
Abstract
While targeting experiments carried out on the genes encoding many cell cycle regulators have challenged our views of cell cycle control, they also suggest that redundancy might not be the only explanation for the observed perplexing phenotypes. Indeed, several observations hint at functions of cyclins and CDK inhibitors that cannot be accounted for by their sole role as kinase regulators. They are found involved in many cellular transactions, depending or not on CDKs that are not directly linked to cell cycle control, but participating to general mechanisms such as transcription, DNA repair or cytoskeleton dynamics. In this review we discuss the roles that these alternative functions might have in cancer cell proliferation and migration that sometime even challenge their definition as proliferation markers.
Introduction
One view of the cell division cycle is that of a cell autonomous mechanism operated by a small number of enzymes, mainly the cyclin-dependent kinases (CDKs), whose major function is to ensure a timely control of its 2 fundamental steps: DNA replication and chromosome segregation. The oscillating activity of CDKs at different phases of the cell cycle results from both reversible phosphorylation events and from their transient association with specific cofactors such as cyclins and CDK inhibitors (CKIs). Cyclins play a dual role as activators and targeting agents toward specific substrates, whereas CKIs act both as architectural and inhibitory components. The complexity of this regulation results from the existence of quality controls (checkpoints) that ensure that each step of the cell division cycle is completed before the next one takes place. Moreover, external cues such as cell-cell or cell-matrix adhesion, or exposure to mitotic spindle or DNA damaging agents, generate signals that modulate the activity of the cell autonomous machinery through specific checkpoints.
This vision of the classical model of the cell division cycle is epitomized in its frequent representation as a closed circle that describes a proliferating cell as undergoing a series of major transitions triggered by specialized CDK-cyclin complexes. The onset of DNA replication defines the beginning of S phase, nuclear envelope breakdown and chromosome condensation, that of M phase, results in the segregation of sister chromatids and culminates with cytokinesis.Citation1,2 This simplistic view was first challenged by the survival of mice bearing individual ablations of genes coding for CDK2, CDK3, CDK4 and CDK6, and then, by the existence of a functional cell division cycle in embryos harboring compound knockoutsCitation3 discussed in.Citation4 At the end, CDK1 appears to be the only essential CDK for the control of the mammalian cell cycle. Similar results were obtained following the ablation of some of the CDK partners such as D- and E-type cyclins.Citation5,6,7,8 Even cyclin A2, whose ablation was initially shown to be embryonically lethal, appeared more recently to be dispensable in adult tissues, with the exception of haematopoietic cells.Citation9
If functional redundancy plays a large part in explaining these observations, it may not provide the full picture. Clearly, CDKs and cyclins reveal their absolute requirements in some specific tissues/cellular processes: CDK6 and cyclin A2 in erythropoiesis and more generally in the haematopoietic lineage, cyclin D3 for lymphocyte development and T cell leukemia,Citation10 CDK4 in the pancreas, and CDK2 in meiosis since mice defective in this CDK have a defective meiosis in both male and female gonads (reviewed inCitation3). But many data hint toward functions not necessarily linked to the cell cycle. An interesting example is that of the CDK activating kinase (CAK). This enzyme, composed of 3 subunits: CDK7, cyclin H and MAT1 (ménage à trois)Citation11,Citation12,13,14,15, is necessary for the full activation of CDKs and its genetic ablation leads to premature aging due to stem cells exhaustion.Citation16 However, it is also part of the general transcription factor TFIIHCitation17 where, by phosphorylating the C-terminal domain of the largest subunit of RNA polymerase II, it is involved in promoter clearance and progression of transcriptionCitation18 (for reviews:Citation19,Citation16,20). The switch between the 2 forms is orchestrated by XPD (xeroderma pigmentosum disorder group D) subunit that is degraded at mitosis after phosphorylation by CDK1-cyclin B, thus silencing transcription during this phase. CAK is also found as a coactivator of some transcription factors such as RARα or the androgen receptorCitation21,22,23 ().
Figure 1. The CAK participates in both CDK-cyclin activation and transcription initiation. CAK has been shown to be required for the activation of CDK1-cyclin B at mitosis and is also recruited into the TFIIH general transcription factor via its XPD subunit and, as part of this complex, it triggers the initiation of RNA polymerase II transcription through phosphorylation of the carboxy terminal domain of its largest subunit. Degradation of XPD, likely triggered after phosphorylation by CDK1-cyclin B, is proposed to be responsible for mitotic silencing of transcription.
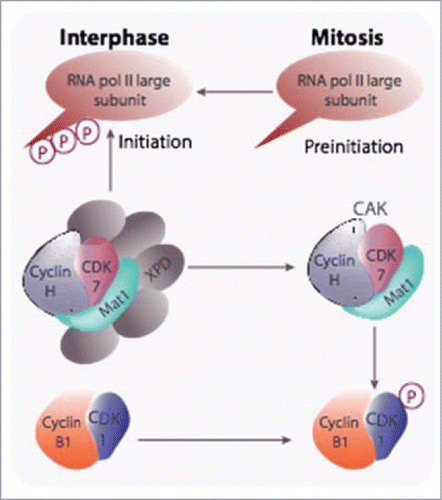
This plurality of roles is also true for many other regulators of the cell division cycle. In this review we will summarize the main data on these unexpected functions of D-, E- and A-type cyclins as well as CKIs such as p21, p27 and p57.
From Transcription Regulation to DNA Damage Control: Cyclins D1 and E1 at the Heart of DNA Transactions
One of the major functions of D-type and E-type cyclins, in association with their cognate CDKs, is the control of the progression from G1 to S, reflected by their accumulation in G1 for D-type, and from the end of G1 through most S phase for E-type cyclins. Among the targets of these cyclins-CDK complexes the retinoblastoma gene product, pRb, occupies a central place since its phosphorylation is a prerequisite to trigger the transcription of E2F-dependent genes whose products are necessary for cell cycle progression. As a consequence, highly phosphorylated forms of pRb are the hallmark of normal proliferating cells. Since its functional inactivation is a prerequisite for cellular proliferation, it is natural to associate pRb to tumor suppression. Consistent with this, its expression is found altered in many cancers, with either allelic losses or methylation of its gene, as well as its hyperphosphorylation leading to the same outcome. Accordingly, a wealth of information has accumulated linking E- and D-types cyclins dysregulation and malignant cell transformation.Citation24,25 Both cyclins are found expressed at a high level in a number of human tumors, frequently resulting from an amplification of their respective loci. Along the same line, transgenic mice expressing cyclin E1 at a high level were prone to develop tumors.Citation26,27 Conversely, mice with ablation in the mammary gland of the gene coding for cyclin D1 are resistant to transformation by Erb-B2/neu and Ras even though they remain sensitive to other transformation pathways.Citation28
Curiously enough we should also stress at this stage the altered regulation of G1 cyclins in human fibroblasts when they enter senescence.Citation29,30 Whereas several cell cycle genes such as cyclins A2 and B1 are down regulated during this irreversible G1 arrest, D- and E-type cyclins are expressed at a high level, and cyclin D1 can be taken, like β-galactosidase, as a good marker of senescent cells.Citation31,32 Thus, a hyper mitogenic drive appears to coexist with mitotic incompetence in senescent cells and the origin of this conflict remains to be explained mainly with respect to putative non-conventional functions of these cyclins.
E-type cyclins: replication origins specification and cell fate determination
The association of cyclin E1 with cell transformation has naturally led people to look for its cooperation with known transforming oncogenes such as constitutively active Ras. Indeed, this cyclin can lead to the transformation of primary rat embryo fibroblasts in the presence of an activated Ha-Ras.Citation33 Surprisingly however, when mutants of cyclin E1 were used in the same transformation assay, those that were unable to form an active cyclin-CDK complex were still active in cooperating with Ras in foci formation.Citation34
In order to analyze E-type cyclins in vivo functions, cyclin Es-null mice have been generatedCitation5,8 and double knock out animals died at mid gestation with placental abnormalities. However, rescued embryos have led to the possibility to grow cyclin E1-/-E2-/- MEFs and thus made possible the study of the molecular defects underlying cyclin Es deficiency.Citation35 In fact, these MEFs are unable to reenter the cell division cycle from a quiescent state when for example serum-starved cells are stimulated with growth factors. Unexpectedly, a cyclin E1 mutant in which residues 188–192 have been changed into alanines and that, as a result, can no longer activate CDK2, was able to restore the ability of deficient cells to leave G0 when stimulated. Moreover, this mutant was also able to restore sensibility to transformation by activated Ras of cyclin E1-/-E2-/- MEFs that were previously shown to be unresponsive to this oncogene (reviewCitation36).
Cyclin E1 has been reported to be required for the loading of MCMs into DNA replication complexes.Citation37 Consistent with this, cyclin E1-/-E2-/- MEFs are unable to do so, and the mutant cyclin E1 behaved as its wild type counterpart in restoring MCM loading. This has led Geng and his colleagues to propose that a chromatin-associated fraction of cyclin E1 facilitates MCMs loading through a physical interaction with these proteins as well as with CDT1 (). Interestingly, a similar function has been proposed for this cyclin as well as for cyclin A2 in the control of centrosome duplication through the recruitment of MCM5 and Orc1.Citation38,39,40
Figure 2. E-type cyclins together with cyclin A2 are involved in the tight linkage between the nuclear and centrosomal cycles. E-type cyclins facilitate MCMs loading through a physical interaction with these proteins as well as with CDT1Citation35. Similarly to chromosomes, centrosomes must be duplicated, and this takes place at the onset of S phase to allow the faithfully duplicated organelles to move to the poles of the duplicating cell and then, to be distributed to the daughter cells. Both cyclins E and cyclin A2 have also been implicated in this phenomenon (Pascreau et al, 2011).
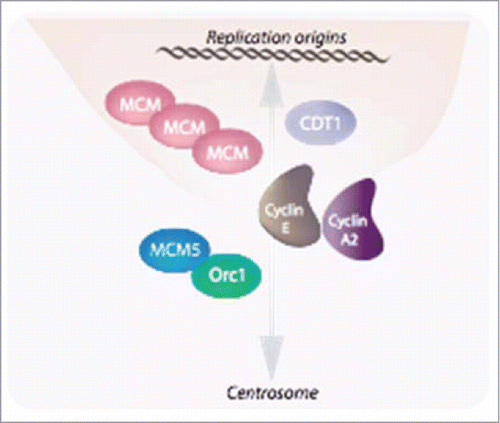
Whereas all other canonical cell-cycle-related functions of cyclin Es can be compensated for by other cyclin-CDK complexes this is not the case for this loading function. Intriguingly, E-type cyclins are required for MCM loading in cells exiting a quiescent state, at a time when, according to the classical model, they are not supposed to be expressed, while they are not in continuously proliferating cells. If this kinase-independent function of cyclin Es could be envisioned, within the scope of cellular transformation, as a mean for the tumor cells to escape from quiescence, it remains to be seen whether it is used during normal development. Moreover, the question is raised of the existence of novel cyclin Es functions, CDK-independent or not, but not necessarily linked to cell cycle progression.
There are hints that this is the case, at least during neural cell fate specification in Drosophila. Whereas the self-renewal capacity in the developing mammalian brain is limited, nearly all neural progenitor cells of the Drosophila central nervous system exhibit self-renewal capacities during its development. Through asymmetric divisions, progenitors, or neuroblasts, give rise to both neurons and glial cells. Whereas in the thoracic segments of the embryonic nervous system neuroblasts divide first asymmetrically, giving rise to both a glial and a neuronal lineage, abdominal neuroblasts divide once symmetrically into 2 glial cells (). Cyclin E was shown to play a critical function in the regulation of asymmetric neuroblasts divisionCitation41 that is independent of its role in cell cycle control.Citation42 In mutant embryos, most thoracic neuroblasts harbor a nuclear localization of Prospero, a transcription factor required for neuronal differentiation, while in a wild type context, Prospero is sequestered into a cortical crescent and, during asymmetric divisions, translocates into the nucleus of glial-producing daughter cells. Interestingly, a mutational analysis allowed delineating 2 distinct domains in the protein, with the deletion of the C-terminal autophosphorylation domain severely affecting its function in cell fate determination, without affecting its cell cycle function. Moreover, this work suggested that cyclin E leads, through a physical interaction, to the cortical localization of Prospero, and as such, is critical for the maintenance of neuroblasts stem cell properties.
Figure 3. Cyclin E is involved in cell fate determination during early neurogenesis in Drosophila. Through asymmetric divisions, progenitors, or neuroblasts, give rise to both neurons and glial cells. Whereas in the thoracic segments of the embryonic nervous system neuroblasts divide first asymmetrically, giving rise to both a glial and a neuronal lineage, abdominal neuroblasts divide once symmetrically into 2 glial cells. Cyclin E is required for maintaining the transcription factor Prospero in a cortical localization in neuronal precursors and is inhibited to do so by the Abdominal-A and Abdominal-B gene products (Abd-A, Abd-B)
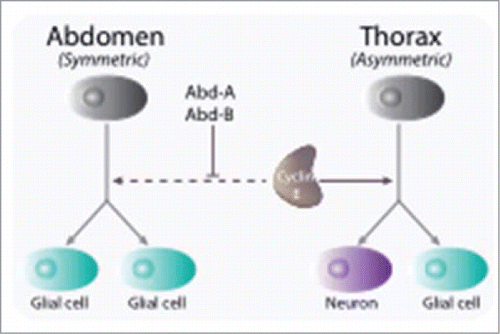
More recently Odajima and collaborators have demonstrated that cyclin E1 forms a catalytically inactive complex with Cdk5 that is proposed to promote synapse formation in quiescent, post mitotic nervous system.Citation43 Interestingly, whereas cyclin E1 is preferentially associated to Cdk1/2 in the developing embryonic brain, it was found to be quantitatively associated to Cdk5 in adult brain. We should point at this level that the exitence of a role in the central nervous system of Drosophila melanogaster is also shared by cyclin A that functions in postmitotic neurons to promote sleep in Drosophila.Citation44
Cyclin D1 in transcription and DNA repair
Cyclin D1 knockout mice are viable albeit developing tissue-restricted abnormalities including retina, nervous system and breast epithelium, which suggests the involvement of this cyclin in other cellular functions, some of which are not necessary linked to cell cycle progression (reviews:Citation45,46). We will examine below its involvement in DNA transactions such as transcription and DNA repair. Within the cell cycle context, D-type cyclins are already indirectly associated with a transcriptional role while associated with CDK4 and CDK6, since upon phosphorylation of pRb, they activate the E2F-dependent pathway. Cell division is associated to variations in cell mass. Interestingly enough, cyclinD1-deficient mice, like CDK4-deficient ones, are smaller than their wild type siblings.Citation47,48 Cyclin D1 is expressed as 2 alternatively spliced isoforms: cyclin D1a and cyclin D1b, that differ in their C-terminus and whose ratio is altered by a polymorphism altering the efficacy of the last intron-exon boundary (reviewed inCitation49). Interestingly, the 2 isoforms have common but also specific subsets of target genes. For example, migration-related genes are regulated by the canonical cyclin D1a but not by cyclin D1b. This includes transcriptional repression of Thrombospondin 1, RhoB and ROCK2 genes and activation of Stathmin.Citation50,51,52
Global gene expression profiling carried out on mice with conditional inactivation of cyclin D1 gene revealed that cyclin D1 inhibits mitochondrial activity and aerobic glycolysis.Citation53 This is likely to occur via a CDK-dependent phosphorylation of nuclear respiratory factor 1 (NRF-1) responsible for the activation of nuclear-encoded mitochondrial genes.Citation54
However, numerous data have accumulated showing that cyclin D1 interacts directly with several transcription factors independently of its ability to complex with CDKsCitation45,46 (). Just to give a few examples it is not only involved, as already mentioned, in the growth of the estrogen-responsive tissues such as the breast epithelium, but also in androgen and secretin signaling. In the first case, cyclin D1 activates estrogen-receptor-mediated transcription in the absence of estrogen via a recruitment of p300/CREB-binding protein-associated factor (P/CAF), probably through a protein kinase A-dependent signaling,Citation55,56 and enhances it in its presenceCitation57 (). In the 2 other cases, cyclin D1 represses both basal and activated transcription. In androgen-responsive tissues such as prostate, it appears to repress a subset of androgen-dependent genes by limiting receptor residence on target promoters.Citation58,59 In the small intestinal epithelium, cyclin D1 is involved in the regulation of the terminal differentiation program of villus cells by preventing secretin gene transcription in the actively dividing cells of the crypt through repression of the BETA2/NeuroB transcription factor.Citation60 Finally, cyclin D1 has also been shown to repress STAT3Citation61 and peroxysome proliferator-activated receptor γ mediated transcription.Citation62
Figure 4. Cyclin D1 plays a central role in transcription. A few examples are given in this figure. Aside from its role in the cell cycle-dependent phosphorylation of pRB, Cyclin D1 harbors both CDK-dependent (NRF-1) and CDK-independent (C/EBPβ, ER) roles in the transcriptional control of genes required for the metabolism.
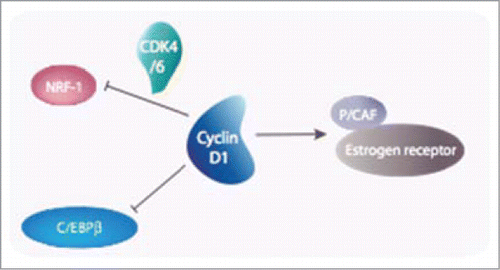
These various examples highlight the central role that cyclin D1 plays in normal physiopathology of metazoan organisms by linking cell cycle progression to cell metabolism (reviewed inCitation63). No surprise to find it again implicated in a large range of tumor types, after chromosomal rearrangements or locus amplification. A search of typical transcriptional signatures that would help understanding cyclin D1 role in cancer revealed C/EBPβ/Nf-IL6 as a major effector of this cyclin in cancer via a CDK-independent activityCitation55 ().
More recently, several genome-wide chromatin immunoprecipitation (ChIP) analyses have reinforced this idea. DNA-bound form of cyclin D1 not only occupies the regulatory region of genes governing mitochondrial biogenesis,Citation64 confirming thus previous data, but also genes involved in maintenance of chromosomal integrity as well. Bienvenu and colleagues have developed knocked-in mice strains expressing tagged cyclin D1 versions that allowed high-throughput mass spectrometry approaches aimed at recovering cyclin D1 interacting proteins in various organs.Citation65 As expected, in addition to cell cycle partners, they observed also many proteins involved in transcription. When paralleled by a genome-wide analysis of chromatin bound factors they were able to show that cyclin D1 resides on the promoters of abundantly expressed genes during development. As an example of the power of this new approach, they describe the recruitment of CREB binding protein (CBP) onto the upstream regulatory region of the Notch 1 gene in the developing retina.
Along the same line, a protein interactome of cyclin D1 established by the same laboratory in different types of human cancer cell lines overexpressing cyclin D1 revealed its participation to a network of proteins involved in DNA repair centered on RAD51.Citation66 RAD51-cyclin D1 binding is induced by radiations and the complex is recruited onto damaged DNA in a BRCA2-dependent manner. Interestingly, only cyclin D1a, the canonical form, is associated with the DNA damage response.Citation67
Cyclins and CKIs: Rho GTPases, Cytoskeleton Dynamics and Cell Motility
Cyclins-CDKs phosphorylate cytoskeleton components
Besides actin monomers, 20-30 proteins constitute actin cytoskeleton. Their assembly and function are highly regulated to allow the formation of actin network. External cues that modulate cell migration and also cell cycle regulate this remodeling. The same is true for intermediate filaments. Indeed, many cytoskeletal, adhesion, and migratory proteins are phosphorylated by cyclins-CDK complexes ( gives a tentative list, even though some of these targets need further validation).
Table 1 Some of cyclin/CDK relevant substrates for cell migration/actin cytoskeleton regulation
The regulation of proteins involved in cell motility includes phosphorylation/dephosphorylation cycles. Cyclin B2-CDK1 activity is specifically induced upon activation of integrins αvβ3 after fibronectin binding, resulting in caldesmon phosphorylation. In these conditions, cell migration is subsequently increased without affecting adhesive properties of LNCaP prostate cancer cells. Similarly, Mouse Embryonic Fibroblasts lacking cyclin B2 showed impaired migration on fibronectin matrix.Citation68
There are few studies suggesting that cyclin B1 plays a role in cancer cell invasion and metastasis of Esophageal Squamous Cell Carcinoma (ESCC). Cyclin B1 expression is increased in the most aggressive ESCC cell lines. Moreover, cyclin B1 ectopic expression enhances invasive abilities of ESCC in vitro and in vivo, this function is more likely involving CDK1 activation.Citation69 In parallel, and in an attempt to identify cyclin B1-CDK1 substrates, Blethrow and colleagues have shown that the complex is responsible of the phosphorylation of ∼70 proteins involved in migration and actin cytoskeleton regulationCitation70 (). Similarly, Chi and coworkers have identified 180 substrates for cyclin A2/CDK2 in cell extracts. Interestingly, among the substrates, while 14 target proteins are involved in cell cycle, 35 proteins are related to cellular structure organization and trafficking.Citation71 Along the same line, Zhong and colleagues identified some cytoskeleton-related proteins that are likely phosphorylated by cyclin D1-CDK4/6. The approach consisted in preventing substrates phosphorylation by the complex in MDA-MB-231 cells, using a peptide derived from p16 inhibitor. Therefore, cyclin D1-CDK4/6 substrates were less phosphorylated in this condition, although this method does not exclude that a kinase downstream from the complex is responsible for these phosphorylations.Citation52 The screens mentioned above helped identify new substrates for cyclin-CDK complexes but further validations are still needed. However, some substrates have been experimentally validated. This includes zyxin phosphorylation by cyclin B1-CDK1,Citation72 and filamin A phosphorylation by cyclin D1-CDK4.Citation52
Figure 5. Cyclins and CKIs regulate cell motility. (A) Cyclins and CKIs target RhoA-ROCK pathway: RhoA, stimulated by its GEF activates ROCK , that in return stimulates LIMK. Cyclins and CKI regulate the pathway at multiple levels. Phosphorylated cytoplasmic p27 interferes between RhoA and its GEF and also inhibit stathmin. Cyclin D1 represses ROCK and Thrombospondin gene transcription and when bound to p27, it interferes with RhoA activation. Furthermore, Cyclin D1 indirectly stabilizes p27 via the regulation of kinases involved in its degradation or its cytoplasmic localization. Cyclin A2, independently of CDKs binding and via direct interaction with RhoA and RhoC, regulates their activity. In contrast to p27, cyclin A2 stimulation RhoA GEFs. How cyclin A2 interferes with RhoC activation during EMT and whether it requires CDKs binding is still unclear. p21 and p57 target kinases downstream of RhoA. While cytoplasmic p21 directly inhibit ROCK activity, p57 sequesters LIMK in the nucleus, away from its substrates. (B) Cyclin B1-CDK1 is not only involved in phosphorylation of substrates involved in cell cycle progression, but also in the phosphorylation of a plethora of other targets including molecules intervening in cell motility.
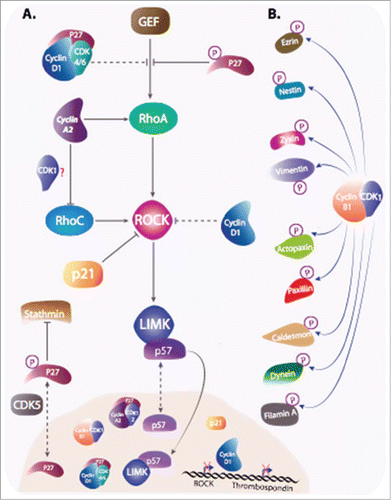
The yin and the yang of the RhoA-ROCK-LIMK pathway
Although many pathways contribute to the regulation of cell motility, it appears that cell cycle proteins are mainly involved in the direct regulation of the RhoA-ROCK-LIMK pathway, and to a lesser extend the Rac pathway.
The Rho family of GTPases, with the archetypal RhoA, RhoB, RhoC, Rac1 and Cdc42 types of proteins, carries out a variety of functions in the cell centered on cytoskeleton structure and dynamics, and therefore play important roles in cell morphogenesis, adhesion, migration and cytokinesis.Citation73,74 They function downstream from various receptors, whether tyrosine kinases or integrins, as molecular switches alternating between a GDP-bound inactive state and a GTP-bound active state. The latter interacts with, and activates effector proteins that will thus translate the peripheral cues into a wide variety of cellular mechanisms. The hydrolysis of GTP is associated to a return of the GTPase to an inactive state, and is promoted by a GTPase-activating protein (GAP). A new cycle of activation requires the exchange of GDP with GTP that is promoted by a guanine-nucleotide-exchange factor (GEF). There are multiple GAPs and GEFs, and several GTPases share many of them, the specificity being brought by both spatial and temporal cues.
The RhoA pathway, along with Rac/Cdc42 pathways, has been extensively studied these last decades (for reviewCitation75,76,77,78). When activated it induces a signaling cascade downstream that includes the activation of Rho-activated kinase (ROCK) that phosphorylates and activates LIM kinase (LIMK). Together, these kinases are responsible for the phosphorylation of a plethora of substrates including cofilin, and myosin light chain (MLC2). Consequently, RhoA pathway activation induces many cellular processes including actin stress fibers formation and actomyosin contractility. The growth of many cells is dependent on interactions with the extracellular matrix through integrin-mediated adhesion that impinges upon cytoskeleton structure. In concert with cell-cell interactions, this has important outcome on mitosis, most particularly in tissue structures such as epithelia, and anchorage-independent proliferation is a hallmark of oncogenic transformation. Loss of adhesion of normal cells results in a G1 arrest, even in the presence of mitogenic signals, with a down regulation of cyclin D1 and an accumulation of CKIs of the CIP/KIP family (p21, p27). Accordingly, cells cannot enter S phase and cyclin A2 is not expressed. Anchorage-dependent Cyclin D1 expression has been proposed to result from the antagonism between Rac1 and Rho-ROCK signalingCitation79,80,81,82, whereas relief of cyclin A2 dependence on anchorage relies on both Ras and Cdc42.Citation83 Rho GTPases appear thus to mediate adhesion/cytoskeleton structure effects on cell cycle regulators expression (reviewed inCitation84).
The first hints that the reverse is also true came from reports on budding yeast Far 1, a CKI which regulates Cdc42 by interfering with its access to its GEF for the establishment of cell polarity during yeast mating.Citation85 p57,Citation86 then p27Citation87 and p21Citation88,89,90 were shown to negatively regulate Rho signaling via inhibitory interactions with different proteins of the cascade (Reviewed inCitation84 ).
The first link between p21 and actin cytoskeleton remodeling has been reported in neural cells and fibroblasts. In these cells, p21 cytoplasmic localization correlated with neurite outgrowth and axonal growth after spinal cord injury or actin cytoskeleton disorganization respectively.Citation89,91 p21 is mainly localized in the nucleus despite shuttling between the latter and cytoplasm. Interestingly, forced cytoplasmic localization of p21 by removing the nuclear localization signal, rendered NIH3T3 fibroblasts unable to form actin stress fibers after serum stimulation. Despite RhoA being active in these conditions, downstream ROCK signaling was impaired. Interestingly, this resulted from a direct inhibitory interaction of cytoplasmic but not nuclear p21 with the catalytic domain of ROCK.Citation89,92
Similarly, p57 inhibits RhoA pathway (for reviewCitation93). Unlike p21, that interacts and inhibits ROCK, p57 binds to LIMK and prevents its normal function. Some studies have shown opposite effects of this binding. Intriguingly, this effect is not the result of the inhibition of the catalytic activity of the kinase by p57. On the one hand, the first study shows that p57 that shuttles between the nucleus and the cytoplasm, piggybacks LIMK to the nucleus, preventing cytoplasmic substrates phosphorylation in HEK293 cells. This is corroborated by the inability of cytoplasmic p57 to inhibit LIMK in these conditions.Citation86 On the other hand, the second study shows that p57-LIMK interaction is exclusively cytoplasmic. Subsequently, LIM kinase activity is enhanced as it increases cofilin phosphorylation on its Ser3. Consequently, this promotes the formation of actin stress fibers, resulting in a decrease of HeLa cells migration. Importantly, this function of p57 is independent of its role in cell cycle, as a p57 mutant defective in CDK inhibition has the same effect reported with the WT protein.Citation94 Similar effects were observed in hepatocellular carcinoma cells where p57 depletion impaired cofilin phosphorylation and increased cell invasiveness.Citation95 Some other groups investigated the role of p57 in neuronal cells migration, although without exploring the functional consequences of this p57-LIMK interaction. Again, these studies demonstrated opposite effects of p57 on cell migration, where Sakai and collaborators have shown that inducible p57 expression in glioblastoma cell lines decreased their ability to migrate and invade.Citation96 On the contrary, Itoh and colleagues have reported that p57 knockdown by RNA interference, while not affecting neuronal differentiation, significantly delayed neuronal migration to the cortical plate of new born mice.Citation97
p27-mediated regulation of cell migration occurs at different levelsCitation84,92,98 and requires specific phosphorylations responsible for its cytoplasmic localization (for reviewCitation84,92,99). Interestingly, despite having a nuclear localization signal, p27 export is enhanced after Ser10/Thr187 and Thr198 phosphorylations by CDK5 and Ribosomal S6 (RSK) kinases respectivelyCitation99,100,101. Elegant experiment demonstrated that p27 forced expression in cancer cells has different effects whether the protein is maintained in the nucleus or in the cytoplasm, showing that nuclear localization correspond to a cell cycle inhibition while a cytoplasmic localization increases cell migration in vitro and in vivo.Citation87,101,102,103,104,105
Nagahara and collaborators made the initial observation linking p27-mediated regulation of actin cytoskeleton and cell migration. The study reported an increase of filopodia and cell scattering of HepG2 hepatocellular carcinoma cells after the overexpression of TAT-p27 fusion protein.Citation104 This effect is correlated with the substantial increase of actin stress fibers and the decrease of migratory abilities of p27-/-MEFs. The phenotype is consistent with an increase of RhoA active forms, as confirmed by a rescue of this phenotype after ROCK inhibition. Mechanistically, p27 interferes with RhoA activation by its GEFs.Citation84,87,92 Interestingly enough, and unlike the mechanistically well-characterized effect of p27 on RhoA, it has been suggested that p27 mediates Rac1 GTPase-dependent cell migration. However, this regulation is poorly understood as the only evidence reported is that the expression of a dominant-negative Rac1 (Rac1-N17) abolishes p27-stimulated cell motility.Citation103
It should be pointed out at this level that some studies have reported that in some cell types, including fibrosarcoma, endothelial and vascular smooth muscle cells, the inhibitory function of p27 on cell migration does not seem to involve the RhoA-ROCK pathway but rather an interaction with the Stathmin-Tubulin network.Citation106,107,108,109
Similarly, cyclin D1-/- macrophages and MEFs show increased focal adhesion structures number and stability, resulting in an increased cell adhesion.Citation110 Cyclin D1 deficiency is also characterized by a decreased motility in 2 and 3 dimensions accompanied by chemotaxis defects.Citation110,111 This phenotype is consistent with sustained ROCK signaling and inhibitory effects of thrombospondin as the expression and activity of these 2 proteins are increased in cyclin D1-/- cells.Citation51,111,112,113,114,115,116
Moreover, cyclin D1 participates also in RhoA-ROCK inhibitory network by binding directly to p27 and thereby preventing interaction of RhoA with its GEF.Citation51,Citation83,Citation115,Citation117 Inhibition of the Rho-ROCK pathway leads to a decrease in stability and/or in number of focal adhesion as well as actin stress fibers, that results in increased cell motility. As a consequence, cyclin D1-/- and p27-/- cells are less motile than their wild type counterparts. Even though the mechanism was not understood, this result was anticipated by an earlier report on HepG2 hepatocellular carcinoma cells transduced with TAT-p27.Citation104
Cyclin D1 also represses Skp2 (S Phase Kinase Associating Protein 2) gene that codes for an F-box protein component of the Skp/Cullin/F-box-type E3 ubiquitin ligase targeting p27, among several cell cycle regulatory proteins, for degradation through the ubiquitin-dependent pathway.Citation117 Indeed, p27 through its stabilization and inhibiton of cyclin D1/CDK plays a crucial role in cyclin D1-mediated cell migration. This function requires the N-terminus of cyclin D1 that mediates its interaction with p27. Accordingly, cyclin D1 deletion mutants unable to bind p27 were not able to stimulate cell motility.Citation117 Interestingly, cyclin D1 also stabilizes CDK5 kinase, which phosphorylates p27, therefore indirectly enhancing the cytoplasmic localization of p27.Citation100,Citation117 This is consistent with the p27 deficiency observed in cyclin D1-/- cells, whose migration defect was rescued by p27 overexpression.Citation117
Recent data pointing to a novel function of Cyclin A2 add another component to this complex regulatory network that involves cell cycle regulators and cytoskeletal structures participating in the control of cell movementCitation118 (reviewed in:Citation119). Surprisingly, depletion of cyclin A2 is sufficient to increase cell motility of fibroblasts in 2D assays and cooperates with oncogenic transformation to increase their invasiveness in 3D collagen matrixes. Cyclin A2-deficient cells contain a perturbed cytoskeleton, where actin filaments are cortical and the distribution of focal adhesions is altered. Interestingly, a cyclin A2 mutant unable to activate its cognate kinases, CDK1 and CDK2, corrects these defects.
This is associated with a down regulation of the RhoA-ROCK pathway and decreased phosphorylation of cofilin, which is involved in the reorganization of actin filaments, consecutively leading to an increased cell migration and invasion. Importantly, cyclin A2 appears to interact directly with RhoA and potentiates, at least in vitro, the exhange activity of its GEFs. An interesting point revealed by this study is the cytoplasmic location of the mutant used. Cyclin A2 has no nuclear localization signal and shuttles between the nucleus and the cytoplasmCitation120 associated with its partners, a capacity that is lost by the mutant.Citation121
This highlights a common denominator shared by cell cycle regulators that appear to play a double jeu with distinct functions in the nucleus and the cytoplasm. Consistently, oncogenic transformation is often associated with their misregulation. Just to take the example of p27, whereas its gene is rarely mutated or deleted, the protein is frequently lost or present at reduced levels. In some malignancies harboring high levels of p27, it is sequestered by cyclin D-CDK4 complexes, or is found in the cytoplasmCitation122,123,124 (reviewed inCitation99). Along the same line, a down regulation of cyclin A2, and/or its cytoplasmic relocalization, is observed in some metastases versus primary tumors in matched sets of human colon adenocarcinomasCitation118 and oral squamous cell carcinoma (OSCC).Citation125
Cyclin A2 and the epithelial to mesenchymal transition (EMT)
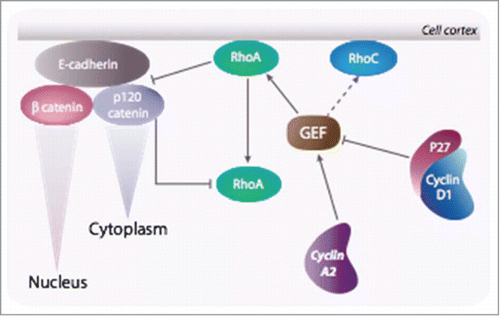
Recent data suggest that cyclin A2 participates in the control of the EMT, a tissue remodeling occurring in early development and reactivated during wound healing and cancer (reviews:Citation126,127,128). Cyclin A2 depletion in epithelial cells leads to loss of cell-to-cell contacts, decreased E-cadherin expression, and a relocalization of p120 catenin to the cytoplasm.Citation129 Strikingly, knockdown of cyclin A2 is associated with a decreased RhoA activity mirrored by an increase in that of RhoC, a situation that is reminiscent of what is observed during the EMT.Citation130 This is consistent with previously published data showing that inhibition of RhoA with C3 toxin or expression of a constitutively active RhoC led to a disruption of adherens junctions.Citation131
These phenotypes are exacerbated upon oncogenic Ras expression, leading to an upregulation of mRNAs coding for mesenchymal markers such as N-Cadherin, the transcriptional regulators Twist2, Slug, E12 and E47 and the metalloproteases MMP-3, -7 and -14. In addition, deficient cells show increased invasive potential in cell culture and increased extravasation in chick embryos. Interestingly, whereas RhoC is dispensable for embryonic and post-natal mouse developmentCitation132 its loss leads to a drastic inhibition of metastasis without affecting tumor development at the primary site. Cyclin A2-deficient cells also exhibited cancer stem cell-like properties and increased capacity to spread in vivo. Overall, these observations establish cyclin A2 as a novel regulator of metastasis through modulation of EMT and stem cell-like properties, leading to a possible scenario. In its presence, adherens junctions are stabilized via RhoA and its effector mDia, whereas in its absence, they are disrupted via RhoC-ROCK signaling (). This situation is even worse when cyclin D1 is overexpressed knowing its repression of RhoA activity via p27 as already mentioned. As a consequence, p120 catenin translocates to the cytoplasm where it forms an inactivating complex with RhoACitation133,134, and β catenin enters the nucleus where it participates in the transcription of genes such as Goosecoid, Slug or Twist whose products are required for the EMT.Citation127
Figure 6. Cyclin A2 controls EMT via a reciprocal regulation of RhoA and RhoC: Cyclin A2 potentiates the loading activity of Rho GEF toward RhoA at the expense of RhoC. Since RhoA is required for the stability of the adherens junction, ablation of cyclin A2 leads to its inhibition and, as a consequence, in the disruption of cell-cell contacts. Accordingly, RhoC activity is increased and contributes further in the instability of the junction. As a result, p120 catenin translocates to the cytoplasm where it forms an inactivating complex with RhoA, and β catenin enters the nucleus where it participates in the transcription of genes required for the EMT. In cells overexpressing cyclin D1 this phenomenon is exacerbated.
According to this model, cyclin A2 resides at the interface between cytoplasmic (cell autonomous) and extracellular cues. in fine it might play a role in the orientation of the mitotic spindle within epithelial structures. When its level is above a given threshold, the 2 daughter cells orientate according to their mother footprint relative to the lamina. Should cyclin A2 level be decreased, then, cell spreading after division is isotropic and cells increase their motility. Within this frame, our observation of a modulation of the GTP loading of RhoA by cyclin A2, and more recently, that a small proportion of cyclin A2 persists beyond metaphase and is degraded by autophagy,Citation135 while consistent with the involvement of the cyclin A2-RhoA interaction in early mitosis, would suggest a novel role of cyclin A2 later during cell division when RhoA participates to the formation of the contractile ring.
Concluding Remarks and Perspectives in Cancer Research
If these observations complete our vision of the intricate relationships entertained by cell cycle regulators and small GTPases, they point to their putative involvement in more general processes governed by the interactions that cells entertain with their surrounding niche. Adapting cell metabolism through a switch in the transcriptional program, determining cell fate during differentiation, or reshaping its cytoskeleton or its motile behavior, are the different facets of the same phenomenom: adaptation of the cell to its environment.
Since the seminal observation of Otto Warburg, advances in our understanding of cancer cell physiopathology accumulated over the last decade has highlighted the pervasive role of the cell environment in tumor progression. However, Warburg's hypothesis envisioned the alteration of cancer cells metabolism as arising from a defect of mitochondrial functions, whereas it appears now that the propensity of cancer cells to “waste” glucose via aerobic glycolysis does not result from defective mitochondria but rather from a true reprogramming of cell metabolism (reviewed in:Citation136,137,138,139). A same change in paradigm has led to the debated notion of cancer stem cell and its controversial link with a latent embryonic program, known as the epithelial to mesenchymal transition, that when reactivated endows cancer cells with resistance to cell death and invasive properties (reviewed in:Citation127,140,141).
Accordingly, within this new pespective, cell cycle regulators can be considered as mediators between external cues and cell autonomous mechanisms such as the cell division cycle. During embryogenesis or wound healing, cells must adapt their energetic metabolism to respond to the demand in new tissue mass. Moreover, organ growth and repair require a spatial and temporal control of cell fate, and in this respect, cell cycle regulators are likely to play a central role in establishing and/or restoring tissue architecture. The control of mitotic spindle orientation within an epithelial structure, and the establishment of cell polarization are good examples for this: remaining a stem cell or embarking into a differentiation program will depend on whether the candidate proliferating cell undergo a symmetric or an asymmetric mode of division, a situation controlled by the niche. No surprise that cancer, with the mediation of cell cycle regulators, is nothing else but the mere cooptation of these essential processes.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Daniel Fisher for his constructing remarks.
Funding
This work was supported by Agence Nationale de la Recherche (08-BLAN-0037-02), Ligue régionale contre le cancer, Fondation ARC pour la Recherche sur le Cancer, and GEFLUC. N.B. was supported by fellowships from the French Ministry of Education and Research and the Fondation pour la Recherche Médicale (FRM).
Related Research Data
References
- Nigg EA. Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol 2001; 2:21-32; PMID:11413462; http://dx.doi.org/10.1038/35048096
- Pines J. Four-dimensional control of the cell cycle. Nat Cell Biol 1999; 1:E73-9; PMID:10559915; http://dx.doi.org/10.1038/11041
- Santamaria D, Barriere C, Cerqueira S, Hunt A, Tardy C, Newton K, Caceres JF, Dubus P, Malumbres M, Barbacid M. Cdk1 is sufficient to drive the mammalian cell cycle. Nature 2007; 448:811-5; PMID:17700700; http://dx.doi.org/10.1038/nature06046
- Hochegger H, Takeda S, Hunt T. Cyclin-dependent kinases and cell-cycle transitions: does one fit all? Nat Rev Mol Cell Biol 2008; 9:910-6; PMID:18813291; http://dx.doi.org/10.1038/nrm2510
- Geng Y, Yu Q, Sicinska E, Das M, Schneider JE, Bhattacharya S, Rideout WM, Bronson RT, Gardner H, Sicinski P. Cyclin E ablation in the mouse. Cell 2003; 114:431-43; PMID:12941272; http://dx.doi.org/10.1016/S0092-8674(03)00645-7
- Kozar K, Ciemerych MA, Rebel VI, Shigematsu H, Zagozdzon A, Sicinska E, Geng Y, Yu Q, Bhattacharya S, Bronson RT, et al. Mouse development and cell proliferation in the absence of D-cyclins. Cell 2004; 118:477-91; PMID:15315760; http://dx.doi.org/10.1016/j.cell.2004.07.025
- Mendez J. Cell proliferation without cyclin E-CDK2. Cell 2003; 114:398-9; PMID:12941268; http://dx.doi.org/10.1016/S0092-8674(03)00649-4
- Parisi T, Beck AR, Rougier N, McNeil T, Lucian L, Werb Z, Amati B. Cyclins E1 and E2 are required for endoreplication in placental trophoblast giant cells. EMBO J 2003; 22:4794-803; PMID:12970191; http://dx.doi.org/10.1093/emboj/cdg482
- Kalaszczynska I, Geng Y, Iino T, Mizuno S, Choi Y, Kondratiuk I, Silver DP, Wolgemuth DJ, Akashi K, Sicinski P. Cyclin A is redundant in fibroblasts but essential in hematopoietic and embryonic stem cells. Cell 2009; 138:352-65; PMID:19592082; http://dx.doi.org/10.1016/j.cell.2009.04.062
- Sicinska E, Aifantis I, Le L Cam, Swat W, Borowski C, Yu Q, Ferrando AA, Levin SD, Geng Y, von Boehmer H, et al. Requirement for cyclin D3 in lymphocyte development and T cell leukemias. Cancer Cell 2003; 4:451-61; PMID:14706337; http://dx.doi.org/10.1016/S1535-6108(03)00301-5
- Darbon JM, Devault A, Taviaux S, Fesquet D, Martinez AM, Galas S, Cavadore JC, Doree M, Blanchard JM. Cloning, expression and subcellular localization of the human homolog of p40MO15 catalytic subunit of cdk-activating kinase. Oncogene 1994; 9:3127-38; PMID:7936635
- Fesquet D, Labbe JC, Derancourt J, Capony JP, Galas S, Girard F, Lorca T, Shuttleworth J, Doree M, Cavadore JC. The MO15 gene encodes the catalytic subunit of a protein kinase that activates cdc2 and other cyclin-dependent kinases (CDKs) through phosphorylation of Thr161 and its homologues. EMBO J 1993; 12:3111-21; PMID:8344251
- Fisher RP, Morgan DO. A novel cyclin associates with MO15/CDK7 to form the CDK-activating kinase. Cell 1994; 78:713-24; PMID:8069918; http://dx.doi.org/10.1016/0092-8674(94)90535-5
- Poon RY, Yamashita K, Adamczewski JP, Hunt T, Shuttleworth J. The cdc2-related protein p40MO15 is the catalytic subunit of a protein kinase that can activate p33cdk2 and p34cdc2. EMBO J 1993; 12:3123-32; PMID:8393783
- Solomon MJ, Harper JW, Shuttleworth J. 1993. CAK, the p34cdc2 activating kinase, contains a protein identical or closely related to p40MO15. EMBO J. 12:3133-42; PMID:8344252
- Ganuza M, Saiz-Ladera C, Canamero M, Gomez G, Schneider R, Blasco MA, Pisano D, Paramio JM, Santamaria D, Barbacid M. Genetic inactivation of Cdk7 leads to cell cycle arrest and induces premature aging due to adult stem cell exhaustion. EMBO J 2012; 31:2498-510; PMID:22505032; http://dx.doi.org/10.1038/emboj.2012.94
- Shiekhattar R, Mermelstein F, Fisher RP, Drapkin R, Dynlacht B, Wessling HC, Morgan DO, Reinberg D. Cdk-activating kinase complex is a component of human transcription factor TFIIH. Nature 1995; 374:283-7; PMID:7533895; http://dx.doi.org/10.1038/374283a0
- Larochelle S, Amat R, Glover-Cutter K, Sanso M, Zhang C, Allen JJ, Shokat KM, Bentley DL, Fisher RP. Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat Struct Mol Biol 2012; 19:1108-15; PMID:23064645; http://dx.doi.org/10.1038/nsmb.2399
- Egly JM, Coin F. A history of TFIIH: two decades of molecular biology on a pivotal transcription/repair factor. DNA Repair (Amst) 2011; 10:714-21; PMID:21592869
- Lolli G, Johnson LN. CAK-Cyclin-dependent Activating Kinase: a key kinase in cell cycle control and a target for drugs? Cell Cycle 2005; 4:572-7; PMID:15876871; http://dx.doi.org/10.4161/cc.4.4.1607
- Bour G, Gaillard E, Bruck N, Lalevee S, Plassat JL, Busso D, Samama JP, Rochette-Egly C. Cyclin H binding to the RARalpha activation function (AF)-2 domain directs phosphorylation of the AF-1 domain by cyclin-dependent kinase 7. Proc Natl Acad Sci U S A 2005; 102:16608-13; PMID:16275922; http://dx.doi.org/10.1073/pnas.0505556102
- Lee DK, Duan HO, Chang C. From androgen receptor to the general transcription factor TFIIH. Identification of cdk activating kinase (CAK) as an androgen receptor NH(2)-terminal associated coactivator. J Biol Chem 2000; 275:9308-13; PMID:10734072; http://dx.doi.org/10.1074/jbc.275.13.9308
- Rochette-Egly C, Adam S, Rossignol M, Egly JM, Chambon P. Stimulation of RAR alpha activation function AF-1 through binding to the general transcription factor TFIIH and phosphorylation by CDK7. Cell 1997; 90:97-107; PMID:9230306; http://dx.doi.org/10.1016/S0092-8674(00)80317-7
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100:57-70; PMID:10647931; http://dx.doi.org/10.1016/S0092-8674(00)81683-9
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646-74; PMID:21376230; http://dx.doi.org/10.1016/j.cell.2011.02.013
- Bortner DM, Rosenberg MP. Induction of mammary gland hyperplasia and carcinomas in transgenic mice expressing human cyclin E. Mol Cell Biol 1997; 17:453-9; PMID:8972226
- Karsunky H, Geisen C, Schmidt T, Haas K, Zevnik B, Gau E, Moroy T. Oncogenic potential of cyclin E in T-cell lymphomagenesis in transgenic mice: evidence for cooperation between cyclin E and Ras but not Myc. Oncogene 1999; 18:7816-24; PMID:10618723; http://dx.doi.org/10.1038/sj.onc.1203205
- Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclin D1 ablation. Nature 2001; 411:1017-21; PMID:11429595; http://dx.doi.org/10.1038/35082500
- Dulic V, Drullinger LF, Lees E, Reed SI, Stein GH. Altered regulation of G1 cyclins in senescent human diploid fibroblasts: accumulation of inactive cyclin E-Cdk2 and cyclin D1-Cdk2 complexes. Proc Natl Acad Sci U S A 1993; 90:11034-8; PMID:8248208; http://dx.doi.org/10.1073/pnas.90.23.11034
- Lucibello FC, Sewing A, Brusselbach S, Burger C, Muller R. Deregulation of cyclins D1 and E and suppression of cdk2 and cdk4 in senescent human fibroblasts. J Cell Sci 1993; 105 (Pt 1):123-33; PMID:8360268
- Leontieva OV, Blagosklonny MV. CDK4/6-inhibiting drug substitutes for p21 and p16 in senescence: duration of cell cycle arrest and MTOR activity determine geroconversion. Cell Cycle 2013; 12:3063-9; PMID:23974099; http://dx.doi.org/10.4161/cc.26130
- Leontieva OV, Natarajan V, Demidenko ZN, Burdelya LG, Gudkov AV, Blagosklonny MV. Hypoxia suppresses conversion from proliferative arrest to cellular senescence. Proc Natl Acad Sci U S A 2012; 109:13314-8; PMID:22847439; http://dx.doi.org/10.1073/pnas.1205690109
- Haas K, Johannes C, Geisen C, Schmidt T, Karsunky H, Blass-Kampmann S, Obe G, Moroy T. Malignant transformation by cyclin E and Ha-Ras correlates with lower sensitivity towards induction of cell death but requires functional Myc and CDK4. Oncogene 1997; 15:2615-23; PMID:9399649; http://dx.doi.org/10.1038/sj.onc.1201434
- Geisen C, Moroy T. The oncogenic activity of cyclin E is not confined to Cdk2 activation alone but relies on several other, distinct functions of the protein. J Biol Chem 2002; 277:39909-18; PMID:12149264; http://dx.doi.org/10.1074/jbc.M205919200
- Geng Y, Lee YM, Welcker M, Swanger J, Zagozdzon A, Winer JD, Roberts JM, Kaldis P, Clurman BE, Sicinski P. Kinase-independent function of cyclin E. Mol Cell 2007; 25:127-39; PMID:17218276; http://dx.doi.org/10.1016/j.molcel.2006.11.029
- Geng Y, Sicinski P. Differences in regulation and function of E-cyclins in human cancer cells. Cell Cycle 2013; 12:1165; PMID:23549169; http://dx.doi.org/10.4161/cc.24487
- Coverley D, Laman H, Laskey RA. Distinct roles for cyclins E and A during DNA replication complex assembly and activation. Nat Cell Biol 2002; 4:523-8; PMID:12080347; http://dx.doi.org/10.1038/ncb813
- Ferguson RL, Maller JL. Centrosomal localization of cyclin E-Cdk2 is required for initiation of DNA synthesis. Curr Biol 2010; 20:856-60; PMID:20399658; http://dx.doi.org/10.1016/j.cub.2010.03.028
- Ferguson RL, Pascreau G, Maller JL. The cyclin A centrosomal localization sequence recruits MCM5 and Orc1 to regulate centrosome reduplication. J Cell Sci 2010; 123:2743-9; PMID:20663915; http://dx.doi.org/10.1242/jcs.073098
- Hemerly AS, Prasanth SG, Siddiqui K, Stillman B. Orc1 controls centriole and centrosome copy number in human cells. Science 2009; 323:789-93; PMID:19197067; http://dx.doi.org/10.1126/science.1166745
- Berger C, Pallavi SK, Prasad M, Shashidhara LS, Technau GM. Cyclin E acts under the control of Hox-genes as a cell fate determinant in the developing central nervous system. Cell Cycle 2005; 4:422-5; PMID:15684605; http://dx.doi.org/10.4161/cc.4.3.1524
- Berger C, Kannan R, Myneni S, Renner S, Shashidhara LS, Technau GM. Cell cycle independent role of Cyclin E during neural cell fate specification in Drosophila is mediated by its regulation of Prospero function. Dev Biol 2010; 337:415-24; PMID:19914234; http://dx.doi.org/10.1016/j.ydbio.2009.11.012
- Odajima J, Wills ZP, Ndassa YM, Terunuma M, Kretschmannova K, Deeb TZ, Geng Y, Gawrzak S, Quadros IM, Newman J, et al. Cyclin E constrains Cdk5 activity to regulate synaptic plasticity and memory formation. Dev Cell 2011; 21:655-68; PMID:21944720; http://dx.doi.org/10.1016/j.devcel.2011.08.009
- Rogulja D, Young MW. Control of sleep by cyclin A and its regulator. Science 2012; 335:1617-21.
- Coqueret O. Linking cyclins to transcriptional control. Gene 2002; 299:35-55; PMID:12459251; http://dx.doi.org/10.1016/S0378-1119(02)01055-7
- Fu M, Wang C, Li Z, Sakamaki T, Pestell RG. Minireview: Cyclin D1: normal and abnormal functions. Endocrinology 2004; 145:5439-47; PMID:15331580; http://dx.doi.org/10.1210/en.2004-0959
- Fantl V, Stamp G, Andrews A, Rosewell I, Dickson C. Mice lacking cyclin D1 are small and show defects in eye and mammary gland development. Genes Dev 1995; 9:2364-72; PMID:7557388; http://dx.doi.org/10.1101/gad.9.19.2364
- Rane SG, Dubus P, Mettus RV, Galbreath EJ, Boden G, Reddy EP, Barbacid M. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat Genet 1999; 22:44-52; PMID:10319860; http://dx.doi.org/10.1038/8751
- Knudsen KE, Diehl JA, Haiman CA, Knudsen ES. Cyclin D1: polymorphism, aberrant splicing and cancer risk. Oncogene 2006; 25:1620-8; PMID:16550162; http://dx.doi.org/10.1038/sj.onc.1209371
- Betticher DC, Thatcher N, Altermatt HJ, Hoban P, Ryder WD, Heighway J. Alternate splicing produces a novel cyclin D1 transcript. Oncogene 1995; 11:1005-11; PMID:7675441
- Li Z, Wang C, Jiao X, Lu Y, Fu M, Quong AA, Dye C, Yang J, Dai M, Ju X, et al. Cyclin D1 regulates cellular migration through the inhibition of thrombospondin 1 and ROCK signaling. Mol Cell Biol 2006b; 26:4240-56; http://dx.doi.org/10.1128/MCB.02124-05
- Zhong Z, Yeow WS, Zou C, Wassell R, Wang C, Pestell RG, Quong JN, Quong AA. Cyclin D1/cyclin-dependent kinase 4 interacts with filamin A and affects the migration and invasion potential of breast cancer cells. Cancer Res 70:2105-14; PMID:20179208; http://dx.doi.org/10.1158/0008-5472.CAN-08-1108
- Sakamaki T, Casimiro MC, Ju X, Quong AA, Katiyar S, Liu M, Jiao X, Li A, Zhang X, Lu Y, et al. Cyclin D1 determines mitochondrial function in vivo. Mol Cell Biol 2006; 26:5449-69; PMID:16809779; http://dx.doi.org/10.1128/MCB.02074-05
- Wang C, Li Z, Lu Y, Du R, Katiyar S, Yang J, Fu M, Leader JE, Quong A, Novikoff PM, Pestell RG. Cyclin D1 repression of nuclear respiratory factor 1 integrates nuclear DNA synthesis and mitochondrial function. Proc Natl Acad Sci U S A 2006; 103:11567-72; PMID:16864783; http://dx.doi.org/10.1073/pnas.0603363103
- Lamb J, Ramaswamy S, Ford HL, Contreras B, Martinez RV, Kittrell FS, Zahnow CA, Patterson N, Golub TR, Ewen ME. A mechanism of cyclin D1 action encoded in the patterns of gene expression in human cancer. Cell 2003; 114:323-34; PMID:12914697; http://dx.doi.org/10.1016/S0092-8674(03)00570-1
- McMahon C, Suthiphongchai T, DiRenzo J, Ewen ME. P/CAF associates with cyclin D1 and potentiates its activation of the estrogen receptor. Proc Natl Acad Sci U S A 1999; 96:5382-7; PMID:10318892; http://dx.doi.org/10.1073/pnas.96.10.5382
- Zwijsen RM, Wientjens E, Klompmaker R, van der Sman J, Bernards R, Michalides RJ. CDK-independent activation of estrogen receptor by cyclin D1. Cell 1997; 88:405-15; PMID:9039267; http://dx.doi.org/10.1016/S0092-8674(00)81879-6
- Comstock CE, Augello MA, Schiewer MJ, Karch J, Burd CJ, Ertel A, Knudsen ES, Jessen WJ, Aronow BJ, Knudsen KE. Cyclin D1 is a selective modifier of androgen-dependent signaling and androgen receptor function. J Biol Chem 2011; 286:8117-27; PMID:21212260; http://dx.doi.org/10.1074/jbc.M110.170720
- Knudsen KE, Cavenee WK, Arden KC. D-type cyclins complex with the androgen receptor and inhibit its transcriptional transactivation ability. Cancer Res 1999; 59:2297-301; PMID:10344732
- Ratineau C, Petry MW, Mutoh H, Leiter AB. Cyclin D1 represses the basic helix-loop-helix transcription factor, BETA2/NeuroD. J Biol Chem 2002; 277:8847-53; PMID:11788592; http://dx.doi.org/10.1074/jbc.M110747200
- Bienvenu F, Gascan H, Coqueret O. Cyclin D1 represses STAT3 activation through a Cdk4-independent mechanism. J Biol Chem 2001; 276:16840-7; PMID:11279133; http://dx.doi.org/10.1074/jbc.M100795200
- Wang C, Pattabiraman N, Zhou JN, Fu M, Sakamaki T, Albanese C, Li Z, Wu K, Hulit J, Neumeister P, et al. Cyclin D1 repression of peroxisome proliferator-activated receptor gamma expression and transactivation. Mol Cell Biol 2003; 23:6159-73; PMID:12917338; http://dx.doi.org/10.1128/MCB.23.17.6159-6173.2003
- Knudsen KE. Cyclin D1 goes metabolic: dual functions of cyclin D1 in regulating lipogenesis. Cell Cycle 2012; 11:3533-4; PMID:22951541; http://dx.doi.org/10.4161/cc.22039
- Casimiro MC, Crosariol M, Loro E, Ertel A, Yu Z, Dampier W, Saria EA, Papanikolaou A, Stanek TJ, Li Z, et al. ChIP sequencing of cyclin D1 reveals a transcriptional role in chromosomal instability in mice. J Clin Invest 2012; 122:833-43; PMID:22307325; http://dx.doi.org/10.1172/JCI60256
- Bienvenu F, Jirawatnotai S, Elias JE, Meyer CA, Mizeracka K, Marson A, Frampton GM, Cole MF, Odom DT, Odajima J, et al. Transcriptional role of cyclin D1 in development revealed by a genetic-proteomic screen. Nature 2010; 463:374-8; PMID:20090754; http://dx.doi.org/10.1038/nature08684
- Jin RJ, Lho Y, Wang Y, Ao M, Revelo MP, Hayward SW, Wills ML, Logan SK, Zhang P, Matusik RJ. Down-regulation of p57Kip2 induces prostate cancer in the mouse. Cancer Res 2008; 68:3601-8; PMID:18483241; http://dx.doi.org/10.1158/0008-5472.CAN-08-0073
- Li Z, Jiao X, Wang C, Shirley LA, Elsaleh H, Dahl O, Wang M, Soutoglou E, Knudsen ES, Pestell RG. Alternative cyclin D1 splice forms differentially regulate the DNA damage response. Cancer Res 2010; 70:8802-11; PMID:20940395; http://dx.doi.org/10.1158/0008-5472.CAN-10-0312
- Manes T, Zheng DQ, Tognin S, Woodard AS, Marchisio PC, Languino LR. Alpha(v)beta3 integrin expression up-regulates cdc2, which modulates cell migration. J Cell Biol 2003; 161:817-26; PMID:12771130; http://dx.doi.org/10.1083/jcb.200212172
- Song Y, Zhao C, Dong L, Fu M, Xue L, Huang Z, Tong T, Zhou Z, Chen A, Yang Z, Lu N, Zhan Q. Overexpression of cyclin B1 in human esophageal squamous cell carcinoma cells induces tumor cell invasive growth and metastasis. Carcinogenesis 2008; 29:307-15; PMID:18048386; http://dx.doi.org/10.1093/carcin/bgm269
- Blethrow JD, Glavy JS, Morgan DO, Shokat KM. Covalent capture of kinase-specific phosphopeptides reveals Cdk1-cyclin B substrates. Proc Natl Acad Sci U S A 2008; 105:1442-7; PMID:18234856; http://dx.doi.org/10.1073/pnas.0708966105
- Chi Y, Welcker M, Hizli AA, Posakony JJ, Aebersold R, Clurman BE. Identification of CDK2 substrates in human cell lysates. Genome Biol 2008; 9:R149
- Hirota T, Morisaki T, Nishiyama Y, Marumoto T, Tada K, Hara T, Masuko N, Inagaki M, Hatakeyama K, Saya H. Zyxin, a regulator of actin filament assembly, targets the mitotic apparatus by interacting with h-warts/LATS1 tumor suppressor. J Cell Biol 2000; 149:1073-86; PMID:10831611; http://dx.doi.org/10.1083/jcb.149.5.1073
- Burridge K, Wennerberg K. Rho and Rac take center stage. Cell 2004; 116:167-79; PMID:14744429; http://dx.doi.org/10.1016/S0092-8674(04)00003-0
- Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol 2005; 21:247-69; PMID:16212495; http://dx.doi.org/10.1146/annurev.cellbio.21.020604.150721
- Hall A. Rho GTPases and the control of cell behaviour. Biochem Soc Trans 2005; 33:891-5; PMID:16246005; http://dx.doi.org/10.1042/BST20050891
- Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol 2008; 9:690-701; PMID:18719708; http://dx.doi.org/10.1038/nrm2476
- Karlsson R, Pedersen ED, Wang Z, Brakebusch C. Rho GTPase function in tumorigenesis. Biochim Biophys Acta 2009; 1796:91-8; PMID:19327386
- Raftopoulou M, Hall A. Cell migration: Rho GTPases lead the way. Dev Biol 2004; 265:23-32; PMID:14697350; http://dx.doi.org/10.1016/j.ydbio.2003.06.003
- Mettouchi A, Klein S, Guo W, Lopez-Lago M, Lemichez E, Westwick JK, Giancotti FG. Integrin-specific activation of Rac controls progression through the G(1) phase of the cell cycle. Mol Cell 2001; 8:115-27; PMID:11511365; http://dx.doi.org/10.1016/S1097-2765(01)00285-4
- Roovers K, Assoian RK. Effects of rho kinase and actin stress fibers on sustained extracellular signal-regulated kinase activity and activation of G(1) phase cyclin-dependent kinases. Mol Cell Biol 2003; 23:4283-94; PMID:12773570; http://dx.doi.org/10.1128/MCB.23.12.4283-4294.2003
- Roovers K, Klein EA, Castagnino P, Assoian RK. Nuclear translocation of LIM kinase mediates Rho-Rho kinase regulation of cyclin D1 expression. Dev Cell 2003; 5:273-84; PMID:12919678; http://dx.doi.org/10.1016/S1534-5807(03)00206-5
- Welsh CF, Roovers K, Villanueva J, Liu Y, Schwartz MA, Assoian RK. Timing of cyclin D1 expression within G1 phase is controlled by Rho. Nat Cell Biol 2001; 3:950-7; PMID:11715015; http://dx.doi.org/10.1038/ncb1101-950
- Philips A, Roux P, Coulon V, Bellanger JM, Vie A, Vignais ML, Blanchard JM. Differential effect of Rac and Cdc42 on p38 kinase activity and cell cycle progression of nonadherent primary mouse fibroblasts. J Biol Chem 2000; 275:5911-7; PMID:10681583; http://dx.doi.org/10.1074/jbc.275.8.5911
- Besson A, Assoian RK, Roberts JM. Regulation of the cytoskeleton: an oncogenic function for CDK inhibitors? Nat Rev Cancer 2004a; 4:948-55; http://dx.doi.org/10.1038/nrc1501
- Shimada Y, Gulli MP, Peter M. Nuclear sequestration of the exchange factor Cdc24 by Far1 regulates cell polarity during yeast mating. Nat Cell Biol 2000; 2:117-24; PMID:10655592; http://dx.doi.org/10.1038/35000073
- Yokoo T, Toyoshima H, Miura M, Wang Y, Iida KT, Suzuki H, Sone H, Shimano H, Gotoda T, Nishimori S, et al. p57Kip2 regulates actin dynamics by binding and translocating LIM-kinase 1 to the nucleus. J Biol Chem 2003; 278:52919-23; PMID:14530263; http://dx.doi.org/10.1074/jbc.M309334200
- Besson A, Gurian-West M, Schmidt A, Hall A, Roberts JM. p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev 2004b; 18:862-76; http://dx.doi.org/10.1101/gad.1185504
- Lee S, Helfman DM. Cytoplasmic p21Cip1 is involved in Ras-induced inhibition of the ROCK/LIMK/cofilin pathway. J Biol Chem 2004; 279:1885-91; PMID:14559914; http://dx.doi.org/10.1074/jbc.M306968200
- Tanaka H, Yamashita T, Asada M, Mizutani S, Yoshikawa H, Tohyama M. Cytoplasmic p21(Cip1/WAF1) regulates neurite remodeling by inhibiting Rho-kinase activity. J Cell Biol 2002; 158:321-9; PMID:12119358; http://dx.doi.org/10.1083/jcb.200202071
- Zhou BP, Liao Y, Xia W, Spohn B, Lee MH, Hung MC. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat Cell Biol 2001; 3:245-52; PMID:11231573; http://dx.doi.org/10.1038/35060032
- Tanaka H, Yamashita T, Yachi K, Fujiwara T, Yoshikawa H, Tohyama M. Cytoplasmic p21(Cip1/WAF1) enhances axonal regeneration and functional recovery after spinal cord injury in rats. Neuroscience 2004; 127:155-64; PMID:15219678; http://dx.doi.org/10.1016/j.neuroscience.2004.05.010
- Besson A, Dowdy SF, Roberts JM. CDK inhibitors: cell cycle regulators and beyond. Dev Cell 2008; 14:159-69; PMID:18267085; http://dx.doi.org/10.1016/j.devcel.2008.01.013
- Guo H, Tian T, Nan K, Wang W. p57: A multifunctional protein in cancer (Review). Int J Oncol 2010; 36:1321-9; PMID:20428755; http://dx.doi.org/10.3892/ijo_00000536
- Vlachos P, Joseph B. The Cdk inhibitor p57(Kip2) controls LIM-kinase 1 activity and regulates actin cytoskeleton dynamics. Oncogene 2009; 28:4175-88; PMID:19734939; http://dx.doi.org/10.1038/onc.2009.269
- Guo H, Lv Y, Tian T, Hu TH, Wang WJ, Sui X, Jiang L, Ruan ZP, Nan KJ. Downregulation of p57 accelerates the growth and invasion of hepatocellular carcinoma. Carcinogenesis 2011; 32:1897-904; PMID:22002319; http://dx.doi.org/10.1093/carcin/bgr220
- Sakai K, Peraud A, Mainprize T, Nakayama J, Tsugu A, Hongo K, Kobayashi S, Rutka JT. Inducible expression of p57KIP2 inhibits glioma cell motility and invasion. J Neurooncol 2004; 68:217-23; PMID:15332324; http://dx.doi.org/10.1023/B:NEON.0000033380.08940.c8
- Itoh Y, Masuyama N, Nakayama K, Nakayama KI, Gotoh Y. The cyclin-dependent kinase inhibitors p57 and p27 regulate neuronal migration in the developing mouse neocortex. J Biol Chem 2007; 282:390-6; PMID:17092932; http://dx.doi.org/10.1074/jbc.M609944200
- Assoian RK. Stopping and going with p27kip1. Dev Cell 2004; 6:458-9; PMID:15068785; http://dx.doi.org/10.1016/S1534-5807(04)00103-0
- Larrea MD, Wander SA, Slingerland JM. p27 as Jekyll and Hyde: regulation of cell cycle and cell motility. Cell Cycle 2009b; 8:3455-61; http://dx.doi.org/10.4161/cc.8.21.9789
- Kawauchi T, Chihama K, Nabeshima Y, Hoshino M. Cdk5 phosphorylates and stabilizes p27kip1 contributing to actin organization and cortical neuronal migration. Nat Cell Biol 2006; 8:17-26; PMID:16341208; http://dx.doi.org/10.1038/ncb1338
- Larrea MD, Hong F, Wander SA, da Silva TG, Helfman D, Lannigan D, Smith JA, Slingerland JM. RSK1 drives p27Kip1 phosphorylation at T198 to promote RhoA inhibition and increase cell motility. Proc Natl Acad Sci U S A 2009a; 106:9268-73; http://dx.doi.org/10.1073/pnas.0805057106
- Denicourt C, Saenz CC, Datnow B, Cui XS, Dowdy SF. Relocalized p27Kip1 tumor suppressor functions as a cytoplasmic metastatic oncogene in melanoma. Cancer Res 2007; 67:9238-43; PMID:17909030; http://dx.doi.org/10.1158/0008-5472.CAN-07-1375
- McAllister SS, Becker-Hapak M, Pintucci G, Pagano M, Dowdy SF. Novel p27(kip1) C-terminal scatter domain mediates Rac-dependent cell migration independent of cell cycle arrest functions. Mol Cell Biol 2003; 23:216-28; PMID:12482975; http://dx.doi.org/10.1128/MCB.23.1.216-228.2003
- Nagahara H, Vocero-Akbani AM, Snyder EL, Ho A, Latham DG, Lissy NA, Becker-Hapak M, Ezhevsky SA, Dowdy SF. Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27Kip1 induces cell migration. Nat Med 1998; 4:1449-52; PMID:9846587; http://dx.doi.org/10.1038/4042
- Wu FY, Wang SE, Sanders ME, Shin I, Rojo F, Baselga J, Arteaga CL. Reduction of cytosolic p27(Kip1) inhibits cancer cell motility, survival, tumorigenicity. Cancer Res 2006; 66:2162-72; PMID:16489017; http://dx.doi.org/10.1158/0008-5472.CAN-05-3304
- Baldassarre G, Belletti B, Nicoloso MS, Schiappacassi M, Vecchione A, Spessotto P, Morrione A, Canzonieri V, Colombatti A. p27(Kip1)-stathmin interaction influences sarcoma cell migration and invasion. Cancer Cell 2005; 7:51-63; PMID:15652749; http://dx.doi.org/10.1016/j.ccr.2004.11.025
- Diez-Juan A, Andres V. Coordinate control of proliferation and migration by the p27Kip1/cyclin-dependent kinase/retinoblastoma pathway in vascular smooth muscle cells and fibroblasts. Circ Res 2003; 92:402-10; PMID:12600894; http://dx.doi.org/10.1161/01.RES.0000059306.71961.ED
- Goukassian D, Diez-Juan A, Asahara T, Schratzberger P, Silver M, Murayama T, Isner JM, Andres V. Overexpression of p27(Kip1) by doxycycline-regulated adenoviral vectors inhibits endothelial cell proliferation and migration and impairs angiogenesis. FASEB J 2001; 15:1877-85; PMID:11532967; http://dx.doi.org/10.1096/fj.01-0065com
- Sun J, Marx SO, Chen HJ, Poon M, Marks AR, Rabbani LE. Role for p27(Kip1) in Vascular Smooth Muscle Cell Migration. Circulation 2001; 103:2967-72; PMID:11413088; http://dx.doi.org/10.1161/01.CIR.103.24.2967
- Neumeister P, Pixley FJ, Xiong Y, Xie H, Wu K, Ashton A, Cammer M, Chan A, Symons M, Stanley ER, et al. Cyclin D1 governs adhesion and motility of macrophages. Mol Biol Cell 2003; 14:2005-15; PMID:12802071; http://dx.doi.org/10.1091/mbc.02-07-0102
- Matsushime H. Macrophage cell cycle control by M-CSF/CSF-1. Rinsho Ketsueki 1995; 36:406-9; PMID:7783343
- Drobnjak M, Osman I, Scher HI, Fazzari M, Cordon-Cardo C. Overexpression of cyclin D1 is associated with metastatic prostate cancer to bone. Clin Cancer Res 2000; 6:1891-5; PMID:10815912
- Ewen ME, Sluss HK, Sherr CJ, Matsushime H, Kato J, Livingston DM. Functional interactions of the retinoblastoma protein with mammalian D-type cyclins. Cell 1993; 73:487-97; PMID:8343202; http://dx.doi.org/10.1016/0092-8674(93)90136-E
- Li Z, Wang C, Jiao X, Katiyar S, Casimiro MC, Prendergast GC, Powell MJ, Pestell RG. Alternate cyclin D1 mRNA splicing modulates p27KIP1 binding and cell migration. J Biol Chem 2008; 283:7007-15; PMID:18180298; http://dx.doi.org/10.1074/jbc.M706992200
- Li Z, Wang C, Prendergast GC, Pestell RG. Cyclin D1 functions in cell migration. Cell Cycle 2006c; 5:2440-2; http://dx.doi.org/10.4161/cc.5.21.3428
- Pestell RG. New roles of cyclin D1. Am J Pathol 2013; 183:3-9; PMID:23790801; http://dx.doi.org/10.1016/j.ajpath.2013.03.001
- Li Z, Jiao X, Wang C, Ju X, Lu Y, Yuan L, Lisanti MP, Katiyar S, Pestell RG. Cyclin D1 induction of cellular migration requires p27(KIP1). Cancer Res 2006a; 66:9986-94; http://dx.doi.org/10.1158/0008-5472.CAN-06-1596
- Arsic N, Bendris N, Peter M, Begon-Pescia C, Rebouissou C, Gadea G, Bouquier N, Bibeau F, Lemmers B, Blanchard JM. A novel function for Cyclin A2: control of cell invasion via RhoA signaling. J Cell Biol 2012; 196:147-62; PMID:22232705; http://dx.doi.org/10.1083/jcb.201102085
- Bendris N, Arsic N, Lemmers B, Blanchard JM. Cyclin A2, Rho GTPases and EMT. Small GTPases 2012; 3:225-8; PMID:22735340; http://dx.doi.org/10.4161/sgtp.20791
- Jackman M, Kubota Y, den Elzen N, Hagting A, Pines J. Cyclin A- and cyclin E-Cdk complexes shuttle between the nucleus and the cytoplasm. Mol Biol Cell 2002; 13:1030-45; PMID:11907280; http://dx.doi.org/10.1091/mbc.01-07-0361
- Bendris N, Lemmers B, Blanchard JM, Arsic N. Cyclin A2 mutagenesis analysis: a new insight into CDK activation and cellular localization requirements. PLoS One 2011; 6:e22879
- Sanchez-Beato M, Camacho FI, Martinez-Montero JC, Saez AI, Villuendas R, Sanchez-Verde L, Garcia JF, Piris MA. Anomalous high p27/KIP1 expression in a subset of aggressive B-cell lymphomas is associated with cyclin D3 overexpression. p27/KIP1-cyclin D3 colocalization in tumor cells. Blood 1999; 94:765-72; PMID:10397744
- Bouchard C, Thieke K, Maier A, Saffrich R, Hanley-Hyde J, Ansorge W, Reed S, Sicinski P, Bartek J, Eilers M. Direct induction of cyclin D2 by Myc contributes to cell cycle progression and sequestration of p27. EMBO J 1999; 18:5321-33; PMID:10508165; http://dx.doi.org/10.1093/emboj/18.19.5321
- Perez-Roger I, Kim SH, Griffiths B, Sewing A, Land H. Cyclins D1 and D2 mediate myc-induced proliferation via sequestration of p27(Kip1) and p21(Cip1). EMBO J 1999; 18:5310-20; PMID:10508164; http://dx.doi.org/10.1093/emboj/18.19.5310
- Wang YF, Chen JY, Chang SY, Chiu JH, Li WY, Chu PY, Tai SK, Wang LS. Nm23-H1 expression of metastatic tumors in the lymph nodes is a prognostic indicator of oral squamous cell carcinoma. Int J Cancer 2008; 122:377-86; PMID:17918157
- Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer 2002; 2:442-54; PMID:12189386; http://dx.doi.org/10.1038/nrc822
- Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell 2008; 14:818-29; PMID:18539112; http://dx.doi.org/10.1016/j.devcel.2008.05.009
- Thiery JP, Lim CT. Tumor dissemination: an EMT affair. Cancer Cell 2013; 23:272-3; PMID:23518345; http://dx.doi.org/10.1016/j.ccr.2013.03.004
- Bendris N, Cheung CT, Leong HS, Lewis JD, Chambers AF, Blanchard JM, Lemmers B. Cyclin A2, a novel regulator of EMT. Cell Mol Life Sci; 2014; PMID:24879294
- Bellovin DI, Simpson KJ, Danilov T, Maynard E, Rimm DL, Oettgen P, Mercurio AM. Reciprocal regulation of RhoA and RhoC characterizes the EMT and identifies RhoC as a prognostic marker of colon carcinoma. Oncogene 2006; 25:6959-67; PMID:16715134; http://dx.doi.org/10.1038/sj.onc.1209682
- Sahai E, Marshall CJ. RHO-GTPases and cancer. Nat Rev Cancer 2002; 2:133-42; PMID:12635176; http://dx.doi.org/10.1038/nrc725
- Hakem A, Sanchez-Sweatman O, You-Ten A, Duncan G, Wakeham A, Khokha R, Mak TW. RhoC is dispensable for embryogenesis and tumor initiation but essential for metastasis. Genes Dev 2005; 19:1974-9; PMID:16107613; http://dx.doi.org/10.1101/gad.1310805
- Anastasiadis PZ, Moon SY, Thoreson MA, Mariner DJ, Crawford HC, Zheng Y, Reynolds AB. Inhibition of RhoA by p120 catenin. Nat Cell Biol 2000; 2:637-44; PMID:10980705; http://dx.doi.org/10.1038/35023588
- Grosheva I, Shtutman M, Elbaum M, Bershadsky AD. p120 catenin affects cell motility via modulation of activity of Rho-family GTPases: a link between cell-cell contact formation and regulation of cell locomotion. J Cell Sci 2001; 114:695-707; PMID:11171375
- Loukil A, Zonca M, Rebouissou C, Baldin V, Coux O, Biard-Piechaczyk M, Blanchard JM, Peter M. High-resolution live-cell imaging reveals novel cyclin A2 degradation foci involving autophagy. J Cell Sci 2014; 127:2145-50; PMID:24634511; http://dx.doi.org/10.1242/jcs.139188
- Barger JF, Plas DR. Balancing biosynthesis and bioenergetics: metabolic programs in oncogenesis. Endocr Relat Cancer 2010; 17:R287-304; PMID:20699334; http://dx.doi.org/10.1677/ERC-10-0106
- Dang CV. Links between metabolism and cancer. Genes Dev 2012; 26:877-90; PMID:22549953; http://dx.doi.org/10.1101/gad.189365.112
- DeBerardinis RJ, Thompson CB. Cellular metabolism and disease: what do metabolic outliers teach us? Cell 2012; 148:1132-44; PMID:22424225; http://dx.doi.org/10.1016/j.cell.2012.02.032
- Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 2012; 21:297-308; PMID:22439925; http://dx.doi.org/10.1016/j.ccr.2012.02.014
- Mehlen P, Puisieux A. Metastasis: a question of life or death. Nat Rev Cancer 2006; 6:449-58; PMID:16723991; http://dx.doi.org/10.1038/nrc1886
- Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol 2006; 7:131-42; PMID:16493418; http://dx.doi.org/10.1038/nrm1835