
Abstract
Stathmin/Oncoprotein 18, a microtubule destabilizing protein, is required for survival of p53-deficient cells. Stathmin-depleted cells are slower to enter mitosis, but whether delayed mitotic entry triggers cell death or whether stathmin has a separate pro-survival function was unknown. To test these possibilities, we abrogated the cell cycle delay by inhibiting Wee1 in synchronized, stathmin-depleted cells and found that apoptosis was reduced to control levels. Synchronized cells treated with a 4 hour pulse of inhibitors to CDK1 or both Aurora A and PLK1 delayed mitotic entry and apoptosis was triggered only in p53-deficient cells. We did not detect mitotic defects downstream of the delayed mitotic entry, indicating that cell death is activated by a mechanism distinct from those activated by prolonged mitotic arrest. Cell death is triggered by initiator caspase 8, based on its cleavage to the active form and by rescue of viability after caspase 8 depletion or treatment with a caspase 8 inhibitor. In contrast, initiator caspase 9, activated by prolonged mitotic arrest, is not activated and is not required for apoptosis under our experimental conditions. P53 upregulates expression of cFLIPL, a protein that blocks caspase 8 activation. cFLIPL levels are lower in cells lacking p53 and these levels are reduced to a greater extent after stathmin depletion. Expression of FLAG-tagged cFLIPL in p53-deficient cells rescues them from apoptosis triggered by stathmin depletion or CDK1 inhibition during G2. These data indicate that a cell cycle delay in G2 activates caspase 8 to initiate apoptosis specifically in p53-deficient cells.
Abbreviations
| AURKA | = | aurora kinase A |
| NT | = | non-targeting |
| STMN | = | stathmin |
Introduction
The microtubule destabilizing protein, stathmin/Oncoprotein 18, is overexpressed in a number of cancers and has therefore has been suggested as a potential therapeutic target.Citation1,2 Stathmin destabilizes microtubules by binding soluble tubulin dimers and by promoting microtubule catastrophe, the switch from polymer growth to shortening states.Citation3 Depletion of stathmin has been shown to slow proliferation and increase cell death in cancer-derived cell linesCitation4-8 and in some studies death was observed only in p53-deficient cells.Citation10-12
The slower proliferation of stathmin-depleted cells is likely the result of slower progression through the cell cycle as well as loss of cells by apoptosis. We previously demonstrated that stathmin-depleted cells spend about 4 hours longer in interphase and that this delay occurs during G2.Citation11,12 Stathmin depletion acts upstream of Aurora A and PLK1 activation at the centrosome, partially reducing the activity of these 2 enzymes, which then reduces the levels of active CDC25 and CDK1, and delays entry into mitosis.Citation13 Depolymerizing interphase microtubules abrogates the delay by restoring PLK1 activity,Citation13 indicating that stathmin depletion slows cell cycle progression at least in part via stabilization of the interphase microtubule cytoskeleton. Once cells enter mitosis, they proceed through mitosis with durations indistinguishable from cells transfected with non-targeting siRNA.Citation11,12 We also did not find a correlation between mitotic duration and the subsequent interphase duration, indicating that the longer time in G2 is not due to a previously slower mitosis.Citation12 The normal mitotic duration observed in stathmin depleted cells is consistent with previous reports that stathmin must be phosphorylated and turned off as a microtubule destabilizer for proper assembly of the mitotic spindleCitation14,15 and indicating that stathmin is not required for mitosis.
While stathmin depletion leads to slower entry into mitosis and cell death in p53-deficient cells,Citation9–13 the mechanism by which stathmin depletion leads to cell death is not yet known, nor is it understood why apoptosis is confined to p53-deficient cells. Here we asked whether stathmin-depletion activates apoptosis by delaying the timing of mitotic entry. We found that inhibiting Wee1 kinase abrogates the G2 delay in stathmin-depleted cells and reduces cell death to background levels. Mimicking the delayed mitotic entry by treating synchronized cells with a 4 hour pulse of inhibitors to either CDK1, or to both Aurora A and PLK1, also induced apoptosis in both Hela and HCT116 p53−/− cells. Cell death occured at random times after the delayed entry into M and not during the G2 delay itself. The delayed entry into M phase did not produce significant mitotic errors, indicating that apoptosis was activated independent of an M phase-induced signal. A prolonged time in mitosis activates the intrinsic apoptosis pathway and the initiator caspase, caspase 9.Citation16,17 Here we show that stathmin depletion, or delayed mitotic entry by CDK1 inhibition during G2, activates initiator caspase 8. Depleting caspase 8 or chemically inhibiting its enzyme activity rescued cells from apoptosis. Together these data provide strong evidence that apoptosis is not activated by a mitotic error caused by the slower time to enter M phase. Apoptosis induced by stathmin depletionCitation10,11 or by enzymatic inhibition of mitotic entry was only observed in cells lacking detectable p53 (Hela and HCT116 p53−/− cells). We tested 2 possible mechanisms responsible for the p53-dependent cell survival in response to stathmin depletion or a mitotic entry delay. Depletion of p21, a CDK inhibitor expressed in response to active p53,Citation18 did not sensitize HCT116 p53+/+ cells to either stathmin depletion or a pulse of a CDK1 inhibitor to delay mitotic entry, indicating that apoptosis of p53-deficient cells is not caused by a lack of p21. Instead, we found that p53-deficient cells express lower levels of cFLIPL, a protein that can form heterodimers with procaspase 8 and inhibit its activation.89 We found that stathmin depletion further lowers cFLIPL level. Expression of FLAG-tagged cFLIPL is sufficient to rescue cells from apoptosis after either stathmin depletion or CDK1 inhibition during G2. Taken together, our results document a novel apoptosis trigger originating from delayed mitotic entry and generated upstream of caspase 8.
Results
A 4 hour delay during G2 is sufficient to induce cell death in p53-deficient cells
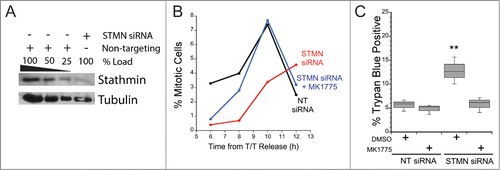
Stathmin depletion has been shown to lengthen G2 by about 4 hoursCitation12,13 and to activate apoptosis in p53-deficient cells.Citation10,11 However these studies did not address whether the 2 phenotypes are linked, where delaying mitotic entry activates cell death, or whether stathmin depletion activates a separate pathway to initiate apoptosis. To differentiate between these possibilities, we asked whether eliminating the cell cycle delay in stathmin-depleted cells is sufficient to rescue cells from death. To eliminate the cell cycle delay at mitotic entry, we reasoned that inhibiting Wee 1 kinase (a kinase that inhibits CDK1 activation) should compensate for the reduced activation of AURKA and PLK1 in these cellsCitation13 and allow CDK1 activation with normal timing.Citation19,20 Hela cells were synchronized and transfected with siRNA targeting stathmin or with a control siRNA as described in Methods (; see also refs 11–13). Cells were released from a double thymidine block and placed into medium supplemented with a Wee 1 kinase inhibitor (10 nM MK1775) or DMSO. Wee 1 kinase inhibition was sufficient to restore the normal timing of mitotic entry in stathmin-depleted cells (), which allowed us to ask whether reduced stathmin level could induce death in the absence of a cell cycle delay. We synchronized control or stathmin-depleted Hela cells, released into medium supplemented with either MK1775 or DMSO, and measured cell viability 48 hours following release. We found that Wee 1 inhibition restored viability in stathmin-depleted cells to that of control cells, indicating that the mitotic entry delay is necessary for stathmin depletion to trigger cell death ().
Figure 1. Stathmin-depleted cells only die if they are delayed in entering mitosis. Hela cells were synchronized with a double thymidine block and transfected with non-targeting siRNA (NT siRNA) or stathmin (STMN) siRNA, and released into medium containing DMSO or the Wee 1 inhibitor MK1775. (A) Western blot of stathmin knockdown, reprobed for tubulin as a loading control. Stathmin level was reduced by approximately 75% compared to non-targeting siRNA transfected cells as shown previously.Citation11–13 (B) Mitotic index was determined at 2 hour intervals for 12 hours following release from the double thymidine block. Wee1 inhibition restored mitotic entry timing in stathmin-depleted cells to that of control treated cells. (C) Viability was assayed at 48 hours after the second thymidine release via trypan blue exclusion. Relieving the mitotic entry delay in stathmin-depleted cells restored viability to that of control treated cells. ** denotes P < 0.01.
To confirm the above conclusion and eliminate the unlikely possibility that Wee1 inhibition is also a pro-survival signal, we asked whether delaying mitotic entry was sufficient to induce apoptosis in synchronized cell populations pulsed with enzyme inhibitors to prevent temporarily entry into M phase. Cells were synchronized and after release from the second thymidine block were incubated in either a combination of S1451 (300 nM) and BI2536 (0.8 nM) to partially inhibit both Aurora kinase A (AURKA) and PLK1, or 10 μM RO3306 to inhibit CDK1. These inhibitor concentrations are sufficient to inhibit mitotic entry.Citation13,21,22 Cells were kept in inhibitors for 4 hrs to delay mitotic entry, then placed in fresh medium to allow cell cycle progression. Cells were followed over the next 72 hours via live cell imaging and the percent of cells that died in each treatment group was determined from the image series. Cell death was marked by cell retraction and formation of apoptotic bodies (). We found that the group of cells treated with inhibitors for 4 hrs had a 3–4-fold increase in cell death (p < 0.01) over DMSO treated control cells (). Additionally, we confirmed that the increase in cell death was due to inhibition of CDK1 prior to mitotic entry and not elsewhere in the cell cycle. We treated asynchronously growing Hela or HCT116 p53−/− cells with the CDK1 inhibitor (RO3306) for 4 hours and assessed viability 48 hours later by trypan blue exclusion (). For asychronous cell populations, incubation in 10 μM RO3306 did not induce cell death over that measured in DMSO treated control cells, indicating that CDK1 inhibition causes cell death only when administered prior to mitotic entry.
Figure 2. A mitotic entry delay triggers cell death in p53-deficient cells. Hela or HCT116 cells were synchronized with a double thymidine block protocol, released and pulsed with either DMSO, a combination of S1451 (300 nM; AURKA inhibitor) and BI2536 (0.8 nM; PLK1 inhibitor), or RO3306 (10 μM; CDK1 inhibitor) for 4 hours beginning 6 hours after the second release. Cell viability was measured by morphological changes recorded from phase contrast images or trypan blue exclusion. (A) Representative phase contrast images of a cell undergoing apoptosis following a mitotic entry delay. Time, in minutes, is given in each frame from an arbitrary point prior to cell retraction. Scale bar is 10 μm. (B) Cells were followed by phase contrast imaging for 48–72 hrs after the 4 hr drug inhibitor pulse and cell death measured by morphological changes as shown in (A). The mitotic entry delay induced by either a combination of 300 nM S1451 and 0.8 nM BI2536 or 10 μM RO33306 significantly increased the percentage of cells that died within 72 hours after the drug pulse. (C) Asynchronously growing Hela or HCT116 p53−/− cells were treated with a 4 hour pulse of inhibitors and followed by live cell recordings as in (A, B). Treatment with the combination of 300 nM S1451 and 0.8 nM BI2536 or with 10 μM RO33306 in asynchronously growing cell populations did not decrease cell viability, demonstrating that the drugs are not simply toxic throughout the cell cycle. (D) Synchronized HCT116 p53+/+ and p53−/− cell lines were pulsed with 10 μM RO3306 as in (B). Viability was assayed 48 hours post inhibitor treatment via trypan blue exclusion. A mitotic entry delay via CDK1 inhibition decreased cell viability only in the p53 knockout cell line. Graphs are representative of at least 3 independent experiments with >300 cells/experiment. ** denotes P < 0.01, *** denotes P < 0.001.
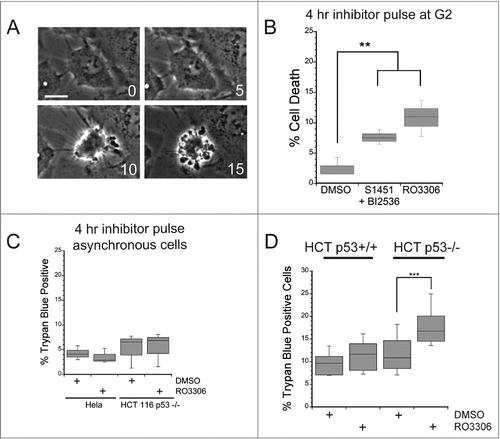
Since it was previously shown that stathmin depletion leads to death only in cells lacking p53,Citation10,11 we next asked whether the death induced by a 4 hour delay in mitotic entry was also dependent on the absence of p53. Using isogenic colorectal cancer cell lines differing in p53 genotype, HCT116 p53+/+ and p53−/−, we synchronized cells with a double thymidine block and pulsed with either DMSO or 10 μM RO3306 for 4 hours beginning 6 hours after release from the block. At 48 hours after the drug pulse, a time at which we observed the most death in the time-lapse imaging experiments in Hela cells, we assayed viability by trypan blue exclusion. We found that only the HCT116 p53−/− line pulsed with 10 μM RO3306 had a significant decrease in viability compared to DMSO pulsed control cells (). We previously showed that stathmin depletion leads to cell death in the HCT116 p53−/− cell line but not in the parental HCT116p53+/+ cell line.Citation11
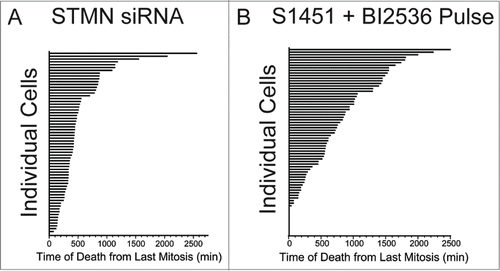
From examination of the live cell image series it appeared that individual cells died asynchronously over the course of several days following drug inhibitor pulses. To quantify this observation and compare to the timing in stathmin depleted cells, we measured the time from the end of the last mitosis to the onset of apoptosis, as measured by morphological changes in cell shape (). Times were measured for both stathmin-depleted Hela cells or for synchronized Hela cells treated with a 4 hr pulse of AURKA (300 nM S1451) and PLK1 (0.8 nM BI2536) inhibitors to lengthen G2. With either experimental treatment we found that cells initiated apoptosis asynchronously and over a wide range of times from their previous mitosis, without any apparent pattern to when death occurs (). A small proportion (7.2%) of synchronized cells pulsed with a combinationa of AURKA and PLK1 inhibitors died during mitosis, which was not observed in stathmin-depleted cells. Microtubule targeting drugs that block cells in mitosis also have a similar phenotype, where many cells die either while blocked in mitosis or at various times after slipping out of the mitotic arrest.Citation23,24
Figure 3. Cell death occurs asynchronously and over several days for cells depleted of stathmin or pulsed with AURKA and PLK1 inhibitors to delay mitotic entry. Time is plotted from the completion of the last mitosis to the initiation of cell death as detected by morphological changes observed from long term live cell recordings. Data shown are for Hela cells treated with (A) stathmin depletion or (B) synchronized and pulsed with a combination of 300 nM S1451 and 0.8 nM BI2536. Each horizontal bar represents an individual cell. Cells dying at a time of zero reflect cells that died during mitosis.
Neither stathmin depletion nor drug inhibitor pulses significantly disrupt mitosis
It was possible that delaying mitotic entry results in subsequent mitotic defects, and that these defects, and not the cell cycle delay itself, are responsible for cell death under our experimental conditions. To address this possibility we examined mitotic timing, spindle structure and mitotic outcomes in cells depleted of stathmin or in synchronized cells pulsed for 4 hrs with inhibitors to either CDK1 or a combination of AURKA and PLK1. We also compared our 4 hr drug pulse in 10 μM RO3306 in synchronized cells to that in cells incubated for 24 hr in 1–3 μM RO3306, conditions recently shown to disrupt bipolar spindle assembly and result in significant mitotic defects.Citation25
We first measured mitotic durations from living cells imaged by phase contrast microscopy at 5 min intervals for 48–72 hrs. As a positive control, continuous incubation in 1 μM RO3306 doubled the time to complete mitosis compared to DMSO-treated controls (), consistent with a recent report.Citation25 As shown in , and consistent with our previously published results,Citation12 stathmin depleted cells completed mitosis with the same kinetics as those transfected with the non-targeting siRNA. Likewise, synchronized cells pulsed for 4 hrs with a combination S1451 (300 nM; AURKA inhibitor) and BI2536 (0.8 nM; PLK1 inhibitor) progressed through mitosis with a timing indistinguishable from DMSO-pulsed cells. Synchronized cells pulsed for 4 hrs with 10 μM RO3306 were slightly slower to move through mitosis, taking on average 13 minutes longer to complete mitosis. Therefore, of the treatments delaying mitotic entry and leading to cell death, only a pulse with 10 μM RO3306 caused a delay in mitotic progression. Given this small delay, we further examined both living and fixed cell populations for possible spindle assembly defects and defects in chromosome segregation.
Figure 4. Neither stathmin depletion nor a delay at mitotic entry result in significant mitotic errors. (A) Mitotic duration times for Hela cells treated as indicated. Times were measured from live cell recordings. As a positive control for delayed mitotic progression, cells were incubated in 1 μM RO3306, a concentration of the CDK1 inhibitor sufficient to allow mitotic entry 25 but shown to produce defects in spindle assembly. Stathmin depletion did not lengthen mitotic duration, consistent with our previous results.Citation12 A 4 hr pulse with a combination of 300 nM S1451 and 0.8 nM BI2536 also did not slow mitosis, but a pulse with 10 μM RO3306 lengthened mitosis by an average of 13 min. Data represent 120–450 mitotic cells per condition from at least 3 experiments. (B, C) Hela cells were fixed and stained to label tubulin (green) and DNA (red). (B) Spindle morphologies were scored for each experimental condition as indicated. None of the experimental conditions used to delay mitotic entry produced significant defects in spindle assembly. In contrast, 24 incubation in 1–3 μM RO3306 produced dramatic changes to spindle shape and chromosome orientations, consistent with McCloy et al (see ref 25). 50–100 mitotics were scored for each condition in each of 3 experiments. (C) Interphase cells were scored for the number of nuclei per cell to assay for errors in completing mitosis/cytokinesis. Neither stathmin depletion nor a 4 hr pulse with 10 μM RO3306 increased the percentage of Hela cells with >1 nucleus. In contrast, 24 hr incubation in 3 μM RO3306 significantly increased the percent multinucleate cells. Approximately 200 cells were scored per condition in each of 3 experiments. (D) Hela cells were depleted of stathmin and treated for 4 hrs with cytochalasin B beginning 44 or 68 hr after transfection, fixed and stained for tubulin and DNA. The number of micronuclei were scored for each binucleate cell, where the binucleate cells reflect those cells that were prevented from completing cytokinesis during the 4 hr drug treatment. Micronuclei reflect defects in segregation that result in 1 or more chromosomes failing to segregate with the rest of the chromosomes.Citation26 Data shown are from 2 experiments with fixation 72 hr after transfection. Results shown are from ∼350 cells per treatment group from 3 independent experiments. Identical results were obtained for cells fixed at 48 hr after transfection. (E) Live cell recordings as in (A) were also used to measure the percent of cells showing aberrant divisions (defined as cells dividing into > 2 daughter cells or in daughter cells containing > 1 nucleus at the completion of cytokinesis).
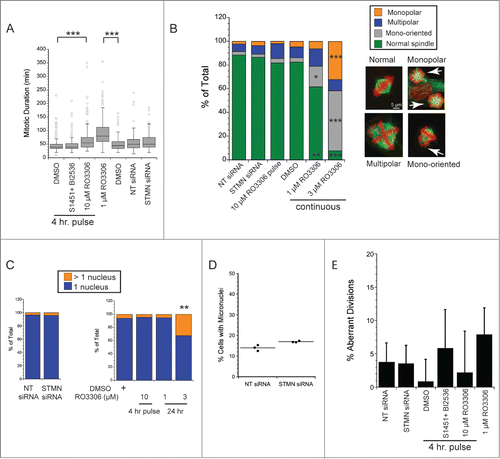
To examine spindle assembly, cells were fixed and stained to localize chromosomes and microtubules 48 hrs after either stathmin depletion from asynchronously growing cells, or a 4 hr pulse with enzyme inhibitors to delay mitotic entry in synchronized cell populations. As shown in , neither stathmin depletion nor a 4 hr pulse with 10 μM RO3306 had any detectable effect on bipolar spindle assembly in Hela cells. The small shifts in the percentage of cells harboring monopolar spindles, multipolar spindles, or mono-oriented chromosomes were not significantly different for experimental treatments compared to their control conditions (non-targeting siRNA or DMSO). In contrast, continous 24 hr incubation in 1–3 μM RO3306 had significant impacts on spindle assembly and chromosome bi-orientation, consistent with a recent report.Citation25 These data demonstrate that our experimental conditions do not cause significant defects in spindle assembly.
To detect possible errors in completing division, we counted the number of Hela cells having >1 nucleus. As shown in , the percentage of multinucleate cells was unchanged relative to control-treatments after either stathmin depletion from asynchronously growing cells or a 4 hr pulse in 10 μM RO3306 to delay mitotic entry of synchronized cells. In contrast, 24 hr incubation in 3 μM RO3306 significantly increased the percentage of multinucleate cells consistent with the aberrant spindles formed at this drug concentration and incubation time. To enhance detection of chromosome segregation defects we also measured micronuclei formation in cytochalasin-B treated cells (4 hr incubation prior to fixation; see ref 26) transfected 72 hr earlier with siRNAs to a non-targeting sequence or to siRNA to deplete stathmin. We again did not detect a significant difference between STMN-depleted cells and the non-targeting siRNA transfected controls, indicating that stathmin depletion did not result in significant chromosome segregation errors.
It is possible that the fixed cell populations shown in reflect only those cells that survive a potential mitotic catastrophe, and therefore we also measured the frequency of aberrant divisions from live cell recordings. Here cell divisions were considered aberrant if they resulted in either >2 daughter cells or in cells harboring >1 nucleus at the completion of cytokinesis and cell respreading. Again, neither stathmin depletion nor 4 hr enzyme inhibitor pulses (in synchronized Hela cells) resulted in a signficant increase in aberrant divisions. In addition, cells completing mitosis and cytokinesis in the presence of 1 μM RO3306 did so without a statistically significant increase in aberrant divisions ().
Finally, we examined whether stathmin depleted cells are slower to correct errors in kinetochore attachments using an Eg5 inhibitor washout assay.Citation27 Here cells are first incubated in 100 μM monastrol to generate monopolar spindles with syntelic attachments between kinetochores and microtubules. The inhibitor is then washed out and the time required to recover from the drug treatment and establish bipolar spindles with bioriented chromosomes used as a readout of kinetchore error correction.Citation27 After monastrol washout, stathmin depleted cells formed bipolar spindles with bi-oriented chromosomes 20 min after drug washout, the shortest time point examined, which was indistinguishable from cells transfected with the non-targeting siRNA. A positive control for loss of error correction, incubation in the Aurora Kinase B inhibitor (10 μM ZM447439), gave the expected slow recovery to a bipolar spindle morphology, where monopolar spindles remained up to 60 min after washout of monastrol (data not shown).
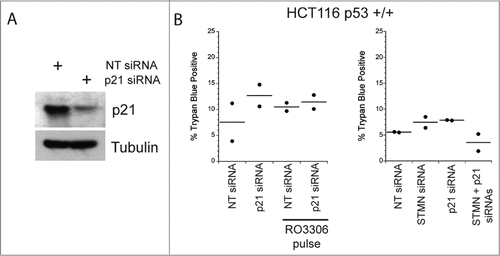
We performed 2 additional experiments to probe whether some well known triggers of cell death were active under our experimental treatments. First we asked whether stathmin depletion activated a DNA damage response. ATM kinase is activated by phosphorylation when DNA damage is detected,Citation28 but we did not see activation of ATM in stathmin depleted cells (). Previous studies demonstrated that a double thymidine block also does not activate ATM in Hela cellsCitation28 making it unlikely that the cell death we measure is due to a DNA damage response. Second, activation of cell death under our experimental conditions is limited to cells lacking detectable p53 expression, raising the possibility that reduced expression of p21, a transcriptional target of p53, prevents these cells from arresting in G1. Such a G1 delay could be necessary to repair errors produced during mitosis that were too subtle to be picked up in our assays of mitotic progression. We addressed this possibility by asking whether p21 depletion rendered p53+/+ cells sensitive to stathmin depletion or to a 4 hr pulse with 10 μM RO3306. As shown in , p21 depletion from HCT116 p53+/+ cells was not sufficient to make these cells susceptible to death after either stathmin depletion or a 4 hr pulse with 10 μM RO3306 in synchronized cells. Taken together, the above data indicate that stathmin depletion is unlikely to activate apoptosis as a downstream consequence of mitotic errors, DNA damage, or from failure to arrest during G1 in response to these potential errors.
Figure 5. ATM is not activated by stathmin depletion. Hela cell lysates were probed with antibodies to phosphorylated ATM (S1981P) to determine whether stathmin depletion induces a DNA damage response. Phosphorylated ATM was readily apparent in doxorubin treated cells, used here as a positive control. In contrast, stathmin depletion did not lead to detectable ATM activation and did not change total ATM concentration.
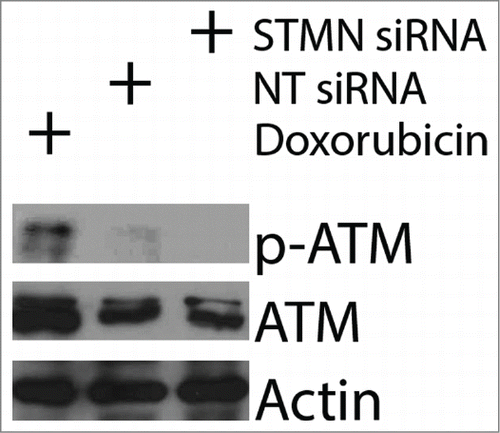
Figure 6. Reducing p21 is not sufficient to activate apoptosis in p53 deficient cells depleted of stathmin or pulsed for 4 hrs with a CDK1 inhibitor. (A) Western blot of p21 depletion from Hela cells. Tubulin is shown as a loading control. (B) Percent trypan blue positive cells for the indicated conditions measured 48 hr after transfection of inhibitor pulse.
A mitotic entry delay induces cell death via caspase 8 and not caspase 9
We next probed the mechanism responsible for cell death in response to either stathmin depletion or to a delay in mitotic entry induced in synchronized cells by a 4 hr incubation in 10 μM RO3306, a CDK1 inhibitor. We, and others, had previously determined that stathmin-depleted cells undergo apoptotic cell death based on cell morphology changes, caspase 3 activation and the presence of cleaved PARP.Citation7,10,11 To determine which initiator caspase, caspase 8 or 9, is activated, we examined procaspase 8 or 9 cleavage to the active form. Cells were either depleted of stathmin or synchronized and then pulsed with 10 μM RO3306 for 4 hrs to delay mitotic entry. Cell lysates were prepared 3–5 d after transfection or the pulse with RO3306 as described in methods. Lysates were then probed with antibodies recognizing cleaved (active) caspase 8 or caspase 9 via western blot. We did not detect caspase 9 activation in either Hela or HCT116 p53−/− cells after either stathmin depletion or a pulse of RO3306 to delay mitotic entry. To confirm that caspase 9 was not required to activate apoptosis in stathmin-depleted cells, we depleted caspase 9 from Hela cells (). Depletion of caspase 9 alone did not promote cell death. Co-depletion of stathmin and caspase 9 resulted in the same amount of cell death as that measured after stathmin depletion alone (). In contrast, an increased amount of cleaved p41/p43-caspase 8 was apparent in stathmin-depleted Hela cells (). We also detected increased cleaved caspase 8 in both Hela cells and HCT116 p53-/- cells delayed for 4 hours at mitotic entry with the CDK1 inhibitor, RO3306 (). Cleaved caspase 8 was undetectable in the HCT116 p53+/+ cell line (), which also does not die in response to stathmin depletionCitation11 or a 4 hr delay in mitotic entry (see ).
Figure 7. Initiator caspase 9 is not required for cell death in stathmin-deleted Hela cells. (A) Western blot demonstrating knockdown of caspase 9 alone or in combination with stathmin depletion. Tubulin was used as a loading control. (B) Percent trypan blue positive cells for the indicated conditions measured 48 hr after transfection. * denotes P < 0.05.
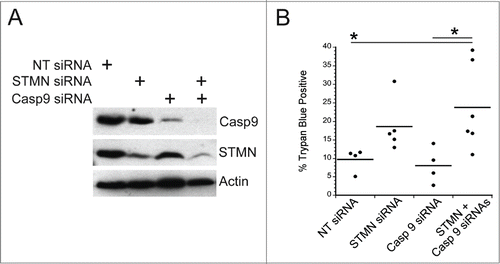
Figure 8. A mitotic entry delay triggers caspase 8 activation. (A) Western blot for cleaved caspase 8, reprobed for tubulin as a loading control for the cell lines treated as indicated. RO3306 pulsed cells were synchronized and incubated in 10 μM RO3306 for 4 hrs to delay mitotic entry. Lysates were prepared 48 hours after transfection or inhibitor pulse. Stathmin depletion or a pulse of CDK1 inhibitor to lengthen G2 increased the level of cleaved caspase 8. (B) Hela cells or HCT116 p53−/− cells were synchronized with a double thymidine block and pulsed with 10 μM RO3306 (CDK1 inhibitor) for 4 hours. Following RO3306 washout, cells were incubated with medium containing DMSO or IETD-CHO (20 μM) for 48 hours and assayed for viability via trypan blue exclusion. Caspase 8 inhibition with IETD-CHO restored viability in RO3306 treated cells. (C) Western blot demonstrating knockdown of caspase 8 alone or in combination with stathmin depletion. Tubulin was used as a loading control. (D) Percent trypan blue positive cells for the indicated conditions. All graphs are representative of at least 3 independent experiments. * denotes P < 0.05, ** denotes P < 0.01, *** denotes P < 0.001.
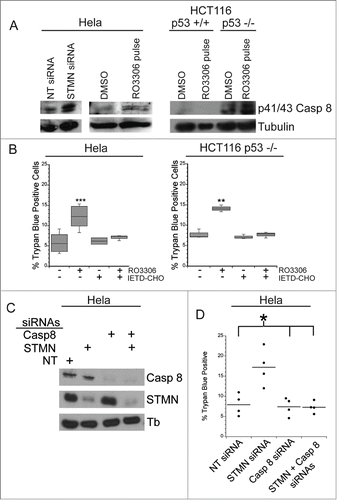
To confirm that caspase 8 activation was triggering cell death under our experimental conditions, we blocked caspase 8 activity with a specific caspase 8 inhibitor, IETD-CHO. We treated Hela and HCT116 p53−/− cells with a 4 hour pulse of either RO3306 or DMSO as described above and then added either 20 μM IETD-CHO or DMSO for 48 hours and measured viability. We found that in both cell lines, treatment of cells with IETD-CHO restored viability in cells pulsed with RO3306 to that of control cells (). In addition, depletion of caspase 8 from Hela cells blocked the cell death normally measured after stathmin depletion (). These data indicate that cell death in p53-deficient cells, triggered by either stathmin depletion or a 4 hr delay during G2, activates apoptosis through initiator caspase 8.
Caspase 8 activation is likely caused by reduced cFLIP level
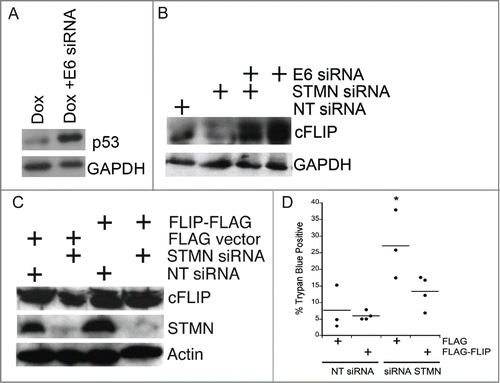
Given that only cells deficient in p53 die after either stathmin depletion or a mitotic entry delay, it is likely that a regulator of caspase 8 activation is influenced by p53 status. Experiments shown in did not support a role for p21 as a potential p53 target gene, whose reduced expression in p53 deficient cells would favor apoptosis. Another candidate, whose expression or stability relies on p53 is cFLIPL, an endogenous caspase 8 inhibitor.Citation29-31 We hypothesized that cFLIPL levels were reduced in p53-deficient cells, favoring caspase 8 activation. We probed cFLIPL levels in Hela cell lysates depleted of stathmin and/or HPV protein E6, a protein expressed by Hela cells that targets p53 for degradation. Depletion of E6 protein from Hela cells restored p53 expression (; see refs 11, 32) and we previously demonstrated that E6 depletion, to restore p53, was sufficient to rescue Hela cells from stathmin-depletion induced apoptosis.Citation11 We found that restoring p53 increased cFLIPL protein level, independent of the level of stathmin (). Others have also reported that the presence or activation of p53 favors increased cFLIP expression in both transformed cell lines and in primary patient tumors.Citation33,34 Surprisingly stathmin depletion reduced the level of cFLIPL in Hela cells () indicating that stathmin normally promotes either cFLIPL expression or protein stability. Co-depletion of stathmin along with E6, to restore p53, resulted in a cFLIPL level above that observed in control transfected cells. Therefore, both p53 and stathmin contribute to regulating cFLIPL level. As a first test of whether the decreased cFLIPL level seen in p53-deficient, stathmin-depleted cells was contributing to caspase 8 activation and therefore cell death, we over-expressed cFLIPL by transient expression of FLAG tagged cFLIPL (). Expression of cFLIPL -FLAG was sufficient to restore viability in stathmin-depleted cells to the level of control cells (). Attempts to deplete cFLIPL from p53 positive cells resulted in excessive cell death (see also refs 35, 36) making it impossible to test directly whether lower cFLIPL levels are responsible for cell death we observe only in p53-deficient cells.
Figure 9. Both p53 deficiency and stathmin depletion reduce cFLIP protein level in Hela cells. (A) HPV E6 depletion from Hela cells restored p53, consistent with our previous results.Citation11,12 Lysates were prepared from Hela cells either untransfected or transfected with siRNA targeting HPV protein E6 and treated with Doxorubicin (included to stabilize p53 and facilitate detection by protein gel blot) for the last 6 hours of a 48 hour incubation post transfection. Doxorubicin was only used in western blot experiments for p53 detection. (B) Hela cells (p53-deficient) have decreased cFLIPL protein levels compared to cells transfected with E6 to restore endogenous p53. Stathmin depletion also reduced the cFLIPL level relative to control siRNA-transfected cells. Western blot for cFLIPL, reprobed with GAPDH as a loading control. (C) Western blots of lysates from Hela cells transfected with FLAG-tagged cFLIP demonstrating FLAG- cFLIPL expression for the conditions indicated. Actin was used as a loading control. (D) Exogenous cFLIPL expression restored viability of stathmin-depleted cells to that of control cells. Hela cells were transfected with non-targeting siRNA or siRNA to stathmin and a plasmid containing FLAG-tagged cFLIPL or empty vector, as noted on the plot. Viability was assayed by trypan blue exclusion at 48 hours following transfection. Graph is a summary of 3 experiments. * denotes P < 0.05.
Discussion
Stathmin's requirement for survival of p53-deficient cancer cells has been documented by several groupsCitation4-7,Citation9,11 but the underlying mechanism linking stathmin depletion to activation of apoptosis has not been studied previously. It was known that stathmin depletion leads to a mitotic entry delay,Citation11-13 as well as cell death,Citation4-7,Citation9,11 particularly in cancer cells deficient in p53,Citation9,11 but whether the cell cycle delay was sufficient to activate apoptosis was not clear. Here we asked whether a mitotic entry delay initiates cell death and which initiator caspase is responsible for activating apoptosis. First, we found that eliminating the cell cycle delay during G2 rescued stathmin-depleted cells from death () and that a 4 hr delay in mitotic entry by treatment with several enzyme inhibitors was sufficient to trigger cell death in p53-deficient cells. We did not find evidence to support a model where the mitotic entry delay acts indirectly by triggering errors in mitosis, which then trigger apoptosis. We were unable to detect a consistent increase in mitotic duration across all treatments, with only a small increase in duration measured in synchronized cells pulsed for 4 hr with 10 μM RO3306 (). We did not detect errors in spindle assembly (), chromosome segregation () or in kinetochore-microtubule error correction (not shown). Reducing p21 level did not render p53+/+ HCT116 cells sensitive to stathmin depletion (), as one would predict if cell survival required arrest in G1 after a defect during M phase. Finally, we established that apoptosis required the initiator caspase 8, and not caspase 9 ( ), while prolonged mitotic delays have been shown to activate caspase 9 via the intrinsic apoptosis pathway.Citation16,17 In sum, our data point to the surprising finding that lengthening G2 and delaying mitotic entry is sufficient to activate apoptosis in p53-deficient cells via the initiator caspase 8.
It is surprising that we identified caspase 8 as the initiator caspase activated by stathmin depletion or by treatment with enzyme inhibitors to delay mitotic entry. Caspase 8 is canonically portrayed as activating apoptosis in response to extracellular signals of the tumor necrosis family, leading to assembly of the death-inducing signal complex at the plasma membrane, but intracellular activation of caspase 8 has also been described.Citation37-40 While there is precedent for intracellular activation of caspase 8, we do not yet know the pathway linking a delay at mitotic entry to activation of caspase 8. Whatever the activation mechanism, it likely requires slow accumulation (or slow loss) of a factor upstream of caspase 8 activation. Such a slow accumulation (or slow loss) would explain the variable time between the cell cycle delay and the onset of apoptosis that we observe ().
Although we have not yet defined a molecular pathway linking a cell cycle delay with caspase 8 activation, our data point to cFLIPL level as a likely contributor to caspase 8 activation under our experimental conditions. cFLIPL has homology to procaspase 8 but lacks catalytic activity and functions to inhibit caspase 8 activation.Citation29 Reduced levels of cFLIPL allow activation of caspase 8.Citation29 We measured reduced cFLIPL levels in Hela cells depleted of stathmin (). Expressing exogenous cFLIPL in these cells was sufficient to prevent apoptosis (). Regardless of the death-triggering mechanism, it is clear that p53-deficient cells are more vulnerable to caspase 8 activation under our experimental conditions and that activation of caspase 8 is due, at least in part, to decreased cFLIPL levels. Note that procaspase 8 is a target of CDK1 phosphorylation, which inhibits its activation as an initiator caspase.Citation41 Stathmin depletion, by delaying mitotic entry, could cause reduced procaspase 8 phosphorylation, possibly contributing to caspase 8 activation.
The level of cFLIPL level may also explain why only p53-deficient cells are sensitive to stathmin depletion or a cell cycle delay prior to mitotic entry. In Hela cells, we find that restoring p53, by E6 depletion, increases cFLIPL level, which should contribute to inhibition of caspase 8 activation. We have not explored whether the higher cFLIPL protein level is due to increased expression or to reduced protein degradation in these cells. It is important to note that conflicting results have been reported in the literature on the role of p53 regulation of cFLIPL expression/stability. In some cell lines p53 is required for increased cFLIPL expression,Citation33,34 while in others p53 expression results in reduced cFLIPL level, possibly by promoting cFLIPL degradation,Citation31,34 and finally, p53 may not regulate cFLIPL under some circumstances.Citation30,40 These differences among cell lines or tumor samples are likely due to a number of additional factors known to regulate cFLIPL expression or protein stability, acting under specific contexts or cell lines.Citation29
Disrupting mitotic entry is an attractive therapeutic strategy since normal cells do not appear to be effected by a small cell cycle delay such as that achieved by partial inhibition of both PLK1 and AURKA. One potential concern of using delayed mitotic entry as a novel target is the low percentage of dead/dying cells that we measured (routinely under 50% and often below 30%). While the percentage of cells that die is relatively small, our results are highly reproducible and the percentage of dead cells that we measure is within the range of that reported by others after treatments with well-accepted apoptosis triggers. For example, heat shock, Anti-Fas, or UV irradiation produce under 50% apoptotic cell death over the first 8–16 hrs after treatment.Citation42 Likewise, cisplatin treatment of chemosensitive ovarian cancer cell lines results in under 40% apoptotic cells over the course of 24 hrs.Citation43 Overall, stathmin inhibition, either via small molecules or RNA interference, would be an ideal means to delay mitotic entry without disrupting normal mitotic progression. Avoiding abnormal mitosis would alleviate some of the harmful effects caused by microtubule disrupting agents such as aneuploidy and cell death in normal tissuesCitation23 and confirm the potential utility of stathmin-inhibiting therapeutics.Citation1,6,8,44
Materials and Methods
Cell Culture and Plasmid Transfections: Hela and HCT116 (p53+/+ and p53−/− lines; gift of Dr. B. Volgelstein, Johns Hopkins School of Medicine) cells were grown in DMEM (Sigma; cat #D5796) supplemented with 10% fetal bovine serum (FBS; Invitogen; cat #16000) and 1X antibiotic/antimycotic (Sigma; cat #A5955). In some experiments, cells were synchronized through a double thymidine block by overnight incubation in 5 mM thymidine (in DMEM), 8 hour release in DMEM after 5 rinses in warm PBS, followed by a 16 hour incubation in 5 mM thymidine (in DMEM). Cells were transferred to DMEM after 5 rinses with warm PBS and used for Trypan Blue exclusion assays and/or cell lysate preparation 48–72 hours following release.
In some experiments Hela cells were transfected with plasmids (300 ng per 35 mm dish) for expression of FLAG-tagged-cFLIPL (pEFrsFLAG; ref 45; a gift from Dr. Marcus Peter, Feinberg School of Medicine, Northwestern University) or empty vectors encoding either FLAG (Addgene cat#20011) or GFP using X-tremeGene HP DNA Transfection Reagent (version 1.0; Roche Diagnostics, Indianapolis, IN) according to the manufacturer's protocol. Cells were transfected approximately 4 hours following siRNA transfections and cell viability was assessed approximately 48 hours following initial transfection.
To detect possible defects in mitotic error correction, cells were transfected with siRNAs targeting stathmin or a control, non-targeting sequence. Approximately 48 hr after transfection cells were incubated for 4 hrs in 100 μM monastrol (Sigma Aldrich; cat#M8515-MG) to cause monopolar spindle assembly and syntelic chromosome attachments.Citation27 Monastrol was then washed out and cells placed in medium supplemented with 5 μM MG132 (EMD Millipore; cat#474790), a protease inhibitor that prevents anaphase onset.Citation27 Cells were fixed at 20 min intervals,Citation27 stained for tubulin, DNA and kinetochores as described below and scored for percent spindles with monopolar or bipolar morphologies. As a positive control, cells were incubated in the Aurora Kinase B inhibitor ZM447439 (10 μM; Selleck Chemicals cat# S1103) during recovery from monastrol. Aurora Kinase B is necessary for correction of kinetchore - microtubule attachment errors and blocked the conversion of monopolar spindles to bipolar spindles.Citation27 Cells were examined by immunofluorescence as described below.
Indirect Immunofluorescence and confocal microscopy: Hela cells were grown on glass coverslips and treated as described above. Cells were fixed in −20°C methanol supplemented with 1 mM EDTA for 10 minutes, rehydrated in PBS, and blocked in 10% FBS in PBS for 30 minutes at 37°C. Coverslips were then incubated in mouse anti-α tubulin (1:1000, Sigma; cat#T5168) for 45 min at 37°C. Cells were then rinsed with PBS 3 times and incubated with goat anti-mouse IgG Alexa 488 (1:50, Life Technologies; cat#A-11029) or and 1.5 μM propidium iodide (Sigma-Aldrich cat# P4864) as a DNA counterstain for an additional 45 minutes at 37°C. Coverslips were then rinsed with PBS 3 times and mounted on slides with Vectashield (Vector Labs; cat#H-1000). Cells were imaged as described previouslyCitation46 using 63X/1.4 numerical aperture plan apo objective on an inverted microscope (Zeiss Axiovert 200M). For synchronized cells, mitotic index was determined by staining with propidium iodide and counting cells with condensed chromatin as a percent of total cells. At least 10 fields (coverslip positions) and >100 cells per time point) were counted for each treatment group for each independent experiment.
Drugs and Reagents: Chemical inhibitors to PLK1 (BI 2536; see ref 47), AURKA (S 1451; see ref 48), and Wee1 (MK1775; see ref 20) were purchased from Selleckchem (cat#'s: S1109 (PLK1), S1451 (AURKA), 51525 (Wee1). Chemical inhibitor to CDK1 (RO3306, cat#4181; see ref 21) and pan-caspase inhibitor, Z-VAD-FMK (cat#2163) were purchased from Tocris Bioscience. Caspase 8 specific inhibitor, IETD-CHO was purchased from EMD Millipore [cat#218773]. All drugs were prepared as stocks in DMSO. All other reagents were from Sigma unless noted otherwise. For CDK1 inhibitor, RO3306, the minimal concentration sufficient to induce a significant 4N peak was determined via FACS analysis and was established at 10 μM, consistent with previous results.Citation49
RNA Interference and Transient Transfection: Cells were grown in 35mm dishes and transfected with siRNAs 1–2 d after plating using GeneSilencer (Genlantis) according to the manufacturer's protocol. Cells were serum starved from the time of transfection to 4 hours post-transfection to improve efficiency. siRNA oligonucleotides were purchased from GE Dharmacon and included: SMTN1, 5′- CGUUUGCGAGAGAAGGAUAdtdt-3′; SMARTpool targeting caspase 8 (cat#L-003466–00); SMARTpool targeting p21 (CDKN1A; cat#L-003471–00–0005); SMARTpool targeting caspase 9 (cat#L-003309–00). SiGenome non-targeting siRNA sequence was used as control siRNA sequences for these experiments.Citation12,13
Western Blotting: Soluble cell extracts were prepared as described previouslyCitation11,13 and protein concentrations were measured by Bradford assay. To enhance detection of active initiator caspases, cells were treated with 50 μM Z-VAD-FMK for 48 hrs prior to lysate preparation. A similar method was described by Tu et al. (see ref 42). Lysates were diluted in PAGE sample buffer, 40 μg total protein per lane was typically loaded and resolved in 10% polyacrylamide gels and transferred to Immobilon Membranes (Millpore, Billerica, MA, cat#IPVH00010). Membranes were blocked with 5% non-fat milk or 5% BSA (depending on manufacturer recommendation) in Tris-buffered saline with 0.1% Tween and then probed with primary antibodies: anti-Caspase 8 (1:000; Santa Cruz cat#sc-56070), anti-cleaved Caspase 8 (1:1000; Cell Signaling cat#18C8), anti-cleaved Caspase 9 (1:1000; Cell Signaling cat#D2D4), anti-caspase 9 (1:1000; Cell Signaling cat#C9), cFLIP (1:1000, Santa Cruz cat#sc-8346), anti-p21 (Cell Signaling cat#12D1), followed by horseradish peroxidase-linked secondary antibodies, anti-mouse (1:2000; Sigma-Aldrich cat#A2554) or anti-rabbit (1:10,000, BioRad cat#170–6515) IgG. Immunoreactive bands were developed using enhanced chemiluminscence (GE Amersham cat#RPN2232). Membranes were reprobed with anti-α-tubulin (1:1000, Sigma-Aldrich cat#T5168), anti-actin (1;1000, Sigma-Aldrich cat#A5060), or GAPDH (1:1000, Abcam) as a loading control.
Live Cell Imaging: To follow cell fates over several days, Hela cells were plated on Mattek dishes (MatTek Corporation, cat#P35G-1.0–14-C) and imaged using a Nikon Biostation IM as described previously.Citation12 Cells were imaged with phase contrast optics using a 20X objective and images were collected at 5 minute intervals for 24–72 hours. Cell fates were tracked from image series. Cell death was scored based on changes in cell morphology characterized by cell retraction and blebbing of the plasma membrane.
Cell Viability Measurements: For synchronization experiments, cells were allowed to grow for 2 d following pulses with kinase inhibitors. For asynchronously growing cell populations transfected with siRNA and/or plasmids, cells were allowed to grow for 3 d following transfection. On the day of measurement, cells were trypsinized and resuspended in PBS with 0.2% Trypan Blue and counted using a hemocytometer to determine viability via Trypan Blue exclusion. Approximately 200 cells were counted per condition per experiment. Each experiment was repeated at least 3 times.
Data Analysis: Statistical analysis of fluorescence intensity and cell cycle durations were performed using unpaired t-tests with either GraphPad Software (www.graphpad.com/quickcalcs/ttest1.cfm) or in Kaliedagraph v4.1 (Synergy Software, Reading PA).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors wish to thank Drs. Bob Skibbens, Linda Lowe-Krentz, Frank Luca and Maureen Murphy for helpful discussions and insightful comments. We also thank Dr. B. Vogelstein, Johns Hopkins School of Medicine for HCT116 cell lines and Dr. Marcus Peter, Feinberg School of Medicine, Northwestern University, for a plasmid encoding FLAG-tagged cFLIP.
Funding
This work supported by a grant from the National Institutes of Health, GM100381, to LC.
References
- Mistry SJ, Atweh GF. Therapeutic interactions between stathmin inhibition and chemotherapeutic agents in prostate cancer. Mol Cancer Ther 2006; 5:3248-57; PMID:17172428; http://dx.doi.org/10.1158/1535-7163.MCT-06-0227
- Belletti B, Baldassarre G. Stathmin: a protein with many tasks. New biomarker and potential target in cancer. Expert Opin Ther Targets 2011; 15:1249-66; PMID:21978024; http://dx.doi.org/10.1517/14728222.2011.620951
- Cassimeris L. The oncoprotein 18/stathmin family of microtubule destabilizers. Curr Opin Cell Biol 2002; 14:18-24; PMID:11792540; http://dx.doi.org/10.1016/S0955-0674(01)00289-7
- Zhang HZ, Wang Y, Gao P, Lin F, Liu L, Yu B, Ren JH, Zhao H, Wang R. Silencing stathmin gene expression by survivin promoter-driven siRNA vector to reverse malignant phenotype of tumor cells. Cancer Biol Ther 2006; 5:1457-61; PMID:17012855; http://dx.doi.org/10.4161/cbt.5.11.3272
- Wang R, Dong K, Lin F, Wang X, Gao P, Wei SH, Cheng SY, Zhang HZ. Inhibiting proliferation and enhancing chemosensitivity to taxanes in osteosarcoma cells by RNA interference-mediated downregulation of stathmin expression. Mol Med 2007; 13:567-75; PMID:17873971; http://dx.doi.org/10.2119/2007-00046.Wang
- Wang X, Ren JH, Lin F, Wei JX, Long M, Yan L, Zhang HZ. Stathmin is involved in arsenic trioxide-induced apoptosis in human cervical cancer cell lines via PI3K linked signal pathway. Cancer Biol Ther 2010; 10:632-43; PMID:20657188; http://dx.doi.org/10.4161/cbt.10.6.12654
- Wang F, Wang LX, Li SL, Li K, He W, Liu HT, Fan QX. Downregulation of stathmin is involved in malignant phenotype reversion and cell apoptosis in esophageal squamous cell carcinoma. J Surg Oncol 2011; 103:704-15; PMID:21360534; http://dx.doi.org/10.1002/jso.21870
- Miceli C, Tejada A, Castaneda A, Mistry SJ. Cell cycle inhibition therapy that targets stathmin in in vitro and in vivo models of breast cancer. Cancer Gene Ther 2013; 20:298-307; PMID:23618950; http://dx.doi.org/10.1038/cgt.2013.21
- Alli E, Bash-Babula J, Yang JM, Hait WN. Effect of stathmin on the sensitivity to antimicrotubule drugs in human breast cancer. Cancer Res 2002; 62:6864-9; PMID:12460900
- Alli E, Yang JM, Hait WN. Silencing of stathmin induces tumor-suppressor function in breast cancer cell lines harboring mutant p53. Oncogene 2007; 26:1003-12; PMID:16909102; http://dx.doi.org/10.1038/sj.onc.1209864
- Carney BK, Cassimeris L. Stathmin/oncoprotein 18, a microtubule regulatory protein, is required for survival of both normal and cancer cell lines lacking the tumor suppressor, p53. Cancer Biol Ther 2010; 9:699-709; PMID:20200495; http://dx.doi.org/10.4161/cbt.9.9.11430
- Carney BK, Caruso Silva V, Cassimeris L. The microtubule cytoskeleton is required for a G2 cell cycle delay in cancer cells lacking stathmin and p53. Cytoskeleton (Hoboken) 2012; 69:278-89; PMID:22407961; http://dx.doi.org/10.1002/cm.21024
- Silva VC, Cassimeris L. Stathmin and microtubules regulate mitotic entry in HeLa cells by controlling activation of both Aurora kinase A and Plk1. Mol Biol Cell 2013; 24:3819-31; http://dx.doi.org/10.1091/mbc.E13-02-0108
- Larsson N, Marklund U, Gradin HM, Brattsand G, Gullberg M. Control of microtubule dynamics by oncoprotein 18: dissection of the regulatory role of multisite phosphorylation during mitosis. Mol Cell Biol 1997; 17:5530-9; PMID:9271428
- Holmfeldt P, Segerman B, Howell B, Morabito J. Cassimeris L, Gullberg M. The catastrophe-promoting activity of ectopic Op18/stathmin is required for disruption of mitotic spindles but not for interphase microtubules. Mol Biol Cell 2001; 12:73-83; PMID:11160824; http://dx.doi.org/10.1091/mbc.12.1.73
- Harley ME, Allan LA, Sanderson HS, Clarke PR. Phosphorylation of Mcl-1 by CDK1-cyclin B1 initiates its Cdc20-dependent destruction during mitotic arrest. EMBO J 2010; 29:2407-20; PMID:20526282; http://dx.doi.org/10.1038/emboj.2010.112
- Wang P, Lindsay J, Owens TW, Mularczyk EJ, Warwood S, Foster F, Streuli CH, Brennan K, Gilmore AP. Phosphorylation of the proapoptotic BH3-only protein Bid primes mitochondria for apoptosis during mitotic arrest. Cell Rep 2014; 7:661-71; PMID:24767991; http://dx.doi.org/10.1016/j.celrep.2014.03.050
- Kreis NN, Louwen F, Yuan J. Less understood issues: p21Cip1 in mitosis and its therapeutic potential. Oncogene 2014;1-10.
- Alli E, Yang JM, Ford JM, Hait WN. Reversal of stathmin-mediated resistance to paclitaxel and vinblastine in human breast carcinoma cells. Mol Pharmacol 2007; 71:1233-40; PMID:17272681; http://dx.doi.org/10.1124/mol.106.029702
- Hirai H, Iwasawa Y, Okada M, Arai T, Nishibata T, Kobayashi M, Kimura T, Kaneko N, Ohtani J, Yamanaka K, et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol Cancer Ther 2009; 8:2992-3000; PMID:19887545; http://dx.doi.org/10.1158/1535-7163.MCT-09-0463
- Vassilev LT, Tovar C, Chen S, Knezevic D, Zhao X, Sun H, Heimbrook DC, Chen L. Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc Natl Acad Sci U S A 2006; 103:10660-5; PMID:16818887; http://dx.doi.org/10.1073/pnas.0600447103
- Van Horn RD, Chu S, Fan L, Yin T, Du J, Beckmann R, Mader M, Zhu G, Toth J, Blanchard K, Ye XS. Cdk1 activity is required for mitotic activation of aurora A during G2/M transition of human cells. J Biol Chem 2010; 285, 21849-57; PMID:20444701; http://dx.doi.org/10.1074/jbc.M110.141010
- Gascoigne KE, Taylor SS. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell 2008; 14:111-22; PMID:18656424; http://dx.doi.org/10.1016/j.ccr.2008.07.002
- Orth JD, Tang Y, Shi J, Loy CT, Amendt C, Wilm C, Zenke FT, Mitchison TJ. Quantitative live imaging of cancer and normal cells treated with Kinesin-5 inhibitors indicates significant differences in phenotypic responses and cell fate. Mol Cancer Ther 2008; 7:3480-9; PMID:18974392; http://dx.doi.org/10.1158/1535-7163.MCT-08-0684
- McCloy RA, Rogers S, Caldon CE, Lorca T, Castro A, Burgess A. Partial inhibition of Cdk1 in G2 phase overrides the SAC and decouples mitotic events. Cell Cycle 2014; 13:1400-12; PMID:24626186; http://dx.doi.org/10.4161/cc.28401
- Fenech, M. The in vitro micronucleus technique. Mutation Res 2012; 455:81-95; http://dx.doi.org/10.1016/S0027-5107(00)00065-8
- DeLuca J G. Kinetochore - microtubule dynamics and attachment stability. Methods Cell Biol 2010; 97:53-79; PMID:20719265; http://dx.doi.org/10.1016/S0091-679X(10)97004-0
- Kubuota S, Fukumoto Y, Ishibashi K, Soeda S, Kubota S, Yuki R, Nakayama Y, Aoyama K, Yamauchi N, Yamaguchi N. Activation of the prereplication complex is blocked by mimosine through reactive oxygen species-activated ataxia telangiectasia mutated (ATM) protein without DNA damage. J Biol Chem 2014; 289:5730-45; PMID:24421316; http://dx.doi.org/10.1074/jbc.M113.546655
- Safa AR. c-FLIP, a master anti-apoptotic regulator. Exp Oncol 2012; 34:176-84; PMID:23070002
- Galligan L, Longley DB, McEwan M, Wilson TR, McLaughlin K, Johnston PG. Chemotherapy and TRAIL-mediated colon cancer cell death: the roles of p53, TRAIL receptors, and c-FLIP. Mol Cancer Ther 2005; 4:2026-36; PMID:16373718; http://dx.doi.org/10.1158/1535-7163.MCT-05-0262
- Fukazawa T, Fujiwara T, Uno F, Teraishi F, Kadowaki Y, Itoshima T, Takata Y, Kagawa S, Roth JA, Tschopp J, et al. Accelerated degradation of cellular FLIP protein through the ubiquitin-proteasome pathway in p53-mediated apoptosis of human cancer cells. Oncogene 2001; 20:5225-31; PMID:11526513; http://dx.doi.org/10.1038/sj.onc.1204673
- Koivusalo R, Krausz E, Helenius H, Hietanen S. Chemotherapy compounds in cervical cancer cells primed by reconstitution of p53 function after short interfering RNA-mediated degradation of human papillomavirus 18 E6 mRNA: opposite effect of siRNA in combination with different drugs. Mol Pharmacol 2005; 68:372-82; PMID:15908516
- Bartke T, Siegmund D, Peters N, Reichwein M, Henkler F, Scheurich P, Wajant H. p53 upregulates cFLIP, inhibits transcription of NF-kappaB-regulated genes and induces caspase-8-independent cell death in DLD-1 cells. Oncogene 2001; 20:571-80; PMID:11313989; http://dx.doi.org/10.1038/sj.onc.1204124
- Zhou XD, Yu JP, Chen HX, Yu HG, Luo HS. Expression of cellular FLICE-inhibitory protein and its association with p53 mutation in colon cancer. WJG 2005; 11:2482-5; PMID:15832422; http://dx.doi.org/10.3748/wjg.v11.i16.2482
- Day TW, Huang S, Safa AR. c-FLIP knockdown induces ligand-independent DR5-, FADD-, caspase-8-, and caspase-9-dependent apoptosis in breast cancer cells. Biochem Pharmacol 2008; 76:1694-704; PMID:18840411; http://dx.doi.org/10.1016/j.bcp.2008.09.007
- Ewald F, Ueffing N, Brockmann L, Hader C, Telieps T, Schuster M, Schulz WA, Schmitz I. The role of c-FLIP splice variants in urothelial tumours. Cell Death Dis 2011; 2:e245; PMID:22190004; http://dx.doi.org/10.1038/cddis.2011.131
- von Haefen C, Wendt J, Semini G, Sifringer M, Belka C, Radetzki S, Reutter W, Daniel PT, Danker K. Synthetic glycosidated phospholipids induce apoptosis through activation of FADD, caspase-8 and the mitochondrial death pathway. Oncogene 2002; 16:636-51
- Wieder T, Essman F, Prokop A, Schmelz K, Schulze-Osthoff K, Beyaert R, Dorken B, Daniel PT. Activation of caspase-8 in drug-induced apoptosis of B-lymphoid cells is independent of CD95/Fas receptor-ligand interaction and occurs downstream of caspase-3. Blood 2001; 97:1378-87; PMID:11222383; http://dx.doi.org/10.1182/blood.V97.5.1378
- Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, Zachariou A, Lopez J, MacFarlane M, Cain K, et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell 2011; 43:432-48; PMID:21737329; http://dx.doi.org/10.1016/j.molcel.2011.06.006
- Wilson TR, McLaughlin KM, McEwan M, Sakai H, Rogers KM, Redmond KM, Johnston PG, Longley DB. c-FLIP: a key regulator of colorectal cancer cell death. Cancer Res 2007; 67:5754-62; PMID:17575142; http://dx.doi.org/10.1158/0008-5472.CAN-06-3585
- Matthess Y, Raab M, Sanhaji M, Lavrik IN, Strebhardt K. Cdk1/cyclin B1 controls Fas-mediated apoptosis by regulating caspase-8 activity. Mol Cell Biol 2010; 30:5726-40; http://dx.doi.org/10.1128/MCB.00731-10
- Tu S, McStay GP, Boucher L-M, Mak T, Beere HM, Green DR. In situ trapping of activated initiator caspases reveals a role for caspase-2 in heat shock-induced apoptosis. Nat Cell Biol 2006; 8:72-7; PMID:16362053; http://dx.doi.org/10.1038/ncb1340
- Abedini MR, Muller EJ, Brun J, Bergeron B, Gray DA, Tsang BK. Cisplatin induces p53-dependent FLICE-like inhibitory protein ubiquitination in ovarian cancer cells. Cancer Res 2008; 68:4511-7; PMID:18559494; http://dx.doi.org/10.1158/0008-5472.CAN-08-0673
- Byrne FL, Yang L, Phillips PA, Hansford LM, Fletcher JI, Ormandy CJ, McCarroll JA, Kavallaris M. RNAi-mediated stathmin suppression reduces lung metastasis in a orthotopic neuroblastoma mouse model. Oncogene 2014; 33:882-90; PMID:23396365; http://dx.doi.org/10.1038/onc.2013.11
- Scaffidi C, Schmitz I, Krammer PH, Peter ME. The role of c-FLIP in modulation of CD95-induced apoptosis. J Biol Chem 1999; 274:1541-8; PMID:9880531; http://dx.doi.org/10.1074/jbc.274.3.1541
- Piehl M, Cassimeris L. Organization and dynamics of growing microtubule plus ends during early mitosis. Mol Biol Cell 2003; 14:916-25; PMID:12631713; http://dx.doi.org/10.1091/mbc.E02-09-0607
- Grinshtein N, Datti A, Fujitani M, Uehling D, Prakesch M, Isaac M, Irwin MS, Wrana JL, Al-Awar R, Kaplan DR. Small molecule kinase inhibitor screen identifies polo-like kinase 1 as a target for neuroblastoma tumor-initiating cells. Cancer Res 2011; 71:1385-95; PMID:21303981; http://dx.doi.org/10.1158/0008-5472.CAN-10-2484
- Yuan H, Wang Z, Zhang H, Roth M, Bhatia R, Chen WY. Overcoming CML acquired resistance by specific inhibition of Aurora A kinase in the KCL-22 cell model. Carcinogenesis 2012; 33:285-93; PMID:22116466; http://dx.doi.org/10.1093/carcin/bgr278
- Ma HT, Tsang YH, Marxer M, Poon RY. Cyclin A2-cyclin-dependent kinase 2 cooperates with the PLK1-SCFbeta-TrCP1-EMI1-anaphase-promoting complex/cyclosome axis to promote genome reduplication in the absence of mitosis. Mol Cell Biol 2009; 29:6500-14; PMID:19822658; http://dx.doi.org/10.1128/MCB.00669-09