
Abstract
Kinase signaling networks are well-established mediators of cell cycle transitions. However, how kinases interact with the ubiquitin proteasome system (UPS) to elicit protein turnover is not fully understood. We sought a means of identifying kinase-substrate interactions to better understand signaling pathways controlling protein degradation. Our prior studies used a luciferase fusion protein to uncover kinase networks controlling protein turnover. In this study, we utilized a similar approach to identify pathways controlling the cell cycle protein p27Kip1. We generated a p27Kip1-luciferase fusion and expressed it in cells incubated with compounds from a library of pharmacologically active compounds. We then compared the relative effects of the compounds on p27Kip1-luciferase fusion stabilization. This was combined with in silico kinome profiling to identify potential kinases inhibited by each compound. This approach effectively uncovered known kinases regulating p27Kip1 turnover. Collectively, our studies suggest that this parallel screening approach is robust and can be applied to fully understand kinase-ubiquitin pathway interactions.
Abbreviations
| PMA | = | Phorbol 12-myristate 13-acetate |
| S/B | = | Signal to background ratio |
| Z′ | = | Z factor |
Introduction
Cell cycle transitions are finely controlled to ensure faithful transmission of genetic material. For instance, cyclin-dependent kinases are regulated to elicit unidirectional entry into S phase or mitosis. Cyclin-dependent kinase (CDK) activity is inhibited via a class of cyclin-dependent kinase inhibitors, or CKIs. CKIs bind to cyclin-dependent kinases, thereby inhibiting CDK-dependent phosphorylation of target proteins. In turn, CKI levels are controlled via ubiquitin-dependent degradation.Citation1-6
p27Kip1 is a key CKI at the G1/S checkpoint that binds and inhibits cyclin-CDK2 and –CDK4 complexes, causing cell cycle arrest.Citation7-9 p27Kip1 protein levels are high during G1/G0, and decrease rapidly upon transition to S phase. Contact inhibition and mitogen deprivation in vitro induce increases in p27Kip1 levels while mitogen stimulation causes p27Kip1 levels to drop.Citation10-17 Overexpression of either a wild-type or a degradation-resistant version of p27Kip1 inhibits cell cycle transit in vitro.Citation8,17-19 By contrast, antisense inhibition of p27Kip1 prevents cell cycle arrest induced by mitogen depletion,Citation14 and full or partial deletion of p27Kip1 in vivo increases cell proliferation and induces organ hypertrophy and tumorigenesis.Citation20-22
Importantly, although p27Kip1 levels have been shown to correlate with prognosis in multiple human cancers,Citation23-28 mutations in the p27Kip1 gene are rare.Citation29-37 This observation led to the realization that p27Kip1 protein levels are likely controlled by translation, protein sequestration, and post-translational modifications, including phosphorylation.Citation7,38 Phosphorylation of p27Kip1 on T187 by CDK2 was shown to be necessary for recognition by Skp2 and subsequent degradation via the ubiquitin-proteasome system (UPS).Citation19,38-40 Similarly, multiple other phosphorylation sites on the p27Kip1 protein have been implicated in modulating protein stability. For instance, phosphorylation at either S10 or T198 has actually been shown to increase the stability of p27Kip1.Citation41,42 In all, 7 phosphorylation sites on the p27Kip1 protein have been described. These include S10, Y74, Y88, Y89, T157, T187, and T198. Given the role of p27Kip1 in controlling the cell cycle and tumorigenesis, uncovering the putative kinases mediating p27Kip1 phosphorylation at these sites is critical.
To identify kinases controlling p27Kip1 turnover, we adapted a protocol we utilized to identify kinases controlling degradation of the cell cycle regulator Wee1.Citation43 Like p27Kip1, phospho-Wee1 is targeted for degradation via an SCF ubiquitin ligase. To identify possible kinases mediating Wee1 recognition by the ligase, we generated a Wee1-luciferase fusion protein, which we expressed in cells treated with putative small molecule kinase inhibitors. We identified one small molecule that selectively stabilized Wee1 relative to other luciferase fusion proteins. When we profiled this compound against 300 kinases, we found that it selectively inhibits CK1δ activity. We then showed that CK1δ phosphorylates Wee1 on N-terminal residues required for Wee1 turnover.Citation43
Thus, our prior studies provided a successful workflow for identifying kinases that interact with cell cycle proteins and induce their subsequent degradation. However, these studies were dependent upon in vitro profiling of small molecules to determine their ability to inhibit or activate kinases. Here we demonstrate that in silico profiling of kinases inhibited by small molecules that also stabilize protein-luciferase fusions can identify signaling pathways controlling turnover of cell cycle proteins. Utilizing this approach we demonstrate that the PKC activator PMA stabilizes p27Kip1-luciferase.
Results
Luciferase fusion proteins are stabilized by proteasomal inhibition
Several studies suggested that luciferase fusion proteins serve as accurate reporters of endogenous proteins.Citation44-48 In the case of cell cycle proteins, the N-terminus of cyclin B1 has been fused to luciferase and utilized in small molecule screens to identify small molecule inhibitors of the ubiquitin ligase APC/C, which controls cyclin B1 turnover.Citation44 Similarly, our prior studies have developed and characterized a luciferase fusion of the cell cycle kinase Wee1 to identify small molecule inhibitors that specifically affect endogenous Wee1 turnover.Citation45 In the case of Wee1, the small molecules inhibited kinases we described as novel regulators of Wee1 turnover.Citation45 Since kinases are known to control degradation of cell cycle proteins, we asked whether we could adapt our prior strategy to identify kinases controlling degradation of p27Kip1. To do this, we created a luciferase fusion of p27Kip1 for use in small molecule screens to identify kinases controlling endogenous p27Kip1 turnover (). We then determined whether this fusion protein is degraded via the UPS by measuring its steady-state level in the presence of the proteasome inhibitor MG132. As we had done previously for Wee1-luciferase, we transfected the construct encoding p27Kip1-luciferase into HeLa cells and added either the proteasome inhibitor MG132 or 0.1% DMSO (vehicle control). As shown in , MG132 increased the steady-state level of p27Kip1-luciferase. Further, we found that the screen was robust since it yielded an average Z′ of 0.58 +/− 0.13 and a S/B of 4.7 +/− 0.4. Only one plate out of 4 did not pass Z′ using Z′ > 0.5 as a validation criteria ().
Figure 1. p27Kip1-luciferase is stabilized by compounds from the Library of Pharmacologically Active Compounds and MG132. (A) Diagram of p27Kip1-luciferase used in our studies. Note that luciferase is attached to the C-terminus of full-length human p27Kip1. (B) HeLa cells were transfected with p27Kip1-PGL4 plasmids and incubated with either MG132 or DMSO (vehicle control). After 24 hours, cells were lysed using BriteLite and the steady-state levels of luciferase measured. Results are from one representative experiment performed in quadruplicate. (C) p27Kip1-luciferase is stabilized by compounds in the Library of Pharmacologically Active Compounds (LOPAC). HeLa cells were transfected with the p27Kip1-luciferase plasmid and incubated with compounds from a library of pharmacologically active compounds (LOPAC). Cells were then lysed after 24 hours and the steady-state levels of luciferase measured. The hit-cutoff based on the average +3 SD calculation method was 73.16%. Based on this cutoff, 21 compounds were designated as hits, i.e., a hit rate of 1.64% (21/1280), which is in the expected range of 1–2% when screening a library of pharmacologically active compounds (otherwise a hit rate of <1% is expected). Further, there were no false positives among the DMSO wells and therefore the assay is not prone to false positives. The plate-to-plate variability of the raw luminescence values was kept below <15% (12% CV for DMSO control from one plate to another, 8% CV for the MG132 controls).
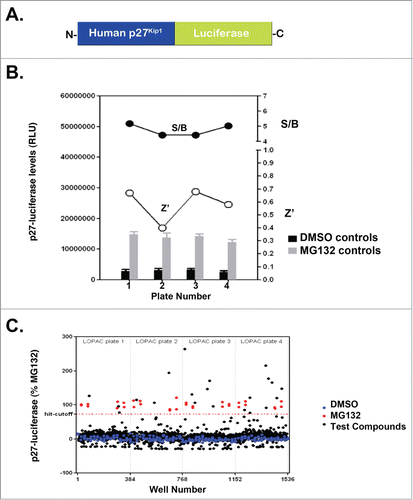
Screening of LOPAC identifies compounds stabilizing luciferase fusion proteins
The observation that MG132 stabilized p27Kip1-luciferase suggested that our screening approach could identify small molecules that inhibit degradation of this luciferase fusion protein. To identify such molecules, we expressed p27Kip1-luciferase in HeLa cells and added small molecules from the library of pharmacologically active compounds (LOPAC). We chose this library since it contains a diverse set of compounds targeting different biological processes. As shown in , several small molecules stabilized p27Kip1-luciferase to the same extent as MG132. When we rank-ordered these compounds based on stabilization relative to MG132, we found that phorbol 12-myristate 13-acetate (PMA) was one of the compounds that stabilized p27Kip1-luciferase to the greatest extent (File S1).
PMA stabilizes multiple luciferase fusion proteins
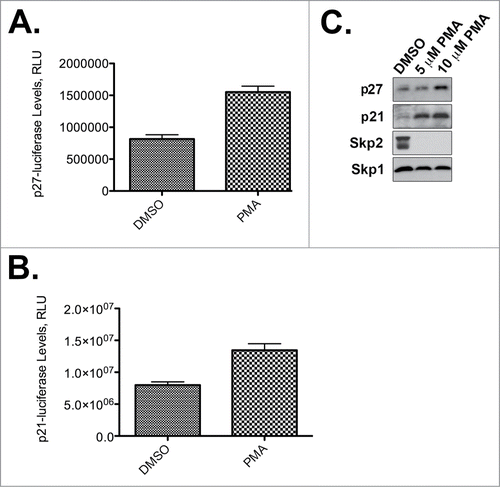
Our screening and kinome prediction data suggest that the PKC activator PMA stabilizes p27Kip1-luciferase. To validate our screening results further, we independently tested the effect of PMA on the steady-state levels of p27Kip1-luciferase. We found that PMA does indeed stabilize p27Kip1-luciferase (), but, importantly, also stabilizes endogenous p27Kip1 (). Furthermore, PMA increases the levels of both p21Cip1-luciferase and endogenous p21Cip1 (). Because Skp2 is one of the E3 ubiquitin ligases for both p27Kip1 and p21Cip1, we investigated whether PMA might modulate Skp2 activity. Interestingly, both 5 mM and 10 mM PMA were sufficient to decrease endogenous Skp2 to almost undetectable levels by Western analysis ().
Figure 2. PMA stabilizes p27Kip1 and p21Cip1. (A) PMA stabilizes p27Kip1-luciferase. Results are from one representative experiment performed in quadruplicate. (B) PMA stabilizes p21Cip1-luciferase. Results are from one representative experiment performed in quadruplicate. (C) PMA stabilizes endogenous p21Cip1 and p27Kip1 and causes degradation of Skp2. HeLa cells were incubated with the indicated concentrations of PMA and processed for p21Cip1, p27Kip1, Skp2, or Skp1 immunoblotting. Skp1 is a loading control.
In silico kinase profiling of LOPAC libraries identifies putative signaling pathways controlling luciferase fusion turnover
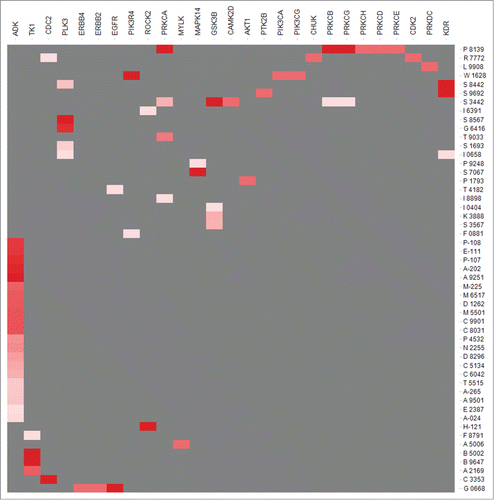
To identify signaling pathways that may modulate the steady-state levels of p27Kip1-luciferase, we measured the probability that compounds in the LOPAC library affect kinase activity. We used our previous predictive algorithm to identify the probability that any one compound either activates or inhibits a particular kinase.Citation49 As shown in and File S1, several compounds in the LOPAC library likely bind and activate or inhibit kinases. Among these, PMA is a compound that is likely to bind PKCa, b, and d. Consistent with this prediction, several studies have shown that PMA potently activates PKCs.Citation41,50,51 Similarly, kenpaullone and indirubin-3′-oxime were predicted to bind GSK3β, on upstream regulator of many cell cycle proteins. Both kenpaullone and indirubin-3′-oxime have previously been implicated in interactions with GSK3b and CDKs.Citation52-56 These results suggest that our prediction algorithm can be supported by experimental results.
Figure 3. Estimated probability of a compound (right) being active (for 51 LOPAC compounds) against a set of kinases (top). The color intensity represents the probability value. Red is highest probability. P8139 refers to PMA.
Discussion
Our studies provide a robust means of identifying kinase pathways controlling degradation of cell cycle proteins. We have demonstrated that luciferase fusions of p27Kip1, p21Cip1 and, previously, Wee1 can be used as reporters of endogenous protein turnover. Each of these fusion proteins was sensitive to MG132 proteasome inhibition, demonstrating that the UPS contributes to their turnover. Furthermore, PMA stabilized p27Kip1-luciferase, which is consistent with prior studies linking PMA to both PKC activation and p27Kip1 stabilization.Citation41,50,51 For instance, in 2012, De Vita et al. demonstrated that PMA activates PKCα to phosphorylate p27Kip1 and thereby increase protein stability.Citation41 Consistent with these findings, an algorithm that predicts the probability that a particular compound will bind a kinase also identified PMA as a likely regulator of PKC.
Our data, however, indicate that PMA may also inhibit degradation of other cell cycle proteins, as it also stabilized p21Cip1-luciferase. Like p27Kip1, p21Cip1 is phosphorylated to promote its proteasomal degradation (). The mechanism involves phosphorylation of the p21Cip1 protein on T145 by Pim-1.Citation57,58 However, PMA induces Pim-1 expression and, therefore, would be expected to promote p21Cip1 degradation, not stabilization as we observed.Citation59 This stabilization may instead be explained by alternative mechanisms of p21Cip1 regulation. Previous studies have suggested that PMA induces upregulation of Kruppel-like transcription factor 6 (KLF6) which, in turn, upregulates transcription of p21Cip1.Citation60 Therefore, PMA-induced increases in p21Cip1 may indicate that transcriptional modulation plays a significant role in regulating p21Cip1activity. However, given our finding that PMA induces significant Skp2 downregulation, it is likely that p21Cip1 protein stabilization is a secondary result of the decline in Skp2 activity. Skp2 is a common E3 ubiquitin ligase for p27Kip1 and p21Cip1. Therefore, a decrease in Skp2 activity would not only stabilize p27Kip1, but also p21Cip1. Whether PMA causes Skp2 downregulation in a p27Kip1-independent manner is not known, but evidence does exist to support the hypothesis that PMA stabilizes p27Kip1 independently of Skp2 inhibition.Citation41 Consistent with this possibility, we find that Skp2 inhibitors do not stabilize p27Kip1-luciferase. This may suggest that the second ubiquitin ligase, KPC, known to modulate p27Kip1 levels may be operational in our system.Citation61
Figure 4. Model of kinase and proteolysis pathways controlling p21Cip1 and p27Kip1 degradation.
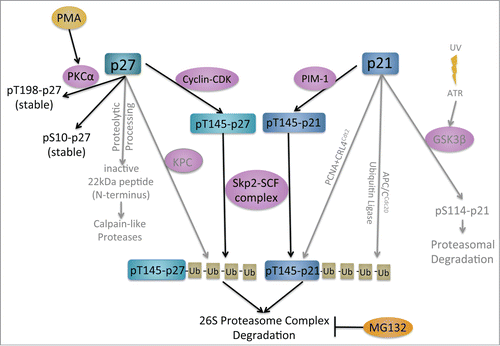
It is possible that PMA activates PKCs to phosphorylate and stabilize p27Kip1. Increased p27Kip1 levels could then cause cell cycle arrest in G1, degradation of Skp2, and stabilization of p21Cip1. Conversely, it is possible that PMA causes the degradation of Skp2, and the loss of Skp2 activity could increase p27Kip1 and p21Cip1 stability. The question of which comes first is difficult to deduce from these cell-based screening approaches because of the interdependence of these proteins and the cell cycle. However, it is known that PMA activates PKCs and PKCα phosphorylates and stabilizes p27Kip1.Citation41 An interaction between PMA and Skp2 has not been reported.
In addition to PMA, the small molecules kenpaullone and indirubin-3′-oxime stabilized p21Cip1-luciferase and p27Kip1-luciferase (File S1). Our in silico prediction suggested that kenpaullone and indirubin-3′-oxime are both potent inhibitors of CDKs and GSK3b. In addition, several studies suggested that kenpaullone and indirubin-3′-oxime are CDK/GSK3b inhibitors.Citation53,54,56,62,63 Further, CDKs have been implicated in p27Kip1 turnover and GSK3b has been shown to control degradation of several cell cycle proteins.Citation52,55 These findings further support that our workflow can identify kinases implicated in cell cycle protein turnover.
One major consideration with our workflow is that compounds may be acting indirectly to stabilize cell cycle protein-luciferase fusions. For instance, one important possibility to rule out is that PMA, kenpaullone, and indirubin-3′-oxime may be acting not on the cell-cycle proteins, but the fused luciferase enzyme. However, these compounds do not affect luciferase as determined after searching the PubChem database. Another caveat with our screening approach is that compounds may arrest cells in a phase where the luciferase fusion proteins are most stable. This would confound the interpretation that the stabilization of the fusion proteins observed with small molecule treatment was due to interference with their degradation. Rather, some other phase-specific factor may be acting independently of the compound itself. However, in the case of PMA, convincing evidence exists that its mechanism of action includes activation of PKCα and phosphorylation of p27Kip1 to decrease its degradation and cause cell cycle arrest.Citation41
One means to identify novel kinases controlling phosphorylation of p27Kip1 is to screen larger compound libraries. Our workflow can be adapted to include more compounds in order to identify multiple kinases inducing protein turnover. The compounds tested in the initial luciferase-fusion protein screen were limited to the 1280 compounds included in the LOPAC. With the screening of increasing numbers of compounds, one may expect to uncover additional interactions. However, by screening known pharmacologically active compounds, the probability of finding interactions is already many-fold greater than libraries that have not been built on known pharmacological activity. Collectively, our studies suggest that similar parallel screenings of cell-cycle fusion proteins may identify kinase cascades controlling protein turnover.
Materials and Methods
BriteLite assays
In a 50 mL conical, 87 uL TransIT-LT1 Transfection Reagent (Mirus Bio, MIR2300) was mixed with 29 ug of luciferase conjugated-protein plasmid DNA and OptiMEM I Reduced-Serum Medium up to a final volume of 12 mL. This mixture incubated at room temperature for 20 minutes to allow complexes to form. In a second conical, 6 × 106 HeLa cells were resuspended in a final volume of 12 mL complete medium (10% FBS, 1% penicillin/streptomycin, DMEM). The two conicals were then combined into a flask and incubated 48 hrs, 37°C, 10% CO2 in cell culture incubator. Transfected cells were then resuspended, passed through a cell filter, and counted. Complete medium was used to dilute the cells to 8.5 × 105 cells/mL and 50 μL of this suspension was added to each well of a 96-well plate. An additional 50 μL of 20 mM drug was added for a final concentration of 10 uM, and the cells were returned to the incubator overnight. Finally, 100 μL of reconstituted BriteLite plus (Perkin Elmer, #6066761) was added to each well and mixed well before reading on a Perkin Elmer EnVision 2104 Multilabel Reader.
In silico kinome profiling
For the purpose of computational kinase profiling, we used Laplacian-corrected naïve Bayesian classification models previously build in house.Citation49 The models were based on the Kinase Knowledge Base (KKB) data, including almost 500,000 tested kinase/compound pairs extracted from literature and patents. Data were subject to rigorous aggregation, standardization, and clustering procedures that resulted in over 180 distinct data sets covering all major groups of the human kinome. For each kinase in the data set, active and inactive compounds were described using extended connectivity fingerprints. Compounds considered inactive in the model training were either known inactives of a given kinase, or taken as the entire set of molecules not tested on that kinase. Rigorous cross-validation and characterization was performed and demonstrated highly predictive classification and quantitative models for the majority of kinase targets if a minimum required number of active compounds or structure-activity data points were available.
For the purposes of this study, we selected models based on 25 data points with 10 being active and applied them to the LOPAC library to predict compound kinase profiles. We identified 72 compounds with the estimated probability that the compound is in the active category (EstPGood) of 0.05 (data provided in Supporting Material), while 51 compounds had an EstPGood value above 0.10. These 51 compounds are shown in .
PAM
In order to identify PAM screening results from PubChem,Citation64 we performed "Phorbol 12-myristate 13-acetate" search in the PubChem compound field. Three unique CIDs were identified with 2 corresponding to the 2 PAM stereoisomers (CID 27924 and CID 122634) while the third compound (CID 4792) has no stereochemistry annotated. All corresponding SIDs were identified and collected. That resulted in the total of 126 unique SIDs. The SIDs were matched to the previously annotated luciferase assaysCitation65,66 and 20 screening data points were identified.
Similarly, kenpaullone was also identified via PubChem compound filed and the search resulted in one compound (CID 3820) and 75 SIDs. They were compared to the luciferase assay data and 69 data points were identified.
For Indirubin-3′-oxime the PubChem CID 5326739 was identified in the same way as described above. The CID corresponded to 101 SIDs and 71 data points in the luciferase assays.
Protein extract preparation, antibodies and western blot
Cells were homogenized and extracts were prepared using lysis buffer (50 mM Tris, 150 mM NaCl, 1 % Triton X- 100, 1× Protease Inhibitor Cocktail, 1 μM Microcystin LR). Cells were lysed by the freeze-thaw method (liquid nitrogen/37°C water bath) and further sonicated. The soluble fraction was recovered by centrifugation at 14,000 RPM for 20 min at 4°C. Protein concentration was measured with the BCA Protein Assay kit (Pierce) and 30 μg of protein from each sample was resolved by SDS-PAGE. The resolved bands were transferred onto a nitrocellulose membrane by Western blotting and then probed with relevant antibodies. Primary Antibodies: anti-p21 (Abcam, ab7960), anti-p27 (Santa Cruz Biotechnology, sc-776), anti-Skp1 p19 antibody (H-163, Santa Cruz Biotechnology, sc-7163); secondary antibody: anti-rabbit IgG-HRP antibody from GE Healthcare (Cat # NA9340V).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
1006987_Supplementary_Materials.zip
Download Zip (143.7 KB)Acknowledgments
We thank all members of the Center for Therapeutic Innovation and the Miami Project to Cure Paralysis for helpful suggestions.
Funding
This work was partly funded by R01NS067289 to NGA and UOIHL11561 to SCS.
Related Research Data
References
- Nurse P. Universal control mechanism regulating onset of M-phase. Nature 1990; 344:503-8; PMID:2138713; http://dx.doi.org/10.1038/344503a0
- Hartwell L. Defects in a cell cycle checkpoint may be responsible for the genomic instability of cancer cells. Cell 1992; 71:543-6; PMID:1423612; http://dx.doi.org/10.1016/0092-8674(92)90586-2
- Reed SI. The role of p34 kinases in the G1 to S-phase transition. Annu Rev Cell Biol 1992; 8:529-61; PMID:1476805; http://dx.doi.org/10.1146/annurev.cb.08.110192.002525
- Sherr CJ. Mammalian G1 cyclins. Cell 1993; 73:1059-65; PMID:8513492; http://dx.doi.org/10.1016/0092-8674(93)90636-5
- Morgan DO. Principles of CDK regulation. Nature 1995; 374:131-4; PMID:7877684; http://dx.doi.org/10.1038/374131a0
- Pines J. Cyclins and their associated cyclin-dependent kinases in the human cell cycle. Biochem Soc Trans 1993; 21:921-5; PMID:8132094
- Polyak K, Kato JY, Solomon MJ, Sherr CJ, Massague J, Roberts JM, Koff A. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev 1994; 8:9-22; PMID:8288131; http://dx.doi.org/10.1101/gad.8.1.9
- Polyak K, Lee MH, Erdjument-Bromage H, Koff A, Roberts JM, Tempst P, Massagué J. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell 1994; 78:59-66; PMID:8033212; http://dx.doi.org/10.1016/0092-8674(94)90572-X
- Russo AA, Jeffrey PD, Patten AK, Massague J, Pavletich NP. Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A-Cdk2 complex. Nature 1996; 382:325-31; PMID:8684460; http://dx.doi.org/10.1038/382325a0
- Nourse J, Firpo E, Flanagan WM, Coats S, Polyak K, Lee MH, Massague J, Crabtree GR, Roberts JM. Interleukin-2-mediated elimination of the p27Kip1 cyclin-dependent kinase inhibitor prevented by rapamycin. Nature 1994; 372:570-3; PMID:7990932; http://dx.doi.org/10.1038/372570a0
- Kato JY, Matsuoka M, Polyak K, Massague J, Sherr CJ. Cyclic AMP-induced G1 phase arrest mediated by an inhibitor (p27Kip1) of cyclin-dependent kinase 4 activation. Cell 1994; 79:487-96; PMID:7954814; http://dx.doi.org/10.1016/0092-8674(94)90257-7
- Halevy O, Novitch BG, Spicer DB, Skapek SX, Rhee J, Hannon GJ, Beach D, Lassar AB. Correlation of terminal cell cycle arrest of skeletal muscle with induction of p21 by MyoD. Science 1995; 267:1018-21; PMID:7863327; http://dx.doi.org/10.1126/science.7863327
- Poon RY, Toyoshima H, Hunter T. Redistribution of the CDK inhibitor p27 between different cyclin.CDK complexes in the mouse fibroblast cell cycle and in cells arrested with lovastatin or ultraviolet irradiation. Mol Biol Cell 1995; 6:1197-213; PMID:8534916; http://dx.doi.org/10.1091/mbc.6.9.1197
- Coats S, Flanagan WM, Nourse J, Roberts JM. Requirement of p27Kip1 for restriction point control of the fibroblast cell cycle. Science 1996; 272:877-80; PMID:8629023; http://dx.doi.org/10.1126/science.272.5263.877
- Hengst L, Reed SI. Translational control of p27Kip1 accumulation during the cell cycle. Science 1996; 271:1861-4; PMID:8596954; http://dx.doi.org/10.1126/science.271.5257.1861
- Rivard N, L'Allemain G, Bartek J, Pouyssegur J. Abrogation of p27Kip1 by cDNA antisense suppresses quiescence (G0 state) in fibroblasts. J Biol Chem 1996; 271:18337-41; PMID:8702474; http://dx.doi.org/10.1074/jbc.271.31.18337
- Vlach J, Hennecke S, Alevizopoulos K, Conti D, Amati B. Growth arrest by the cyclin-dependent kinase inhibitor p27Kip1 is abrogated by c-Myc. EMBO J 1996; 15:6595-604; PMID:8978686
- Toyoshima H, Hunter T. p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell 1994; 78:67-74; PMID:8033213; http://dx.doi.org/10.1016/0092-8674(94)90573-8
- Sutterluty H, Chatelain E, Marti A, Wirbelauer C, Senften M, Muller U, Krek W. p45SKP2 promotes p27Kip1 degradation and induces S phase in quiescent cells. Nat Cell Biol 1999; 1:207-14; PMID:10559918; http://dx.doi.org/10.1038/12027
- Fero ML, Rivkin M, Tasch M, Porter P, Carow CE, Firpo E, Polyak K, Tsai LH, Broudy V, Perlmutter RM, et al. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)-deficient mice. Cell 1996; 85:733-44; PMID:8646781; http://dx.doi.org/10.1016/S0092-8674(00)81239-8
- Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, Horii I, Loh DY, Nakayama K. Mice lacking p27(Kip1) display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell 1996; 85:707-20; PMID:8646779; http://dx.doi.org/10.1016/S0092-8674(00)81237-4
- Fero ML, Randel E, Gurley KE, Roberts JM, Kemp CJ. The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature 1998; 396:177-80; PMID:9823898; http://dx.doi.org/10.1038/24179
- Porter PL, Malone KE, Heagerty PJ, Alexander GM, Gatti LA, Firpo EJ, Daling JR, Roberts JM. Expression of cell-cycle regulators p27Kip1 and cyclin E, alone and in combination, correlate with survival in young breast cancer patients. Nat Med 1997; 3:222-5; PMID:9018243; http://dx.doi.org/10.1038/nm0297-222
- Catzavelos C, Bhattacharya N, Ung YC, Wilson JA, Roncari L, Sandhu C, Shaw P, Yeger H, Morava-Protzner I, Kapusta L, et al. Decreased levels of the cell-cycle inhibitor p27Kip1 protein: prognostic implications in primary breast cancer. Nat Med 1997; 3:227-30; PMID:9018244; http://dx.doi.org/10.1038/nm0297-227
- Mori M, Mimori K, Shiraishi T, Tanaka S, Ueo H, Sugimachi K, Akiyoshi T. p27 expression and gastric carcinoma. Nat Med 1997; 3:593; PMID:9176477; http://dx.doi.org/10.1038/nm0697-593
- Fredersdorf S, Burns J, Milne AM, Packham G, Fallis L, Gillett CE, Royds JA, Peston D, Hall PA, Hanby AM, et al. High level expression of p27(kip1) and cyclin D1 in some human breast cancer cells: inverse correlation between the expression of p27(kip1) and degree of malignancy in human breast and colorectal cancers. Proc Nat Acad Sci U S A 1997; 94:6380-5; PMID:9177226
- Tan P, Cady B, Wanner M, Worland P, Cukor B, Magi-Galluzzi C, Lavin P, Draetta G, Pagano M, Loda M. The cell cycle inhibitor p27 is an independent prognostic marker in small (T1a,b) invasive breast carcinomas. Cancer Res 1997; 57:1259-63; PMID:9102210
- Kawana H, Tamaru J, Tanaka T, Hirai A, Saito Y, Kitagawa M, Mikata A, Harigaya K, Kuriyama T. Role of p27Kip1 and cyclin-dependent kinase 2 in the proliferation of non-small cell lung cancer. Am J Pathol 1998; 153:505-13; PMID:9708810; http://dx.doi.org/10.1016/S0002-9440(10)65593-9
- Hunter T, Pines J. Cyclins and cancer. II: Cyclin D and CDK inhibitors come of age. Cell 1994; 79:573-82; PMID:7954824; http://dx.doi.org/10.1016/0092-8674(94)90543-6
- Ferrando AA, Balbin M, Pendas AM, Vizoso F, Velasco G, Lopez-Otin C. Mutational analysis of the human cyclin-dependent kinase inhibitor p27kip1 in primary breast carcinomas. Hum Genet 1996; 97:91-4; PMID:8557269; http://dx.doi.org/10.1007/BF00218840
- Ponce-Castaneda MV, Lee MH, Latres E, Polyak K, Lacombe L, Montgomery K, Mathew S, Krauter K, Sheinfeld J, Massague J, et al. p27Kip1: chromosomal mapping to 12p12-12p13.1 and absence of mutations in human tumors. Cancer Res 1995; 55:1211-4; PMID:7882310
- Pietenpol JA, Bohlander SK, Sato Y, Papadopoulos N, Liu B, Friedman C, Trask BJ, Roberts JM, Kinzler KW, Rowley JD, et al. Assignment of the human p27Kip1 gene to 12p13 and its analysis in leukemias. Cancer Res 1995; 55:1206-10; PMID:7882309
- Kawamata N, Morosetti R, Miller CW, Park D, Spirin KS, Nakamaki T, Takeuchi S, Hatta Y, Simpson J, Wilcyznski S, et al. Molecular analysis of the cyclin-dependent kinase inhibitor gene p27/Kip1 in human malignancies. Cancer Res 1995; 55:2266-9; PMID:7757974
- Kawamata N, Seriu T, Koeffler HP, Bartram CR. Molecular analysis of the cyclin-dependent kinase inhibitor family: p16(CDKN2/MTS1/INK4A), p18(INK4C) and p27(Kip1) genes in neuroblastomas. Cancer 1996; 77:570-5; PMID:8630967; http://dx.doi.org/10.1002/(SICI)1097-0142(19960201)77:3<570::AID-CNCR21>3.0.CO;2-0
- Morosetti R, Kawamata N, Gombart AF, Miller CW, Hatta Y, Hirama T, Said JW, Tomonaga M, Koeffler HP. Alterations of the p27KIP1 gene in non-Hodgkin's lymphomas and adult T-cell leukemia/lymphoma. Blood 1995; 86:1924-30; PMID:7655021
- Spirin KS, Simpson JF, Takeuchi S, Kawamata N, Miller CW, Koeffler HP. p27/Kip1 mutation found in breast cancer. Cancer Res 1996; 56:2400-4; PMID:8625318
- Stegmaier K, Takeuchi S, Golub TR, Bohlander SK, Bartram CR, Koeffler HP. Mutational analysis of the candidate tumor suppressor genes TEL and KIP1 in childhood acute lymphoblastic leukemia. Cancer Res 1996; 56:1413-7; PMID:8640833
- Vlach J, Hennecke S, Amati B. Phosphorylation-dependent degradation of the cyclin-dependent kinase inhibitor p27. EMBO J 1997; 16:5334-44; PMID:9311993; http://dx.doi.org/10.1093/emboj/16.17.5334
- Sheaff RJ, Groudine M, Gordon M, Roberts JM, Clurman BE. Cyclin E-CDK2 is a regulator of p27Kip1. Genes Dev 1997; 11:1464-78; PMID:9192873; http://dx.doi.org/10.1101/gad.11.11.1464
- Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol 1999; 1:193-9; PMID:10559916; http://dx.doi.org/10.1038/12013
- De Vita F, Riccardi M, Malanga D, Scrima M, De Marco C, Viglietto G. PKC-dependent phosphorylation of p27 at T198 contributes to p27 stabilization and cell cycle arrest. Cell Cycle 2012; 11:1583-92; PMID:22441823; http://dx.doi.org/10.4161/cc.20003
- Ishida N, Kitagawa M, Hatakeyama S, Nakayama K. Phosphorylation at serine 10, a major phosphorylation site of p27(Kip1), increases its protein stability. J Biol Chem 2000; 275:25146-54; PMID:10831586; http://dx.doi.org/10.1074/jbc.M001144200
- Penas C, Ramachandran V, Simanski S, Lee C, Madoux F, Rahaim RJ, Chauhan R, Barnaby O, Schurer S, Hodder P, et al. Casein kinase 1delta-dependent Wee1 protein degradation. J Biol Chem 2014; 289:18893-903; PMID:24817118; http://dx.doi.org/10.1074/jbc.M114.547661
- Verma R, Peters NR, D'Onofrio M, Tochtrop GP, Sakamoto KM, Varadan R, Zhang M, Coffino P, Fushman D, Deshaies RJ, et al. Ubistatins inhibit proteasome-dependent degradation by binding the ubiquitin chain. Science 2004; 306:117-20; PMID:15459393; http://dx.doi.org/10.1126/science.1100946
- Penas C, Ramachandran V, Simanski S, Lee C, Madoux F, Rahaim RJ, Chauhan R, Barnaby O, Schurer S, Hodder P, et al. Casein kinase 1delta-dependent Wee1 protein degradation. J Biol Chem 2014; 289:18893-903; PMID:24817118; http://dx.doi.org/10.1074/jbc.M114.547661
- Madoux F, Simanski S, Chase P, Mishra JK, Roush WR, Ayad NG, Hodder P. An ultra-high throughput cell-based screen for wee1 degradation inhibitors. J Biomol Screen 2010; 15:907-17; PMID:20660794; http://dx.doi.org/10.1177/1087057110375848
- Herman AG, Hayano M, Poyurovsky MV, Shimada K, Skouta R, Prives C, Stockwell BR. Discovery of Mdm2-MdmX E3 ligase inhibitors using a cell-based ubiquitination assay. Cancer Discov 2011; 1:312-25; PMID:22586610; http://dx.doi.org/10.1158/2159-8290.CD-11-0104
- Zhang GJ, Safran M, Wei W, Sorensen E, Lassota P, Zhelev N, Neuberg DS, Shapiro G, Kaelin WG Jr. Bioluminescent imaging of Cdk2 inhibition in vivo. Nat Med 2004; 10:643-8; PMID:15122251; http://dx.doi.org/10.1038/nm1047
- Schurer SC, Muskal SM. Kinome-wide activity modeling from diverse public high-quality data sets. J Chem Inform Model 2013; 53:27-38; PMID:23259810; http://dx.doi.org/10.1021/ci300403k
- Ballester R, Rosen OM. Fate of immunoprecipitable protein kinase C in GH3 cells treated with phorbol 12-myristate 13-acetate. J Biol Chem 1985; 260:15194-9; PMID:3905792
- Nishizuka Y, Nakamura S. Lipid mediators and protein kinase C for intracellular signalling. Clin Exp Pharmacol Physiol Suppl 1995; 22:S202-3; PMID:9072357; http://dx.doi.org/10.1111/j.1440-1681.1995.tb02883.x
- Ryves WJ, Harwood AJ. The interaction of glycogen synthase kinase-3 (GSK-3) with the cell cycle. Prog Cell Cycle Res 2003; 5:489-95; PMID:14593744
- Leclerc S, Garnier M, Hoessel R, Marko D, Bibb JA, Snyder GL, Greengard P, Biernat J, Wu YZ, Mandelkow EM, et al. Indirubins inhibit glycogen synthase kinase-3 beta and CDK5/p25, two protein kinases involved in abnormal tau phosphorylation in Alzheimer's disease. A property common to most cyclin-dependent kinase inhibitors? J Biol Chem 2001; 276:251-60; PMID:11013232; http://dx.doi.org/10.1074/jbc.M002466200
- Damiens E, Baratte B, Marie D, Eisenbrand G, Meijer L. Anti-mitotic properties of indirubin-3'-monoxime, a CDK/GSK-3 inhibitor: induction of endoreplication following prophase arrest. Oncogene 2001; 20:3786-97; PMID:11439342; http://dx.doi.org/10.1038/sj.onc.1204503
- Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev 1998; 12:3499-511; PMID:9832503; http://dx.doi.org/10.1101/gad.12.22.3499
- Kunick C, Lauenroth K, Leost M, Meijer L, Lemcke T. 1-Azakenpaullone is a selective inhibitor of glycogen synthase kinase-3 beta. Bioorg Med Chem Lett 2004; 14:413-6; PMID:14698171; http://dx.doi.org/10.1016/j.bmcl.2003.10.062
- Wang Z, Bhattacharya N, Mixter PF, Wei W, Sedivy J, Magnuson NS. Phosphorylation of the cell cycle inhibitor p21Cip1/WAF1 by Pim-1 kinase. Biochimica et Biophysica Acta 2002; 1593:45-55; PMID:12431783; http://dx.doi.org/10.1016/S0167-4889(02)00347-6
- Zhang Y, Wang Z, Magnuson NS. Pim-1 kinase-dependent phosphorylation of p21Cip1/WAF1 regulates its stability and cellular localization in H1299 cells. Mol Cancer Research: MCR 2007; 5:909-22; PMID:17855660; http://dx.doi.org/10.1158/1541-7786.MCR-06-0388
- Wang Z, Petersen K, Weaver MS, Magnuson NS. cDNA cloning, sequencing and characterization of bovine pim-1. Vet Immunol Immunopathol 2001; 78:177-95; PMID:11182156; http://dx.doi.org/10.1016/S0165-2427(00)00259-2
- Tahara E, Kadara H, Lacroix L, Lotan D, Lotan R. Activation of protein kinase C by phorbol 12-myristate 13-acetate suppresses the growth of lung cancer cells through KLF6 induction. Cancer Biol Ther 2009; 8:801-7; PMID:19333010; http://dx.doi.org/10.4161/cbt.8.9.8186
- Kamura T, Hara T, Matsumoto M, Ishida N, Okumura F, Hatakeyama S, Yoshida M, Nakayama K, Nakayama KI. Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27(Kip1) at G1 phase. Nat Cell Biol 2004; 6:1229-35; PMID:15531880; http://dx.doi.org/10.1038/ncb1194
- Bain J, McLauchlan H, Elliott M, Cohen P. The specificities of protein kinase inhibitors: an update. Biochem J 2003; 371:199-204; PMID:12534346; http://dx.doi.org/10.1042/BJ20021535
- Hoessel R, Leclerc S, Endicott JA, Nobel ME, Lawrie A, Tunnah P, Leost M, Damiens E, Marie D, Marko D, et al. Indirubin, the active constituent of a Chinese antileukaemia medicine, inhibits cyclin-dependent kinases. Nat Cell Biol 1999; 1:60-7; PMID:10559866; http://dx.doi.org/10.1038/9035
- Banerjee S, Lu J, Cai Q, Sun Z, Jha HC, Robertson ES. EBNA3C augments Pim-1 mediated phosphorylation and degradation of p21 to promote B-cell proliferation. PLoS Pathogens 2014; 10:e1004304; PMID:25121590; http://dx.doi.org/10.1371/journal.ppat.1004304
- Schurer SC, Vempati U, Smith R, Southern M, Lemmon V. BioAssay ontology annotations facilitate cross-analysis of diverse high-throughput screening data sets. J Biomol Scr 2011; 16:415-26; PMID:21471461; http://dx.doi.org/10.1177/1087057111400191
- Vempati UD, Przydzial MJ, Chung C, Abeyruwan S, Mir A, Sakurai K, Visser U, Lemmon VP, Schürer SC. Formalization, annotation and analysis of diverse drug and probe screening assay datasets using the BioAssay Ontology (BAO). PloS One 2012; 7:e49198; PMID:23155465; http://dx.doi.org/10.1371/journal.pone.0049198