
Abstract
The p53, p63, and p73 proteins belong to the p53 family of transcription factors, which play key roles in tumor suppression. Although the transactivation domains (TADs) of the p53 family are intrinsically disordered, these domains are commonly involved in the regulatory interactions with mouse double minute 2 (MDM2). In this study, we determined the solution structure of the p73TAD peptide in complex with MDM2 using NMR spectroscopy and biophysically characterized the interactions between the p53 family TAD peptides and MDM2. In combination with mutagenesis data, the complex structures revealed remarkably close mimicry of the MDM2 recognition mechanism among the p53 family TADs. Upon binding with MDM2, the intrinsically disordered p73TAD and p63TAD peptides adopt an amphipathic α-helical conformation, which is similar to the conformation of p53TAD, although the α-helical content induced by MDM2 binding varies. With isothermal titration calorimetry (ITC) and circular dichroism (CD) data, our biophysical characterization showed that p73TAD resembles p53TAD more closely than p63TAD in terms of helical stability, MDM2 binding affinity, and phosphorylation effects on MDM2 binding. Therefore, our structural information may be useful in establishing alternative anticancer strategies that exploit the activation of the p73 pathway against human tumors bearing p53 mutations.
Abbreviations
| AIRs | = | Ambiguous interaction restraints |
| HSQC | = | heteronuclear single quantum coherence |
| IDP | = | Intrinsically disordered protein |
| ITC | = | Isothermal titration calorimetry |
| MDM2 | = | mouse double minute 2 |
| NMR | = | nuclear magnetic resonance |
| NOESY | = | nuclear Overhauser effect spectroscopy |
| RDC | = | Residual dipolar coupling |
| TAD | = | transactivation domain |
Introduction
The p53, p63, and p73 proteins comprise the p53 family of transcription factors, and have distinct functions in tumor suppression. The tumor suppressor p53 mediates cell cycle arrest, DNA repair, apoptosis, and senescence in response to a variety of stress stimuli, such as DNA damage, ultraviolet light, hypoxia, oncogene activation, and viral infection. The p73 and p63 proteins can mediate p53-like activities through the transactivation of p53-responsive genes.Citation1,2 However, in contrast with p53, p73 is involved in neuronal development and differentiation, and p63 is essential for limb, craniofacial, and epithelial development.Citation3-Citation6 The p53 protein family contains homologous domains, including a disordered N-terminal transactivation domain (TAD), a central DNA-binding domain (DBD), and a C-terminal oligomerization domain (OD) (). Moreover, p73 and p63 possess additional domains including a sterile α motif (SAM) and a transcription inhibitory domain (TID) in their C-terminal portion, which are not present in p53. Unlike p53, p73 and p63 exist in various functionally distinct isoforms, which could be classified as isoforms with a TAD and isoforms lacking a TAD. The presence or absence of a TAD determines the protein activity in cells. The TADs of the p53 family are intrinsically disordered, and these domains are able to bind diverse partners. This binding is regulated via post-translational modifications such as phosphorylation.
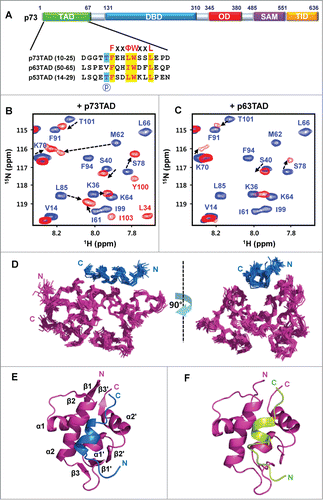
Figure 1. Structure determination of the MDM2/p73TAD peptide and the MDM2-p63TAD peptide complexes. (A) Sequence alignment of the MDM2 binding motifs in the p53 family TADs. “P” indicates the site for phosphorylation. (B and C) The binding of the p73TAD and p63TAD peptides to MDM2. Overlaid 2D 1H-15N HSQC spectra for 15N-labeled MDM2 in the absence (blue) or in the presence (red) of the p73TAD (B) and p63TAD peptides (C). (D) Overlay of the 20 lowest-energy structures of the complex between MDM2 (magenta) and the p73TAD (residues 10–25) peptide (blue). (E) Ribbon representation of the lowest-energy structure of the MDM2/p73TAD (residues 10–25) peptide complex. (F) A refined structural model for the complex between MDM2 (magenta) and the p63TAD (residues 51–65) peptide (green).
Under normal conditions, the cellular level of p53 is precisely controlled by mouse double minute 2 (MDM2). MDM2 is an E3 ubiquitin ligase that negatively regulates p53 stability through ubiquitin-dependent degradation and inhibits the transcriptional activity of p53 through steric occlusion of the p53TAD via direct interaction.Citation7,8 Similar to p53, MDM2 binding impairs the transcriptional activity of p73 by preventing the p73TAD from binding the transcriptional coactivators p300 and CBPCitation9-Citation12; however, MDM2 does not lead to ubiquitin-dependent degradation of p73.Citation9,10 The physiological relevance of the regulatory binding of p53TAD by MDM2 is well recognized because its aberrant regulation is known to induce tumors.Citation13-Citation17 However, little is known about the structural basis for the MDM2 binding mechanisms of p73TAD and p63TAD.
Reactivating the p53 pathway in cancers has been an attractive strategy for cancer therapy.Citation18 Previous mouse model studies demonstrated that restoration of wild-type p53 function induces the regression of established tumors in vivo via apoptosis and/or senescence.Citation19,20 Using this therapeutic strategy, pharmaceutical and academic studies have been performed to identify lead compounds for cancer drug discovery. However, p53 is mutated or inactivated in greater than 50% of human cancers, Citation21 which limits the applications of reactivating the p53 pathway in cancer therapy. In contrast, p73 and p63 are rarely mutated or lost (fewer than 0.5% for p73) in human cancers.Citation22 Additionally, the transcriptionally active forms of p73 are capable of inducing apoptosis independently of the p53 status.Citation23,24 Thus, activation of the p73-mediated pathway in p53-deficient cancer cells may provide a promising anticancer strategy.Citation23 In particular, inhibition of the interaction between p73TAD and MDM2 to activate the p73 pathway could be an attractive target for cancer therapy.
In this study, we determined the solution structure of a p73TAD peptide in complex with MDM2 and provided a refined structural model of the p63TAD peptide in complex with MDM2 using nuclear magnetic resonance (NMR) spectroscopy. The complex structures revealed that the p73TAD and p63TAD peptides bind to MDM2 with a similar mode of binding as the p53TAD peptide. The mutagenesis data show that the highly conserved hydrophobic triads of residues in the p53 family TAD motifs are major determinants of MDM2 binding. Using isothermal titration calorimetry (ITC) and circular dichroism (CD), we characterized the MDM2-binding affinity, the α-helical content induced by MDM2 binding, and the effect of phosphorylation on MDM2 binding among the p53 family TAD motifs. Our complex structures may be useful for the structure-based design of anticancer therapeutics to activate the p73 pathway against many human tumors that have p53 mutations.
Results
Interactions between p73TAD and p63TAD and MDM2
Despite a lack of high sequence homology, sequence alignments showed that the FxxΦWxxL (Φ: Leu or Ile, x: any amino acid) sequence motif is highly conserved in p53TAD, p63TAD, and p73TAD (). Based on this finding, we designed minimal MDM2-binding motifs of p73TAD and p63TAD, which are homologous to the previously known MDM2-binding motif of p53TAD (residues 15–29). To examine whether each minimal binding motif could interact with the N-terminal domain of MDM2 (residues 3–109), we monitored the chemical shift perturbations of 15N-labeled MDM2 titrated with p73TAD (residues 10–25) and p63TAD (residues 51–65) peptides using NMR spectroscopy (; Fig. S1).
Upon binding to the p73TAD peptide, the majority of the MDM2 cross-peaks underwent a slow chemical exchange between the free and bound states on the NMR chemical shift time scale in a manner similar to what was observed for the p53TAD peptide (; Fig. S1A). However, when bound to the p63TAD peptide, the majority of equivalent MDM2 cross-peaks disappeared because of severe line-broadening, indicating an intermediate exchange on the NMR chemical shift time scale (; Fig. S1B). The chemical shift perturbations of MDM2 induced by the p73TAD and p63TAD peptides are similar to the perturbations induced by the p53TAD peptide.Citation25 These results suggest that the minimal binding motifs of the p73TAD and p63TAD peptides could bind to the same binding site of MDM2 to which the p53TAD peptide binds. Consequently, the p53 family TADs with conserved sequence motifs may share the same site for MDM2 binding.
Structure determination of the MDM2-p73TAD and MDM2-p63TAD complexes
To understand the structural basis for the regulatory binding mechanism of p73TAD by MDM2, we determined the solution structure of MDM2 (residues 3–109) in complex with the p73TAD peptide using NMR spectroscopy (; ). The p73TAD-bound MDM2 structure forms an α/β-fold, resembling the structure of the p53TAD-bound MDM2.Citation26 Two subdomains comprising 2 α-helices and one 3-stranded β-sheet are related by a dyad axis of symmetry. A cleft between the subdomains is formed with the side-walls made up of the 2 longer helices (α2 and α2') and the floor made up of the 2 shorter helices (α1 and α1'). The cleft is closed by 2 antiparallel β-sheets at the opposite ends. A singular amphipathic α-helix is formed within residues 15–22 of the bound p73TAD peptide. Because the MDM2-p63TAD complex demonstrates severe line broadening for the majority of the interfacial resonances (; Fig. S1B), it was difficult to observe unambiguous intermolecular nuclear Overhauser effects (NOEs) between MDM2 and the p63TAD peptide. Nevertheless, the chemical shift perturbations of the residues in the binding interface were sufficient experimental restraints for NMR data-driven docking calculation. Thus, we calculated a refined structural model for the MDM2-p63TAD peptide complex using the program HADDOCK 2.1 (). The overall structure of the MDM2-p63TAD peptide complex is similar to structures of the MDM2-p53TAD peptideCitation26 and MDM2-p73TAD peptide complexes ().
Table 1. Structural statistics for the MDM2/p73TAD peptide complexa
Table 2. Structural statistics for the MDM2/p63TAD peptide complex
Structural comparison of the complexes formed between the p53 family TADs and MDM2
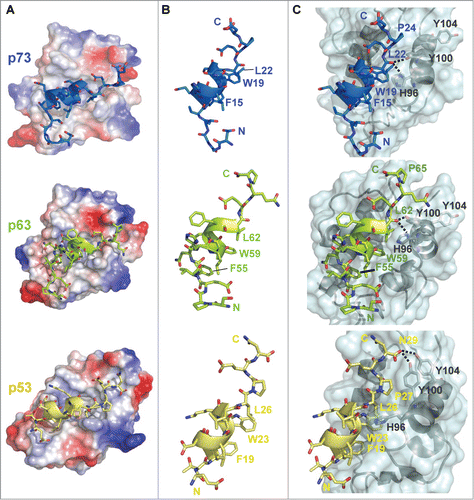
The structures of the p73TAD and p63TAD peptides bound to MDM2 were superimposed on the structure of the MDM2-bound p53TAD peptide (PDB code: 1YCR), resulting in root mean square deviations (RMSDs) of 0.88 Å and 1.32 Å, respectively. Structural comparisons of the 3 p53 family TAD-MDM2 complexes revealed that the p73TAD and p63TAD peptides occupy the hydrophobic groove of MDM2 in a manner similar to the p53TAD peptide (). The p73TAD and p63TAD peptides bind to the MDM2 groove, forming an interface with the α2 helix on one side and with the ends of the middle β-sheet on the opposite side. As shown for p53TAD, the binding cleft for p73TAD and p63TAD on MDM2 is composed of many conserved hydrophobic core residues: Met50, Val53, Leu54, Phe55, Leu57, Ile61, Met62, Tyr67, Ile74, Val75, Leu82, Phe86, Phe91, Val93, Ile99, Tyr100, Met102, and Ile103. Although p73TAD and p63TAD lack high sequence homology with p53TAD, the equivalent triads of hydrophobic residues comprising the FxxΦWxxL motif (i.e., Phe15/Trp19/Leu22 in p73TAD and Phe55/ Trp59/Leu62 in p63TAD) are aligned on one face of the amphipathic α-helices of the p73TAD and p63TAD peptides (). The bulky hydrophobic and aromatic side-chains of the triad of residues snugly fit into the hydrophobic binding pocket of MDM2 in the same fashion as the Phe19, Trp23, and Leu26 residues of the p53TAD (). Thus, the hydrophobic characteristic of the interaction between MDM2 and the triad of hydrophobic residues of the TADs is highly conserved among p53TAD, p63TAD, and p73TAD. This similarity among the complex structures between the TADs and MDM2 demonstrates that the p53 family TADs share a highly conserved binding mode with MDM2.
Figure 2. Structural comparisons of the complexes formed between the p53 family TADs and MDM2. (A) Electrostatic surface representation of the MDM2/p73TAD (upper), MDM2/p63TAD (middle), and MDM2/p53TAD complexes (PDB code: 1YCR) (lower). The p53 family TAD peptides are represented as ribbons. The positively and negatively charged regions of MDM2 are depicted in blue and red, respectively. (B) Structural comparisons of the p73TAD (blue), p63TAD (green), and p53TAD (yellow) peptides bound to MDM2. (C) Comparisons of the intermolecular interactions between the p53 family TAD peptides and MDM2 at the interfaces of the complexes. The structure of MDM2 is shown as a ribbon within the transparent molecular surface (cyan), and the p53 family TAD peptides are represented as ribbons. Intermolecular hydrogen bonds are shown as dotted lines.
Mutational studies of the interaction between p73TAD and MDM2
To determine the critical binding determinants and evaluate the relative contributions of the p73TAD residues to MDM2 binding, we performed NMR binding experiments using mutant p73TAD peptides with alanine substitutions (i.e., F15A, L18A, W19A, and L22A) (). As observed in the 2D 1H-15N HSQC spectra of 15N-labeled MDM2 with mutant p73TAD peptides, alanine mutations at Phe15 and Trp19 completely abolished the NMR chemical shift perturbations observed in the binding with the wild-type p73TAD peptide, whereas the L18A mutation had a negligible effect. Although wild-type p73TAD peptide underwent slow exchange on the NMR time-scale with MDM2 (Fig. S1; ), the L22A mutant peptide underwent intermediate exchange (), indicating the significant decrease in binding affinity caused by the L22A mutation. In addition, the binding of MDM2 with the mutant p73TAD peptides, with the exception of L18A, could not be determined by ITC measurements (). These data indicate that the triad of hydrophobic residues of p73TAD are essential for the interaction between MDM2 and the p73TAD. Previous p53TAD mutagenesis results showed that mutations at Phe19, Leu22, Trp23, and Leu26 are the most disruptive to MDM2 binding.Citation27 Thus, the major binding determinants for MDM2 and p73TAD are identical to those for MDM2 and p53TAD.
Table 3. Comparison of the Kd values for the binding of p53 family TAD peptides and MDM2 using ITC
Figure 3. Mutational analysis of the interactions between p73TAD and MDM2. (A) Binding site mapping of the p73TAD peptide on the molecular surface of MDM2. Weighted chemical shift perturbations were calculated using the equation ΔCS = [(Δ1H)Citation2 + 0.2(Δ15N)Citation2]0.5, in which Δ1H and Δ15N represent the chemical shift changes on the 1H and 15N dimensions. The MDM2 residues showing chemical shift changes of ΔCS > 0.5 ppm and 0.25 ppm < ΔCS < 0.5 ppm are colored red and yellow, respectively. (B–F) Chemical shift perturbations of the MDM2 residues by binding the wild-type (WT) (B) and mutant p73TAD peptides (F15A, L18A, W19A, and L22A) (C–F). Resonance assignment of free MDM2 was performed using previously reported assignments (BMRB ID 6612) .Citation29 Weighted ΔCS values were plotted against the MDM2 residue numbers. Resonances that disappeared upon binding are shown as gray bars.
![Figure 3. Mutational analysis of the interactions between p73TAD and MDM2. (A) Binding site mapping of the p73TAD peptide on the molecular surface of MDM2. Weighted chemical shift perturbations were calculated using the equation ΔCS = [(Δ1H)Citation2 + 0.2(Δ15N)Citation2]0.5, in which Δ1H and Δ15N represent the chemical shift changes on the 1H and 15N dimensions. The MDM2 residues showing chemical shift changes of ΔCS > 0.5 ppm and 0.25 ppm < ΔCS < 0.5 ppm are colored red and yellow, respectively. (B–F) Chemical shift perturbations of the MDM2 residues by binding the wild-type (WT) (B) and mutant p73TAD peptides (F15A, L18A, W19A, and L22A) (C–F). Resonance assignment of free MDM2 was performed using previously reported assignments (BMRB ID 6612) .Citation29 Weighted ΔCS values were plotted against the MDM2 residue numbers. Resonances that disappeared upon binding are shown as gray bars.](/cms/asset/5c33da83-c329-47ee-8085-61112053c6ad/kccy_a_998056_f0003_oc.gif)
Comparison of the binding interface
Despite their conserved binding mode with shared binding determinants, noticeable differences are found in the binding interface between the p53 family TADs and MDM2. In the MDM2-p53TAD complex structure, the backbone carbonyl atom of Leu26 in p53TAD points out from the MDM2 binding cleft without establishing any interactions (). In contrast, the backbone carbonyl atoms of Leu22 and Leu62 in equivalent positions in p73TAD and p63TAD, respectively, point in toward the MDM2 binding cleft, therein forming 2 inter-molecular hydrogen bonds with the NH atom of His96 and the OH atom of Tyr100, respectively (). Unlike Pro24 in p73TAD and Pro65 in p63TAD, the Pro-27 side-chain in p53TAD induces distortion of the p53TAD backbone to some degree to prevent collisions with the MDM2 binding pocket. Instead, Asn29 in p53TAD is positioned toward MDM2 to form 2 intermolecular hydrogen bonds with the OH atoms of Tyr100 and Tyr104 in MDM2 ().
Dynamics of the MDM2-p73TAD complex
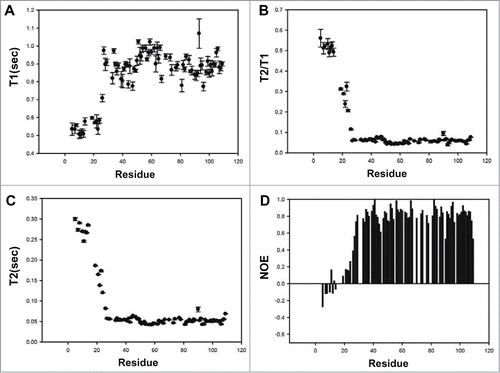
Structural comparisons of unbound MDM2 and p73TAD-bound MDM2 showed that the hydrophobic binding pocket of MDM2 is covered by its N-terminal lid (residues 18–24) in the absence of p73TAD, but its surface is exposed upon binding the p73TAD peptide, followed by a rigidity change in the MDM2 lid. In unbound MDM2, the Ile19 side-chain on the lid occupies the space filled by the p53 family TAD peptides.Citation28,29 The phenyl ring of Tyr100 interacts with Leu54 and Ile99 to make the entrance of the cleft narrower in the unbound MDM2.Citation29 However, in the p73TAD peptide-bound MDM2, the Tyr100 ring forms a hydrogen bond with the backbone carbonyl atom of Leu22 in p73TAD or Asn27 in p53TAD (). Previous NMR dynamics studies showed that the N-terminal lid in unbound MDM2 slowly interconverts between a “closed” state that lids over the binding pocket and an “open” state that is highly flexible.Citation30 Thus, it is possible that the p73TAD peptide binds to only the open state of MDM2 without largely perturbing the N-terminal lid, which may explain no much larger chemical shift perturbations in the lid upon binding to the p73TAD peptide (). To monitor the backbone dynamics of MDM2 upon binding to p73TAD, we measured the 15N T1 and T2 relaxation times and the heteronuclear NOEs of the p73TAD-bound MDM2 (). The p73TAD-bound MDM2 residues (28—108) are well ordered, however, the N-terminal lid residues (5–25) are flexible in a fast motion time scale (i.e., ps-ns). In particular, the rigidity of residues Ile19–Leu27 is significantly less than the rigidity of the corresponding residues in the unbound MDM2, Citation29 indicating that the N-terminal lid of MDM2 is displaced by p73TAD binding and undergoes a rigidity change similar to that observed for p53TAD binding. These backbone dynamics data support the conserved MDM2 recognition mechanism of p73TAD and p53TAD accompanied by a rigidity change in the N-terminal lid of MDM2.
Figure 4. NMR relaxation data of the complex formed between MDM2 and the p73TAD peptide. Measurements of T1 (A), T2 (B), T2/T1 (C) and heteronuclear NOE (D).
Helical induction of the p53 family TADs upon MDM2 interaction
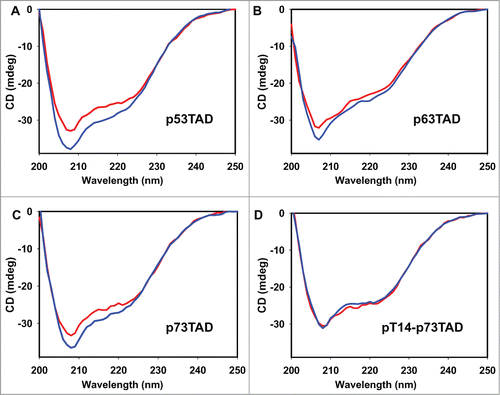
To compare the α-helical content of the p53 family TADs induced by MDM2 binding, we analyzed the secondary structures of the 3 p53 family TAD-MDM2 complexes using CD spectroscopy (). Compared with the sum of the individual molecule signals, a decrease in the CD ellipticities of the complexes at 208–222 nm showed that the α-helical content of the TAD peptides increased with complex formation with MDM2. However, there was a noticeable difference among the p53 family TADs in the degree of α-helical induction caused by MDM2 binding, with p53TAD ≥ p73TAD > p63TAD (). This result is supported by previous computational data showing the distinct helical propensity of the p53 family TAD peptides.Citation31 In addition, this is consistent with structural comparison among the p53 family TAD peptides in complex with MDM2, which reveals that p73TAD and p53TAD peptides form a 2-turn α-helix, whereas the p63TAD peptide adopts only a one-turn helix (). According to the ITC data, the dissociation constant (Kd) values for the MDM2 interactions with the p53TAD, p73TAD, and p63TAD peptides were determined to be 1.6, 3.9, and 33.5 μM, respectively (). Thus, the MDM2-binding affinity of p73TAD is comparable to the MDM2-binding affinity of p53TAD, whereas p63TAD binds to MDM2 more weakly. Taken together, our results suggest that the stronger binding affinity of p53TAD and p73TAD with MDM2 compared with that of p63TAD arises from the increased helical induction caused by MDM2 binding.
Figure 5. Comparison of the α-helical content of the p53 family TAD peptides induced by MDM2 binding. The CD spectra of the MDM2/p53TAD peptide (A), MDM2/p63TAD peptide (B), MDM2/p73TAD peptide (C), and MDM2/Thr14-phosphorylated p73TAD peptide complexes (D) (blue) are compared with the sums of the individual CD ellipticities of MDM2 and the p53 family TAD peptides (red).
Effects of phosphorylation of the p53 family TADs on MDM2 interaction
The Thr18 phosphorylation site in p53TAD is well aligned with Thr14 in p73TAD (), which is highly conserved among all of the known p73 proteins, suggesting a similar regulatory role for phosphorylation in p73TAD. To test this hypothesis, we investigated the effects of the phosphorylation of Thr14 in p73TAD on MDM2-binding affinity. The ITC measurements showed that the Kd value for the interaction between MDM2 and the Thr14-phosphorylated p73TAD peptide was 37.5 μM (). Phosphorylation of the p73TAD peptide decreased the MDM2-binding affinity by approximately one order of magnitude, indicating that phosphorylation of Thr14 in p73TAD is responsible for abrogating the interaction between MDM2 and p73TAD. To monitor the effect of p73TAD Thr14 phosphorylation on the α-helical induction, we performed a CD experiment on the phosphorylated p73TAD-MDM2 complex (). The CD data showed that phosphorylation of the p73TAD Thr14 abrogated the α-helical induction by MDM2 binding that was observed for the unphosphorylated p73TAD. Thus, Thr14 phosphorylation in p73TAD appears to inhibit MDM2 binding by preventing α-helix formation.
Discussion
Despite the lack of high sequence homology and well defined 3-dimensional structure, p53 family TADs exhibit functional conservation in their regulatory interaction with MDM2. In this study, our structural and biophysical data revealed a convergent mechanism for MDM2 recognition among the intrinsically disordered p53 family TADs (p53TAD, p63TAD, and p73TAD). First, the same conserved “FxxΦWxxL” motif in the p53 family TADs is utilized for MDM2 recognition. Mutagenesis data for p73TAD and p53TAD supported that the triads of hydrophobic residues (Phe15, Trp19, and Leu22 in p73TAD; Phe19, Trp23, and Leu26 in p53TAD) are critical for MDM2 binding. This observation is consistent with a unique characteristic of intrinsically disordered proteins (IDPs) that specific motifs, rather than whole sequences, act as mediators of protein-protein interactions. Second, there is a noticeably similar mimicry in the bound conformation of the p53 family TAD motifs induced by MDM2 binding. Although all of the p53 family TAD motifs are largely disordered in the free state, they adopt a structurally homologous α-helix upon binding to MDM2. The p53 family TADs and MDM2 complex structures showed that the triad of hydrophobic residues align on one face of the amphipathic α-helix and point into the elongated hydrophobic groove of MDM2. In particular, as found with Trp23 in p53TAD, the aromatic ring of Trp19 in p73TAD fits snugly into the hydrophobic binding pocket, thereby making the largest contribution to the complex formation. This conserved mode of interaction is reminiscent of other complex structures of intrinsically disordered TADs. Overall, the manner in which p53 family TADs interact with MDM2 mimics what has been observed for the binding of VP16 TAD to hTAFII31, Citation32 the binding of c-Myb TAD to the CBP Kix domain, Citation33 and the binding of p53TAD to RPA and Bcl-XL/Bcl-2.Citation34-Citation36 This may represent a highly conserved mechanism underlying protein recognition by IDPs.
Despite a highly conserved mode of binding, there is a noticeable difference between the complex structures of the p53 family TADs and MDM2. After forming a complex with MDM2, the p73TAD peptide forms an α-helix with a similar length to that formed with the p53TAD peptide, whereas the p63TAD peptide generates only a one-turn helix spanning Phe55 to Ile58, which is shorter than the helices formed with the p53TAD and p73TAD peptides. While the inter-molecular hydrophobic interactions between the p53 family TADs and MDM2 are well conserved, the intra-molecular interactions in p73TAD and p63TAD are different from the interactions in p53TAD. In the MDM2-bound structures, the side-chain hydrogen bonds between Thr18 and Asp21 in p53TAD and between Thr14 and Glu16 in p73TAD make significant contribution to stabilization of the α-helix. However, no such hydrogen bond is found at the corresponding position in p63TAD. Thus, the α-helices of p53TAD and p73TAD appear to be more stable than the helix of p63TAD. This finding is supported by our observation of higher α-helical induction in p53TAD and p73TAD than in p63TAD using CD spectroscopy (). Combined with the CD and ITC data, our results showed that the p53TAD and p73TAD motifs with more stable helical structures might bind to MDM2 more tightly than the p63TAD motif.
Phosphorylation could be an alternative way of regulating the binding specificity of IDPs. In response to stress signals such as DNA damage, the phosphorylation of Thr18 in p53TAD serves to modulate the protein-protein interaction of p53, because the phosphorylation results in dissociation from MDM2 and association with the transcriptional coactivator p300/CBP.Citation25,37 Our results showed that, similar to Thr18 in p53TAD, the phosphorylation of Thr14 in p73TAD weakens the binding to MDM2 by approximately 10-fold (). Because the OG2 side-chain atoms of Thr14 in p73TAD and Thr18 in p53TAD involve intra-molecular hydrogen bonds to stabilize the α-helices in the MDM2-bound TAD structures, phosphorylation of Thr14 in p73TAD or Thr18 in p53TAD would disrupt such hydrogen bonds, and destabilize the α-helices. In contrast, phosphorylation of Thr14 in p73TAD was shown to increase the affinity to p300 Taz2 domain by 10-fold.Citation38 Therefore, similar to the situation in p53TAD, phosphorylation of p73TAD may modulate the helical stability to regulate interactions with binding partners. However, such binding modulation by phosphorylation would be impossible for p63TAD because the corresponding phosphorylation site in the FxxWΦxxL motif is replaced with a valine (Val54).
Because the p73 isoform lacking p73TAD (ΔNp73) and the full-length p63 carrying p63TAD were unable to replace p53 in p53-deficient breast cancer cells, Citation24 it has been suggested that p73TAD, not p63TAD, is critical for exerting the p53-like functions that trigger apoptosis and cell cycle arrest. With our finding that p73TAD, rather than p63TAD, closely mimics p53TAD at the 3-dimensional structural level, the noticeable differences between p73TAD and p63TAD in terms of helical stability, MDM2-binding affinity, and phosphorylation regulation might explain why p73, and not p63, is able to function as a substitute for p53 in p53-deficient cancer cells.Citation24,39 Inhibition of the interaction between p73TAD and MDM2 could trigger activation of the p73 pathway, leading to an increase in chemosensitivity to apoptogenic drugs. Thus, the 3-dimensional structural information of the MDM2-p73TAD complex could contribute to rational drug design and chemotherapy for many human tumors bearing p53 mutations such as cholangiocarcinoma.
In summary, we demonstrated the structural basis for the conserved binding mechanism of MDM2 with the TADs of the p53 family, which play important roles in tumor suppression in response to cellular stress. Together with the mutagenesis, dynamics, CD, and ITC data, our NMR complex structures reveal a close mimicry in the recognition of MDM2 between p53TAD and p73TAD, which might have important implications in the development of novel p73-based therapies for many tumors bearing p53 mutations.
Materials and Methods
Preparation of proteins and peptides
13C,15N-labeled p73TAD (residues 10–25) was expressed as glutathione-S-transferase (GST) fusion protein in E. coli BL21(DE3) RIL cells grown on M9 minimal media with 15NH4Cl and 13C-glucose as the only nitrogen and carbon sources. The fusion protein was purified by GST affinity chromatography and was thrombin-digested. Following cleavage, the p73TAD peptide was further purified with a size-exclusion gel-filtration column (HiLoad 16/60 Superdex G75, Pharmacia) and a desalting Sep-Pak PlusTM C18 cartridge (Waters). The N-terminal domain of MDM2, corresponding to residues 3–109, was expressed and purified as previously reported.Citation25 p63TAD (residues 51–65), Thr14-phosphorylated p73TAD (residues 10–25), and wild-type and mutant p73TAD (residues 10–25) peptides were chemically synthesized and purified by Peptron Inc.
NMR epectroscopy
All of the NMR data were acquired at 10°C on a Bruker Avance II 800 spectrometer equipped with a cryogenic probe at the Korea Basic Science Institute. For the NMR studies of the MDM2-p73TAD (residues 10–25) complex, we used a sample containing 0.9 mM of the complex in a 1:1 ratio (13C,15N-MDM2:p73TAD and 13C,15N-p73TAD:MDM2) in 20 mM MES (pH 6.5), 150 mM NaCl, 0.1 mM PMSF, 2 mM DTT, 0.1 mM EDTA, 0.1 mM benzamidine, and 90% H2O/10% D2O. For the 2D 1H-15N HSQC experiments, we used a sample containing 0.1 mM 15N-MDM2 in 20 mM MES (pH 6.5), 150 mM NaCl, 0.1 mM PMSF, 2 mM DTT, 0.1 mM EDTA, 0.1 mM benzamidine, and 90% H2O/10% D2O; unlabeled p73TAD (residues 10–25) or p63TAD (residues 51–65) peptides were added to a final ratio of 1:1. All of the NMR data were processed and analyzed using NMRPipe/nmrDraw Citation40 and Sparky software. NMR resonance assignment was performed using HNCACB, CBCA(CO)NH, HNCO, HN(CA)CO, HBHA(CO)NH, HCCH-TOCSY, HCCH-COSY, and CCH-TOCSY spectra.
Residual dipolar coupling (RDC) measurements
The 13C,15N-labeled MDM2/unlabeled p73TAD peptide and unlabeled MDM2/13C,15N –labeled p73TAD peptide were aligned in bacteriophage Pf1, purchased from ASLA Biotech (Riga, Latvia). Pf1 was added to a final concentration of 17 mg/mL and the NaCl concentration was 150 mM. Couplings were obtained from 2D 1H-15N IPAP HSQC spectra.
Structure calculation
Interproton distance restraints were derived from the 3D 13C-edited and 15N-edited NOESY-HSQC spectra. Dihedral angle restraints were generated from the secondary chemical shifts using the program TALOS+. The solution structures of the MDM2-p73TAD complex were calculated with CYANA 2.1.Citation41 The resulting structures were refined with RDC restraints with a water refinement protocol using XPLOR-NIH.Citation42 The quality of the refined structures was validated using PROCHECK-NMRCitation43 and Molprobity.Citation44
Molecular docking
The structure of the MDM2-p63TAD (51–65) complex was calculated using the HADDOCK 2.1 program.Citation45 Ambiguous interaction restraints (AIRs) were defined based on the NMR chemical shift perturbation data. The “active” residues in MDM2 were defined as the residues that disappeared upon binding to the p63TAD peptide with relative solvent accessibility larger than 40% for all of the atoms. All of the surrounding surface residues of MDM2 near the active residues were defined as “passive” residues. The initial structure of the p63TAD peptide was homology-modeled based on the p73TAD peptide structure determined in this study. Beginning with the structure of MDM2, 1,000 rigid-body docking solutions were obtained. The best 200 structures were selected for semi-flexible refinement and were finally refined in explicit water. The clustering was performed with a 1.5 Å cut-off and the complex models were selected for visualization by the RMSD from the best energy structure and HADDOCK energy score. Figures of the complex model were drawn using the PyMOL software package.
NMR dynamics
For measurements of 15N relaxation times, a series of 2D 1H-15N HSQC spectra were acquired with different relaxation delays. T1 values were measured in a series of spectra with relaxation delays of 50, 100, 150, 200, 300, 500, 700, 900, and 1200 ms. T2 measurements were made with relaxation delays of 10, 30, 50, 70, 90, 110, 130, 150, 170, and 190 ms. The relaxation delay for T1 and T2 measurements was 1.5 s. The 1H-15N heteronuclear steadystate NOEs were measured from a pair of spectra recorded with and without a proton presaturation. The proton saturation period was 3 s.
ITC measurements
The ITC measurements were performed at 25°C in a buffer containing 20 mM MES (pH 6.5), 150 mM NaCl, 0.1 mM PMSF, and 0.1 mM benzamidine, using an Auto-iTC200 MicroCalorimeter in the Korea Basic Science Institute. For the ITC measurements of p53TAD (15–29), p63TAD (51–65), Thr14-phosphorylated p73TAD (10–25), and wild-type and mutant p73TAD (10–25) peptides, the peptides of 400–600 μM were titrated into a calorimeter cell containing 30 μM MDM2 (3–109) with 2 μL injections. The ITC data were analyzed using MicroCal OriginTM software.
CD spectroscopy
The CD spectra were recorded at 25°C using a Jasco-815 spectropolarimeter in 20 mM sodium phosphate (pH 6.5) and 50 mM NaCl. The concentrations of MDM2 and the p53TAD, p63TAD, and p73TAD peptides were 20 μM. All of the CD spectra obtained were the average of 5 consecutive scans from 250 to 200 nm.
Accession codes
The atomic coordinates for the MDM2 (3–109)/p73TAD (10–25) complex (code 2MPS) have been deposited in the Protein Data Bank (http://wwpdb.org/). The NMR chemical shifts assignments and restraints used for structure calculation of the MDM2 (3–109)/p73TAD (10–25) complex were deposited to the Biological Magnetic Resonance Bank database under BMRB ID 18876.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
998056_Supplementary_Materials.zip
Download Zip (220.3 KB)Funding
This work was supported by a grant from the National R&D Program for Cancer Control, Ministry of Health & Welfare, Republic of Korea (1220150) and the National Research Foundation of Korea (NRF) grants funded by the Korea government (MSIP) (NRF-2013M3A9C4078135 and NRF-2014R1A2A2A05002961). This study made use of the NMR facility at Korea Basic Science Institute, which is supported by Advanced MR Technology Program of the Korean Ministry of Education, Science and Technology.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Related Research Data
References
- Jost CA, Marin MC, Kaelin WG, Jr. p73 is a simian [correction of human] p53-related protein that can induce apoptosis. Nature 1997; 389:191-4; PMID:9296498
- Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, Dotsch V, Andrews NC, Caput D, McKeon F. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell 1998; 2:305-16; PMID:9774969
- Mills AA, Zheng B, Wang XJ, Vogel H, Roop DR, Bradley A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 1999; 398:708-13; PMID:10227293; http://dx.doi.org/10.1038/19531
- Yang A, Schweitzer R, Sun D, Kaghad M, Walker N, Bronson RT, Tabin C, Sharpe A, Caput D, Crum C, et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 1999; 398:714-8; PMID:10227294; http://dx.doi.org/10.1038/19539
- Yang A, Walker N, Bronson R, Kaghad M, Oosterwegel M, Bonnin J, Vagner C, Bonnet H, Dikkes P, Sharpe A, et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature 2000; 404:99-103; PMID:10716451; http://dx.doi.org/10.1038/35003607
- Danilova N, Sakamoto KM, Lin S. p53 family in development. Mech Dev 2008; 125:919-31; PMID:18835440; http://dx.doi.org/10.1016/j.mod.2008.09.003
- Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992; 69:1237-45; PMID:1535557; http://dx.doi.org/10.1016/0092-8674(92)90644-R
- Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, Vogelstein B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature 1993; 362:857-60; PMID:8479525; http://dx.doi.org/10.1038/362857a0
- Balint E, Bates S, Vousden KH. Mdm2 binds p73 alpha without targeting degradation. Oncogene 1999; 18:3923-9; PMID:10435614; http://dx.doi.org/10.1038/sj.onc.1202781
- Zeng X, Chen L, Jost CA, Maya R, Keller D, Wang X, Kaelin WG, Jr., Oren M, Chen J, Lu H. MDM2 suppresses p73 function without promoting p73 degradation. Mol Cell Biol 1999; 19:3257-66; PMID:10207051
- Kadakia M, Slader C, Berberich SJ. Regulation of p63 function by Mdm2 and MdmX. DNA Cell Biol 2001; 20:321-30; PMID:11445003; http://dx.doi.org/10.1089/10445490152122433
- Zdzalik M, Pustelny K, Kedracka-Krok S, Huben K, Pecak A, Wladyka B, Jankowski S, Dubin A, Potempa J, Dubin G. Interaction of regulators Mdm2 and Mdmx with transcription factors p53, p63 and p73. Cell Cycle 2010; 9:4584-91; PMID:21088494; http://dx.doi.org/10.4161/cc.9.22.13871
- Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature 1997; 387:299-303; PMID:9153396; http://dx.doi.org/10.1038/387299a0
- Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature 1997; 387:296-9; PMID:9153395; http://dx.doi.org/10.1038/387296a0
- Okamoto K, Taya Y, Nakagama H. Mdmx enhances p53 ubiquitination by altering the substrate preference of the Mdm2 ubiquitin ligase. FEBS Lett 2009; 583:2710-4; PMID:19619542; http://dx.doi.org/10.1016/j.febslet.2009.07.021
- Danovi D, Meulmeester E, Pasini D, Migliorini D, Capra M, Frenk R, de Graaf P, Francoz S, Gasparini P, Gobbi A, et al. Amplification of Mdmx (or Mdm4) directly contributes to tumor formation by inhibiting p53 tumor suppressor activity. Mol Cell Biol 2004; 24:5835-43; PMID:15199139; http://dx.doi.org/10.1128/MCB.24.13.5835-5843.2004
- Linke K, Mace PD, Smith CA, Vaux DL, Silke J, Day CL. Structure of the MDM2/MDMX RING domain heterodimer reveals dimerization is required for their ubiquitylation in trans. Cell Death and Differ 2008; 15:841-8; PMID:18219319; http://dx.doi.org/10.1038/sj.cdd.4402309
- Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: drugging the p53 pathway. Nature Rev Cancer 2009; 9:862-73; PMID:19935675; http://dx.doi.org/10.1038/nrc2763
- Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, Jacks T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007; 445:661-5; PMID:17251932; http://dx.doi.org/10.1038/nature05541
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007; 445:656-60; PMID:17251933; http://dx.doi.org/10.1038/nature05529
- Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science 1991; 253:49-53; PMID:1905840; http://dx.doi.org/10.1126/science.1905840
- Melino G, De Laurenzi V, Vousden KH. p73: Friend or foe in tumorigenesis. Nature Rev Cancer 2002; 2:605-15; PMID:12154353; http://dx.doi.org/10.1038/nrc861
- Zawacka-Pankau J, Kostecka A, Sznarkowska A, Hedstrom E, Kawiak A. p73 tumor suppressor protein: a close relative of p53 not only in structure but also in anti-cancer approach? Cell Cycle 2010; 9:720-8; PMID:20160513; http://dx.doi.org/10.4161/cc.9.4.10668
- Vayssade M, Haddada H, Faridoni-Laurens L, Tourpin S, Valent A, Benard J, Ahomadegbe JC. P73 functionally replaces p53 in Adriamycin-treated, p53-deficient breast cancer cells. Int J Cancer 2005; 116:860-9; PMID:15849742; http://dx.doi.org/10.1002/ijc.21033
- Schon O, Friedler A, Bycroft M, Freund SM, Fersht AR. Molecular mechanism of the interaction between MDM2 and p53. J Mol Biol 2002; 323:491-501; PMID:12381304; http://dx.doi.org/10.1016/S0022-2836(02)00852-5
- Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996; 274:948-53; PMID:8875929; http://dx.doi.org/10.1126/science.274.5289.948
- Picksley SM, Vojtesek B, Sparks A, Lane DP. Immunochemical analysis of the interaction of p53 with MDM2;–fine mapping of the MDM2 binding site on p53 using synthetic peptides. Oncogene 1994; 9:2523-9; PMID:8058315
- McCoy MA, Gesell JJ, Senior MM, Wyss DF. Flexible lid to the p53-binding domain of human Mdm2: implications for p53 regulation. Proc Natl Acad Sci USA 2003; 100:1645-8; PMID:12552135
- Uhrinova S, Uhrin D, Powers H, Watt K, Zheleva D, Fischer P, McInnes C, Barlow PN. Structure of free MDM2 N-terminal domain reveals conformational adjustments that accompany p53-binding. J Mol Biol 2005; 350:587-98; PMID:15953616; http://dx.doi.org/10.1016/j.jmb.2005.05.010
- Showalter SA, Bruschweiler-Li L, Johnson E, Zhang F, Bruschweiler R. Quantitative lid dynamics of MDM2 reveals differential ligand binding modes of the p53-binding cleft. J Am Chem Soc 2008; 130:6472-8; PMID:18435534; http://dx.doi.org/10.1021/ja800201j
- Mavinahalli JN, Madhumalar A, Beuerman RW, Lane DP, Verma C. Differences in the transactivation domains of p53 family members: a computational study. BMC Genomics 2010; 11 Suppl 1:S5; PMID:20158876
- Uesugi M, Nyanguile O, Lu H, Levine AJ, Verdine GL. Induced alpha helix in the VP16 activation domain upon binding to a human TAF. Science 1997; 277:1310-3; PMID:9271577; http://dx.doi.org/10.1126/science.277.5330.1310
- Zor T, De Guzman RN, Dyson HJ, Wright PE. Solution structure of the KIX domain of CBP bound to the transactivation domain of c-Myb. J Mol Biol 2004; 337:521-34; PMID:15019774; http://dx.doi.org/10.1016/j.jmb.2004.01.038
- Ha JH, Shin JS, Yoon MK, Lee MS, He F, Bae KH, Yoon HS, Lee CK, Park SG, Muto Y, et al. Dual-site interactions of p53 protein transactivation domain with anti-apoptotic Bcl-2 family proteins reveal a highly convergent mechanism of divergent p53 pathways. J Biol Chem 2013; 288:7387-98; PMID:23316052; http://dx.doi.org/10.1074/jbc.M112.400754
- Ha JH, Won EY, Shin JS, Jang M, Ryu KS, Bae KH, Park SG, Park BC, Yoon HS, Chi SW. Molecular mimicry-based repositioning of nutlin-3 to anti-apoptotic Bcl-2 family proteins. J Am Chem Soc 2011; 133:1244-7; PMID:21210687; http://dx.doi.org/10.1021/ja109521f
- Bochkareva E, Kaustov L, Ayed A, Yi GS, Lu Y, Pineda-Lucena A, Liao JC, Okorokov AL, Milner J, Arrowsmith CH, et al. Single-stranded DNA mimicry in the p53 transactivation domain interaction with replication protein A. Proc Natl Acad Sci USA 2005; 102:15412-7.
- Teufel DP, Bycroft M, Fersht AR. Regulation by phosphorylation of the relative affinities of the N-terminal transactivation domains of p53 for p300 domains and Mdm2. Oncogene 2009; 28:2112-8; PMID:19363523; http://dx.doi.org/10.1038/onc.2009.71
- Burge S, Teufel DP, Townsley FM, Freund SM, Bycroft M, Fersht AR. Molecular basis of the interactions between the p73 N terminus and p300: effects on transactivation and modulation by phosphorylation. Proc Natl Acad Sci USA 2009; 106:3142-7.
- Zheng T, Wang J, Chen X, Meng X, Song X, Lu Z, Jiang H, Liu L. Disruption of p73-MDM2 binding synergizes with gemcitabine to induce apoptosis in HuCCT1 cholangiocarcinoma cell line with p53 mutation. Tumour Biol 2010; 31:287-95; PMID:20422343; http://dx.doi.org/10.1007/s13277-010-0035-7
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 1995; 6:277-93; PMID:8520220; http://dx.doi.org/10.1007/BF00197809
- Herrmann T, Guntert P, Wuthrich K. Protein NMR structure determination with automated NOE-identification in the NOESY spectra using the new software ATNOS. J Biomol NMR 2002; 24:171-89; PMID:12522306; http://dx.doi.org/10.1023/A:1021614115432
- Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Reson 2003; 160:65-73; PMID:12565051; http://dx.doi.org/10.1016/S1090-7807(02)00014-9
- Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR 1996; 8:477-86; PMID:9008363; http://dx.doi.org/10.1007/BF00228148
- Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 2007; 35:W375-83; PMID:17452350; http://dx.doi.org/10.1093/nar/gkm216
- Dominguez C, Boelens R, Bonvin AM. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J Am Chem Soc 2003; 125:1731-7; PMID:12580598; http://dx.doi.org/10.1021/ja026939x
- Fernandez-Recio J, Totrov M, Abagyan R. Identification of protein-protein interaction sites from docking energy landscapes. J Mol Biol 2004; 335:843-65; PMID:14687579; http://dx.doi.org/10.1016/j.jmb.2003.10.069