by DMC β−interstrand crosslink DNA damage structures and specific post-translational modifications of proteins to promote DDR signaling (See for a flow model). Our data showed that in the unperturbed cells ATR associated with chromatin and that in response to DMC treatment the ATR was released from the chromatin. Following DMC, but not MC, treatment the ATR was evicted from the chromatin and activated nuclear Chk1 and induced a rapid increase in gadd45α mRNA (). Moreover, our previous data has shown that DMC treatment first activates Chk1 and then subsequently down-regulates Chk1 by causing increased ubiquitination and subsequent degradation by the proteasome in the later stages to stimulate cell death.Citation5 Our real time cell viability analysis showed that at the early treatment point of 4 hours cells were viable. Cell death was not complete until after 9 hours of DMC drug treatment. We observed that after 4 hours of DMC treatment Chk1 was phosphorylated and Rad51 was recruited to chromatin. Our findings suggest that multistep pathways induce association of Rad51 and chromatin eviction of ATR.
Post-translational modifications and post-transcriptional regulation: a multi-layered DNA damage response
The autophosphorylation of ATR is crucial for its kinase activity and this works as a molecular switch for checkpoint activation.Citation33 Our observation of DMC induced Chk1 phosphorylation at ser-345 implicates the phosphorylation of Chk1 in affecting Rad51 binding to damaged sites to accomplish HR repair. It has been reported that Chk1 interacts with Rad51 and Rad51 is phosphorylated on Thr 309 after hydroxyurea treatment.Citation34 Human Rad51 has been shown to be phosphorylated at Ser-14 by Plk1 in response to DNA damage leading to direct binding to the MRN component Nbs, and this process helps Rad51 to be recruited to DNA damage sites, thus allowing accurate HR.Citation35
In our work, DMC induced high levels of gadd45 mRNA presumably because of p53-independent ATR/p38 MAPK mediated gadd45 mRNA stabilization. In addition to gadd45 being a p53 transcriptional target, the gadd45 mRNA induction occurs through BRCA1 and a p53-independent mechanism through a BRCA1 binding site located in the first intron of the gadd45 gene.Citation36 Our data showed that p53 was not required for the DMC-mediated activation of gadd45. The stability of mRNA plays an important role in maintaining genome integrity in response to DNA damage.Citation37 RNA-binding proteins (RBPs) can modulate mRNA half-life, spliceosome targeting, nuclear export and translatability in cells with genotoxic stress.Citation37 However transcription is generally inhibited in response to DNA damage, for example, an ATM-dependent transcriptional silencing occurs in response to DNA double strand breaks.Citation38 Our finding of p53-independent gadd45 activation suggests ATR is involved in multiple pathways to regulate the DNA damage response.
Prospective application of synthetic lethality in cancer treatment
The efficient repair of ICLs is critical for the maintenance of genome integrity. Once the crosslinked adduct is removed by error-free repair conducted by Rad51 mediated homologous recombination, the DNA replication fork can be restored.Citation39 However, cells with the deficient checkpoint control may have genomic instability that will promote cancer development. The observation that DMC treatment activates the recruitment of Rad51 to chromatin and the formation of Rad51 foci indicates orchestrated and ordered protein-signaling pathways in the homologous recombination proficient cells. In addition, several overlapping DNA damage repair pathways were activated in DMC treated cells to allow them to control cell cycle progression. Our RT-PCR data suggested that the gadd45α stabilization upon DMC-mediated DNA damage was an early event. Activation of ATR chromatin eviction, Chk1 phosphorylation, and Rad51 foci formation were all induced significantly earlier than MCF7 cell death. Replication catastrophe has been associated with the depletion of ATR and RPA exhaustion.Citation23 As we found that ATR was dissociated from chromatin, and because there was a delay in cell death induction it suggests that DMC might also cause a later phase RPA exhaustion.
Our results suggest inhibition of the ATR pathway in cancers with mutant p53 has the potential of improving the efficacy of MC treatments in breast cancer cells. Preliminary evidence predicts promise for cancer treatment in humans with targeting ATR and Chk1 inhibition.Citation10 Cancer cells defective in the ATM-p53 pathway rely on the S and G2/M checkpoint controlled by the ATR-Chk1 pathway and therefore Chk1 inhibition in combination with chemotherapy has been tested in preclinical studies for the treatment of p53-mutated tumors to induce “mitotic cell death."Citation19 A Chk1 inhibitor is currently undergoing phase 1 and 2 clinical trials and ATM, Rad51 and ATR inhibitors are also in preclinical testing.Citation40 Future studies on how the inhibition of ATR and Chk1 activity might enable the death of cancer are promising and our work has highlighted regulation of chromatin eviction as an additional pathway for targeted treatment development.
Recent studies on the use of combination treatments suggest that inhibiting the Chk1 pathway, or the ATR pathway, in combination with more standard therapeutics can be highly efficacious.Citation41,42 Combination treatment of colon cancers with mutations in the APC tumor suppressor gene with a Chk1 kinase inhibitor and standard 5-fluorouricil is more efficacious than either drug alone.Citation42 Moreover, combining the standard-of care platinum-based chemotherapic drugs with VX-970 (the first clinical ATR inhibitor) in a lung cancer model dramatically increases the efficacy of cisplatin for tumor growth inhibition of previously cisplatin insensitive cancers.Citation41 It is possible that combining the standard chemotherapeutic mitomycin with drugs that inhibit either ATR or Chk1 would improve efficacy for killing cancers with mutations in p53. Such combination treatments of mtp53 expressing cancers could provoke synergistic death outcomes. The synergism of mitomycin treatment with inhibition of ATR or Chk1 might recapitulate the DMC-induced p53-independent cell death.
Materials and Methods
Materials
Mitomycin C was obtained from Bristol-Myers Squibb Co., Wallingford, CT and from Kyowa Hakko Kogyo Co. Ltd., Japan. The procedure used for the synthesis of 10-decarbamoyl mitomycin C was as described previously.Citation43 Growth medium and penicillin−streptomycin solution were obtained from Mediatech Inc.., Herndon, VA. Fetal bovine serum (FBS) was purchased from Gemini Bio-Products, West Sacramento, CA.
Cell culture, drugs, and plasmids
MCF7 human breast cancer cells were obtained from American Type Culture Collection. p53 shRNA MCF7 cells were constructed as described previously.Citation31 To induce shRNA expression, cells were treated with 2 μg/ml doxycycline (DOX) for 7 d MCF7, MCF7.p53shRNA cells were grown in 90% DMEM (Invitrogen, Carlsbad, CA, USA)/10% fetal bovine serum and penicillin (50 units/mL)/streptomycin (50 μg/mL) solution. MC and DMC were dissolved in 30% methanol/70% water.
Cell lysates
PBS-washed cell pellets were gently resuspended in a hypotonic lysis buffer (10 mM HEPES pH 7.8, 10 mM KCl, 1.5 mM MgCl2, 0.5 mM DTT, 1 mM PMSF, 8.5 μg/ml Aprotinin, 2 μg/ml Leupeptin. and phosphatase inhibitors) and Triton X-100 (0.1%). After incubation of the cells on ice for 10 min, the cellular extracts were then centrifuged at 800 × g for 10 min at 4°C and the supernatant was collected as the cytoplasmic fraction and further clarified by high-speed centrifugation (5 min, 12,000rpm, 4°C) to remove cell debris and insoluble aggregates. The nuclei-enriched fraction was washed once with hypotonic buffer and resuspended in a nuclear extraction buffer (20 mM HEPES, pH 7.8, 1.5 mM MgCl2, 420 mM NaCl, 25% v/v glycerol, 0.2 mM EDTA, 0.5 mM DTT, 1 mM PMSF, 8.5 μg/ml Aprotinin, 2 μg/ml Leupeptin. and phosphatase inhibitors). Nuclear proteins were extracted via agitation for 30 min at 4°C and centrifuged at 13,000 rpm and 4°C for 30 min. The supernatants containing solubilized nuclear proteins were stored at −80°C.
Cell fractionation
Described previously by Méndez and Stillman.Citation44 To isolate chromatin, cells were resuspended in buffer A (10 mM HEPES, pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM DTT, 1 mM PMSF, 8.5 μg/ml Aprotinin, 2 μg/ml Leupeptin at 4 × 107cells/ml). Triton X-100 (0.1%) was added, and the cells were incubated for 5 min on ice. Nuclei were collected in pellet 1 (P1) by low-speed centrifugation (5 min, 3600rpm, 4°C). The supernatant (S1) was further clarified by high-speed centrifugation (5 min, 13,000 rpm, 4°C) to remove cell debris and insoluble aggregates. Nuclei were washed once in buffer A, and then lysed in buffer B (3 mM EDTA, 0.2 mM EGTA, 1 mM DTT, protease inhibitors as described above). Insoluble chromatin was collected by centrifugation (5 min, 4,000 rpm, 4°C), washed once in buffer B, and centrifuged again under the same conditions. The final chromatin pellet (P3) was resuspended in buffer B and sonicated 3 times for 30 s each in sonicator at 95% amplitude.
Western blot analysis
50 μg of protein samples were electrophoresised in 4–12% SDS denaturing poly acrylamide gels (Life technologies) and electrotransferred to nitrocellulose membranes (Amersham). The blots were incubated with the following primary antibodies: p53 specific monoclonal antibodies (1:1:1 mixture of 421, 240, and 1801 antibodies) as described, anti-ATR, Rad51 (Abcam), anti-RPA70 (EMD/ Calbiochem), anti-Chk1, anti-phopho-ser345 Chk1(Cell signaling) and the polyclonal anti- Actin antibody (Sigma) as the manufacturer's recommendation. The membranes were then incubated in anti-mouse or anti-rabbit secondary antibodies (Sigma), and the signals were visualized by chemiluminescence. Densitometry analysis was performed using NIH ImageJ, and values were normalized to Actin.
Immunofluorescence
Cells were grown and treated with 10 μM MC or DMC on coverslips. Untreated and treated cells were fixed with 4% Formaldehyde and permeabilized with 0.5% Triton-X-100. Immunohistochemistry was done with ATR (Abcam, ab2905) and Lamin A/C (Millipore, MAB3211) antibodies followed by incubation with FITC-conjugated anti-Rabbit (Invitrgen A11037) and Alexa-conjugated anti-mouse (Invitrogen A11058). Coverslips were mounted onto slides using Vectashield mounting medium with DAPI (Fisher Scientific NC9524612, Pittsburgh, PA, USA). Images were collected by PerkinElmer UltraVIEW ERS Spinning Disc Microscope, Waltham, MA, USA. All images were captured using a 100× objective.
Chromatin immunoprecipitation (ChIP)
MCF7 cells left untreated, treated with10 μM MC or DMC for 4 hours or UV (50J/m2) irradiated. After UV exposure, cells were re-incubated for 1 hour before Formaldehyde crosslink. Cells were incubated with 1% Formaldehyde for 30 minutes at 5% CO2 37°C humidified incubator, followed by 0.125 M Glycine treatment for 5 minutes. Cells were lysed in RIPA buffer with 1 mM PMSF, 8.5 μg/ml Aprotinin, 2 μg/ml Leupeptin and Phosphatase Inhibitor Cocktail 1 (Sigma). Lysates were sonicated 15 times (one minute pulse and one minute rest) in a Branson Digital Sonifier, Danbury, CT, USA and spun down for 30 minutes 13,000 rpm at 4°C. 400 μg of protein from cell lysates were subjected to overnight incubation at 4°C with 2 μg of ATR antibody (Abcam), or non-specific IgG (Santa Cruz, IgG mouse sc-2025, IgG rabbit sc-2027). 50 μl of 25% beads slurry of protein A/G Plus Agarose beads (Santa Cruz sc-2003), pre-blocked with 0.3 mg/ml sheared herring sperm DNA (Invitrogen, 15634–017), were added to immunoprecipitation samples for 2 hours at 4°C, followed by washes: (1) 0.1% SDS, 1% Triton-X-100, 20 mM Tris pH8.1, 150 mM NaCl; (2) 0.1% SDS, 1% Triton-X-100, 20 mM Tris pH8.1, 500 mM NaCl; (3) 0.25 M LiCl, 1% NP-40, 1% Deoxycholate, 1 mM EDTA, 10 mM Tris pH8; and (4) twice with TE pH8. Immunoprecipitated chromatin was de-crosslinked overnight at 65°C with 1 mg/ml ProteinaseK, 1% SDS and 0.1M NaHCO3. For total DNA input, 40 μg were similarly de-crosslinked. DNA fragments were purified using Qiagen QiaQuick kit (Qiagen) and amplified by real-time quantitative PCR in 7500 Sequence Detection System (Applied Biosystems). Primers sequences are below:
p53 intron 7 forward primer: 5′-CCTCTTACCGAT-TTCTTCCA.
p53 intron 7 reverse primer, 5′-GCAAGAGGCAGTAAGGAA-3′.
Quantitative reverse transcription-PCR (qRT-PCR)
RNA was isolated using QIAshredder columns and RNeasy Mini Kit (Qiagen, Valencia, CA, USA). A total of 5 μg of RNA was used for cDNA synthesis using High Capacity cDNA Archive Kit reagents (Applied Biosystems, Foster City, CA, USA). 150 ng of cDNA was combined with Taqman Universal Master Mix (Applied Biosystems, Foster City, CA, USA) and Applied Biosystems Assays on Demand primers/probes for Gadd45 (Hs_Hs00169255_m1), puma (Hs00248075_m1), p21 (Hs00355782_m1) or gapdh (433764–1207037). PCR reaction was carried out in 7500 Sequence Detection System (Applied Biosystems). Fluorescence activated cell sorting (FACS).
Fluorescence activated cell sorting (FACS)
FACS was performed on a FACScan (BD Biosciences, San Jose, CA, USA). After treatments, cells were harvested, washed, resuspended in PBS containing 2% bovine serum albumin, 0.1% sodium azide, fixed in 30% ethanol. Before sorting, propidium iodide staining and RNase treatment were performed for 30 minutes at 37°C.
Preparation of mono-nucleosomal DNA and EMSA
Three × 106 MCF7 cells were treated with 100 units Micrococcal Nuclease (New England Biolabs) for 10 minutes and followed by proteinase k digestion. Mononucleosomal DNA was extracted and labeled with32P-gamma-ATP. Reaction mixtures for EMSA experiments (30 μl) were composed of 0.1 pmol of mononucleosomal DNA, 20 mM HEPES (pH 7.8), 100 mM KCl, 1 mM EDTA, 1 mM DTT, 1 μg of sonicated salmon sperm DNA, and 10% glycerol. Two μg of untreated, MC or DMC treated nuclear protein extract was added to each reaction. All samples were incubated at room temperature for 20 min. The protein-DNA complexes were resolved on a 5% acrylamide gel.
xCELLigence real-time cell analysis (RTCA) of cytotoxicity
xCELLigence cell index (CI) impedance measurements were performed according to the instructions of the supplier (Roche Diagnostics GmbH, Mannheim, Germany). Cells were resuspended in medium and subsequently adjusted to 50,000cells/mL. Six hours after seeding 100μL of the cell suspensions into the wells of the E-plate, cells were treated or untreated with 10 μM DMC and were monitored every 15min for a period of up to 26hr by the xCELLigence system.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Bargonetti laboratory members for critical discussions and Dr. Nataly Shtraizent and Alla Polotskaia for their technical assistance.
Funding
This work was supported by NIH SCORE Grant (1SC1CA137843) and The Breast Cancer Research Foundation.
References
- Garewal HS. (1988) Mitomycin C in the chemotherapy of advanced breast cancer, Semin Oncol 15:74-9; PMID:3134698
- Bradner WT. (2001) Mitomycin C: a clinical update, Cancer Treat Rev 27:35-50; PMID:11237776; http://dx.doi.org/10.1053/ctrv.2000.0202
- Bargonetti J, Champeil E, and Tomasz M. (2010) Differential toxicity of DNA adducts of mitomycin C, J Nucleic Acids 2010; PMID:20798760; http://dx.doi.org/10.4061/2010/698960
- Lawley PD, Phillips DH. (1996) DNA adducts from chemotherapeutic agents, Mutat Res 355:13-40; PMID:8781575; http://dx.doi.org/10.1016/0027-5107(96)00020-6
- Boamah EK, Brekman A, Tomasz M, Myeku N, Figueiredo-Pereira M, Hunter S, Meyer J, Bhosle RC, Bargonetti J. (2010) DNA adducts of decarbamoyl mitomycin C efficiently kill cells without wild-type p53 resulting from proteasome-mediated degradation of checkpoint protein 1, Chem Res Toxicol 23:1151-62; PMID:20536192; http://dx.doi.org/10.1021/tx900420k
- Boamah EK, White DE, Talbott KE, Arva NC, Berman D, Tomasz M, Bargonetti J. (2007) Mitomycin-DNA adducts induce p53-dependent and p53-independent cell death pathways, ACS Chem Biol 2:399-407; PMID:17530733; http://dx.doi.org/10.1021/cb700060t
- Abbas T, Olivier M, Lopez J, Houser S, Xiao G, Kumar GS, Tomasz M, Bargonetti J. (2002) Differential activation of p53 by the various adducts of mitomycin C, J Biol Chem 277:40513-9; PMID:12183457; http://dx.doi.org/10.1074/jbc.M205495200
- Hussain SP, Harris CC. (1998) Molecular epidemiology of human cancer: contribution of mutation spectra studies of tumor suppressor genes, Cancer Res 58:4023-37; PMID:9751603
- (2012) Comprehensive molecular portraits of human breast tumours, Nature 490:61-70; PMID:23000897; http://dx.doi.org/10.1038/nature11412
- Reaper PM, Griffiths MR, Long JM, Charrier JD, Maccormick S, Charlton PA, Golec JM, Pollard JR. (2011) Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR, Nat Chem Biol 7:428-30; PMID:21490603; http://dx.doi.org/10.1038/nchembio.573
- Xie L, Gazin C, Park SM, Zhu LJ, Debily MA, Kittler EL, Zapp ML, Lapointe D, Gobeil S, Virbasius CM, et al. (2012) A synthetic interaction screen identifies factors selectively required for proliferation and TERT transcription in p53-deficient human cancer cells, PLoS Genet 8:e1003151; PMID:23284306; http://dx.doi.org/10.1371/journal.pgen.1003151
- Deans AJ, West SC. (2011) DNA interstrand crosslink repair and cancer, Nat Rev Cancer 11:467-80; PMID:21701511; http://dx.doi.org/10.1038/nrc3088
- O'Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA. (2003) A splicing mutation affecting expression of ataxia-telangiectasia and rad3-related protein (ATR) results in seckel syndrome, Nat Genet 33:497-501; PMID:12640452; http://dx.doi.org/10.1038/ng1129
- Brown EJ, Baltimore D. (2000) ATR disruption leads to chromosomal fragmentation and early embryonic lethality, Genes Dev 14:397-402; PMID:10691732
- Tibbetts RS, Cortez D, Brumbaugh KM, Scully R, Livingston D, Elledge SJ, Abraham RT. (2000) Functional interactions between BRCA1 and the checkpoint kinase ATR during genotoxic stress, Genes Dev 14:2989-3002; PMID:11114888; http://dx.doi.org/10.1101/gad.851000
- Brown AD, Sager BW, Gorthi A, Tonapi SS, Brown EJ, Bishop AJ. (2014) ATR suppresses endogenous DNA damage and allows completion of homologous recombination repair, PLoS One 9:e91222; PMID:24675793; http://dx.doi.org/10.1371/journal.pone.0091222
- Reinhardt HC, Aslanian AS, Lees JA, Yaffe MB. (2007) p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage, Cancer Cell 11:175-89; PMID:17292828; http://dx.doi.org/10.1016/j.ccr.2006.11.024
- Reinhardt HC, Hasskamp P, Schmedding I, Morandell S, van Vugt MA, Wang X, Linding R, Ong SE, Weaver D, Carr SA, et al. (2010) DNA damage activates a spatially distinct late cytoplasmic cell-cycle checkpoint network controlled by MK2-mediated RNA stabilization, Mol Cell 40:34-49; PMID:20932473; http://dx.doi.org/10.1016/j.molcel.2010.09.018
- Tamura RE, de Vasconcellos JF, Sarkar D, Libermann TA, Fisher PB, Zerbini LF. (2012) GADD45 proteins: central players in tumorigenesis, Curr Mol Med 12:634-51; PMID:22515981; http://dx.doi.org/10.2174/156652412800619978
- Zhao H, Piwnica-Worms H. (2001) ATR-mediated checkpoint pathways regulate phosphorylation and activation of human chk1, Mol Cell Biol 21:4129-39; PMID:11390642; http://dx.doi.org/10.1128/MCB.21.13.4129-4139.2001
- Zou L, Elledge SJ. (2003) Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes, Science 300:1542-8; PMID:12791985; http://dx.doi.org/10.1126/science.1083430
- Pabla N, Ma Z, McIlhatton MA, Fishel R, Dong Z. (2011) hMSH2 recruits ATR to DNA damage sites for activation during DNA damage-induced apoptosis, J Biol Chem 286:10411-8; PMID:21285353; http://dx.doi.org/10.1074/jbc.M110.210989
- Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, Bekker-Jensen S, Mailand N, Bartek J, Lukas J. (2013) ATR prohibits replication catastrophe by preventing global exhaustion of RPA, Cell 155:1088-103; PMID:24267891; http://dx.doi.org/10.1016/j.cell.2013.10.043
- Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW, Cortez D. (2011) Analysis of protein dynamics at active, stalled, and collapsed replication forks, Genes Dev 25:1320-7; PMID:21685366; http://dx.doi.org/10.1101/gad.2053211
- Polo SE, Jackson SP. (2011) Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications, Genes Dev 25, 409-33; PMID:21363960; http://dx.doi.org/10.1101/gad.2021311
- Stone HR, Morris JR. (2014) DNA damage emergency: cellular garbage disposal to the rescue?, Oncogene 33:805-13; PMID:23503465; http://dx.doi.org/10.1038/onc.2013.60
- Raderschall E, Golub EI, Haaf T. (1999) Nuclear foci of mammalian recombination proteins are located at single-stranded DNA regions formed after DNA damage, Proc Natl Acad Sci U S A 96:1921-6; PMID:10051570; http://dx.doi.org/10.1073/pnas.96.5.1921
- Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM. (2003) p53 has a direct apoptogenic role at the mitochondria, Mol Cell 11:577-90; PMID:12667443; http://dx.doi.org/10.1016/S1097-2765(03)00050-9
- Dart DA, Adams KE, Akerman I, Lakin ND. (2004) Recruitment of the cell cycle checkpoint kinase ATR to chromatin during S-phase, J Biol Chem 279:16433-40; PMID:14871897; http://dx.doi.org/10.1074/jbc.M314212200
- Jiang G, Sancar A. (2006) Recruitment of DNA damage checkpoint proteins to damage in transcribed and nontranscribed sequences, Mol Cell Biol 26:39-49; PMID:16354678; http://dx.doi.org/10.1128/MCB.26.1.39-49.2006
- Brekman A, Singh KE, Polotskaia A, Kundu N, Bargonetti J. (2011) A p53-independent role of mdm2 in estrogen-mediated activation of breast cancer cell proliferation, Breast Cancer Res 13:R3; PMID:21223569; http://dx.doi.org/10.1186/bcr2804
- Pang B, Qiao X, Janssen L, Velds A, Groothuis T, Kerkhoven R, Nieuwland M, Ovaa H, Rottenberg S, van Tellingen O, et al. (2013) Drug-induced histone eviction from open chromatin contributes to the chemotherapeutic effects of doxorubicin, Nat Commun 4:1908; PMID:23715267; http://dx.doi.org/10.1038/ncomms2921
- Liu S, Shiotani B, Lahiri M, Marechal A, Tse A, Leung CC, Glover JN, Yang XH, Zou L. (2011) ATR autophosphorylation as a molecular switch for checkpoint activation, Mol Cell 43:192-202; PMID:21777809; http://dx.doi.org/10.1016/j.molcel.2011.06.019
- Sorensen CS, Hansen LT, Dziegielewski J, Syljuasen RG, Lundin C, Bartek J, Helleday T. (2005) The cell-cycle checkpoint kinase chk1 is required for mammalian homologous recombination repair, Nat Cell Biol 7:195-201; PMID:15665856; http://dx.doi.org/10.1038/ncb1212
- Krejci L, Altmannova V, Spirek M, Zhao X. (2012) Homologous recombination and its regulation, Nucleic Acids Res 40:5795-818; PMID:22467216; http://dx.doi.org/10.1093/nar/gks270
- Jin S, Zhao H, Fan F, Blanck P, Fan W, Colchagie AB, Fornace AJ Jr., Zhan Q. (2000) BRCA1 activation of the GADD45 promoter, Oncogene 19:4050-7; PMID:10962562; http://dx.doi.org/10.1038/sj.onc.1203759
- Reinhardt HC, Cannell IG, Morandell S, Yaffe MB. (2011) Is post-transcriptional stabilization, splicing and translation of selective mRNAs a key to the DNA damage response?, Cell Cycle 10:23-7; PMID:21173571; http://dx.doi.org/10.4161/cc.10.1.14351
- Shanbhag NM, Rafalska-Metcalf IU, Balane-Bolivar C, Janicki SM, Greenberg RA. (2010) ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks, Cell 141:970-81; PMID:20550933; http://dx.doi.org/10.1016/j.cell.2010.04.038
- Helleday T. (2003) Pathways for mitotic homologous recombination in mammalian cells, Mutat Res 532:103-15; PMID:14643432; http://dx.doi.org/10.1016/j.mrfmmm.2003.08.013
- Canaani D. (2013) Application of the concept synthetic lethality toward anticancer therapy: a promise fulfilled?, Cancer Lett 352(1):59-65; PMID:23962561; http://dx.doi.org/10.1016/j.canlet.2013.08.019
- Hall AB, Newsome D, Wang Y, Boucher DM, Eustace B, Gu Y, Hare B, Johnson MA, Milton S, Murphy CE. et al. (2014) Potentiation of tumor responses to DNA damaging therapy by the selective ATR inhibitor VX-970, Oncotarget 5:5674-85; PMID:25010037
- Martino-Echarri E, Henderson BR, Brocardo MG. (2014) Targeting the DNA replication checkpoint by pharmacologic inhibition of chk1 kinase: a strategy to sensitize APC mutant colon cancer cells to 5-fluorouracil chemotherapy, Oncotarget 5:9889-900; PMID:25301724
- Kinoshita S, Uzu K, Nakano K, Takahashi T. (1971) Mitomycin derivatives. Two. derivatives of decarbamoylmitosane and decarbamoylmitosene, J Med Chem 14:109-12; PMID:4993458; http://dx.doi.org/10.1021/jm00284a006
- Mendez J, Stillman B. (2000) Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis, Mol Cell Biol 20:8602-12; PMID:11046155; http://dx.doi.org/10.1128/MCB.20.22.8602-8612.2000