
Abstract
During DNA damage response (DDR), histone ubiquitination by RNF168 is a critical event, which orchestrates the recruitment of downstream DDR factors, e.g. BRCA1 and 53BP1. Here, we report USP7 deubiquitinase regulates the stability of RNF168. We showed that USP7 disruption impairs H2A and ultraviolet radiation (UVR)-induced γH2AX monoubiquitination, and decreases the levels of pBmi1, Bmi1, RNF168 and BRCA1. The effect of USP7 disruption was recapitulated by siRNA-mediated USP7 depletion. The USP7 disruption also compromises the formation of UVR-induced foci (UVRIF) and ionizing radiation-induced foci (IRIF) of monoubiquitinated H2A (uH2A) and polyubiquitinated H2AX/A, and subsequently affects UVRIF and IRIF of BRCA1 as well as the IRIF of 53BP1. USP7 was shown to physically bind RNF168 in vitro and in vivo. Overexpression of wild-type USP7, but not its interaction-defective mutant, prevents UVR-induced RNF168 degradation. The USP7 mutant is unable to cleave Ub-conjugates of RNF168 in vivo. Importantly, ectopic expression of RNF168, or both RNF8 and RNF168 together in USP7-disrupted cells, significantly rescue the formation of UVRIF and IRIF of polyubiquitinated H2A and BRCA1. Taken together, these findings reveal an important role of USP7 in regulating ubiquitin-dependent signaling via stabilization of RNF168.
Introduction
Eukaryotic cells evoke a sophisticated DNA damage response (DDR) to preserve the integrity of the genome. DDR is responsible for transiently arresting the cell cycle and allowing faithful DNA repair. In DDR, DNA damage induces a re-localization of damage sensing, signaling and repair factors into distinct foci at damage sites. Phosphorylation of variant H2A (H2AX) at the sites of DNA breaks by Ataxia telangiectasia mutated (ATM), ATM and Rad3-related (ATR) and DNA-dependent protein kinase (DNA-PK) is an early and well characterized event.Citation1,2 The phosphorylated H2AX (γH2AX) mark can spread to megabase DNA segments of chromatin.
Accumulation of γH2AX in the vicinity of DNA double-/single-strand breaks (DSBs/SSBs) is instrumental for the recruitment and retention of mediators and repair factors, such as MDC1, BRCA1 and 53BP1.Citation3-5 The mediator MDC1 is recruited to DSBs/SSBs and serves as a platform for recruiting 2 RING-type ubiquitin (Ub) ligases, RNF8 and RNF168, to modify γH2AX and H2A.Citation6-8 Ubiquitination of H2A and γH2AX by the concerted action of RNF168 and RNF8 generates not only the monoubiquitinated H2A and γH2AX, but also the polyubiquitin conjugates with lysine 63 (K63)-linked Ub chains.Citation7-10 More rigorous experimentation has revealed that lysine 13 and 15 (K13/15) in H2A/X are ubiquitinated by RNF168 and the K63-Ub chains are then extended by RNF8.Citation11 H2A and γH2AX ubiquitination is considered to be important in DDR,Citation12-14 because this foremost ubiquitination event allows accumulation of the key repair factors BRCA1 and 53BP1,Citation11 which respectively determine the choice between homologous recombination (HR) and non-homologous end joining (NHEJ) repair pathways.Citation15-17 When BRCA1 function is lost, RNF168 becomes a crucial determinant of the repair pathway choice, channelling DNA damage through NHEJ.Citation18 In essence, the sequentially coordinated ubiquitination activities of RNF168 and RNF8 are pivotal for proper DSB repair.Citation8,9,19
Besides RNF168 and RNF8, Polycomb repressive complex 1 (PRC1) is also known to participate in histone ubiquitination. The PRC1-mediated ubiquitination generates monoubiquitinated H2A at K119 (uH2A).Citation20 This prevalent modification can constitute up to 10% of cellular H2A in chromatin as a result of ubiquitination by E3 ligase Ring1B present in PRC1, which plays a key role in transcription silencing.Citation21-23 The Polycomb proteins Ring1B and Bmi1 form an active heterodimeric Ub ligase in which Bmi1 acts as an activator for Ring1B. Ubiquitination of H2A at K119 also occurs locally at sites of UV lesions and DSBs.Citation24-27 It has been shown that Ring1B is required for UV-induced H2A K119-ubiquitination.Citation24 Recently, emerging evidence shows that Ring1B/Bmi1 E3 ligase also regulates H2AX K118/119 monoubiquitination at DSBs.Citation28-31 Both Ring1B and Bmi1 are recruited to DSBs and, the Bmi1 recruitment requires its RING domain.Citation30,31 Loss or ablation of Bmi1 function by knockout or siRNA-mediated depletion decreases H2AX K118/119 monoubiquitination,Citation28,30 and in functional terms leads to impaired repair of DSBsCitation31 as well as decreased cell survival after ionizing radiation (IR).Citation30,31 Nevertheless, how H2AX K118/119 monoubiquitination by Ring1B/Bmi1 regulates H2AX polyubiquitination and subsequent recruitment of repair factor BRCA1 and 53BP1 is still unclear and being debated.Citation30,31
Despite the importance of RNF8, RNF168 and Ring1B/Bmi1 E3 ligases in DDR, cellular regulation of these potent E3 ligases is poorly understood. Ub specific peptidase 7 (USP7), a known p53 and mdm2 deubiquitinating enzyme,Citation32-34 was identified as a regulator for Ring1B.Citation35 USP7 associates with Polycomb proteins and disassembles K48-linked Ub chain from ubiquitinated Ring1B in regulation of Ring1B stability. Similarly, USP7 was shown to co-purify with PRC1 complex, interact with Bmi1 and regulate ubiquitination status of Bmi1.Citation36 Computational modeling predicts a bistable switch and oscillation in Ring1B/Bmi1 ubiquitination systems and, USP7 underexpression confers the ubiquitination system more prone to be bistable, which can lead to all-or-none histone H2A monoubiquitination.Citation37
In this study, we have extensively built on our previous finding that USP7 disruption severely compromises the formation of UV radiation (UVR)-induced foci (UVRIF) of polyubiquitinated H2A/X.Citation38 We examined the potential role of USP7 in regulation of H2A/X ubiquitination and factor recruitment driven by Ub-dependent signaling. Our observations reveal that USP7 plays an important role in regulating H2A/X ubiquitination via monitoring the stability of RNF168, in addition to Ring1B and Bmi1.
Results
USP7 regulates H2A and γH2AX monoubiquitination
We previously found that the formation of UVR-induced polyubiquitinated H2A/X foci is unexpectedly compromised by USP7 disruption.Citation38 Since polyubiquititination of H2A/X is a critical event in Ub-dependent signaling, we further compared the H2A and γH2AX monoubiquitination in HCT116 vs. HCT116-USP7−/− cells post-UVR exposure. The results showed that UVR leads to a decrease in global levels of uH2A but not H2A in parental HCT116 cells (). USP7 disruption affected the H2A level only slightly but quite noticeably reduced the inherent cellular uH2A levels, which was further abolished upon exposure to UVR. In contrast, γH2AX monoubiquitination in the presence of USP7 was induced by UVR from 2 to 24 h post-UVR time periods. However, the induction of γH2AX monoubiquitination was diminished by USP7 disruption despite a robust H2AX phosphorylation in the absence of USP7. Given that USP7 is a deubiquitinating enzyme and monoubiquitination does not drive the substrate proteolysis, these results suggested that uH2A and monoubiquitinated γH2AX may not be the direct substrates of USP7. Thus, we next probed the changes in Ring1B/Bmi1 E3 ligases of PRC1 which play a major role in maintaining the cellular uH2A abundance.Citation39 UVR did not affect Ring1B levels in HCT116 and, USP7 disruption led only to a slight, if any, decrease in Ring1B in HCT116-USP7−/− cells (). On the contrary, USP7 disruption led to a clear decrease in both the native and phosphorylated form of Bmi1. The latter was induced by UVR in HCT116, but the induction appeared to be compromised in HCT116-USP7−/− cells. It is noteworthy that phosphorylation enhances Bmi1/Ring1B E3 activity and stimulates H2A monoubiquitination.Citation40 As predicted by computational modeling, underexpression of USP7 confers Ring1B/Bmi1 ubiquitination system more prone to be bistable, which may lead to an all-or-none behavior of H2A monoubiquitination.Citation37 Thus, the decrease in both native and phosphorylated Bmi1 may explain the extremely low levels of uH2A in HCT116-USP7−/− cells.
Figure 1. USP7 regulates H2A and γH2AX ubiquitination in DNA damage response. (A) HCT116 and HCT116-USP7−/− cells were exposed to 20-J/m2 UV and the cell extracts were prepared at indicated time points following UVR. The uH2A and ubiquitinated γH2AX were examined by Western blotting using anti-uH2A and γH2AX antibodies. (B) The cells were treated as in . Ring1B and Bmi1 were examined by Western blotting. (C) The cells were subjected to UVR and then treated with or without proteasome inhibitor MG132 and the cell extracts were examined by Western blotting. “Long Exp." indicates longer exposure for chemiluminescent detection (D) Anti-γH2AX blots were overexposed to show low level of γH2AX ubiquitination in HCT116-USP7−/− cells. (E) HeLa cells were transfected with USP7 siRNA or control siRNA for 2 rounds. The transfected cells were irradiated with UV and processed as in . Ubiquitinated γH2AX, uH2A, USP7 and other DDR factors were examined by Western blotting. (F) HCT116 cells were exposed to UVR, and then incubated with USP7 inhibitor HBX 411108 at indicated concentration for 8 h. The Ub-γH2AX/γH2AX ratio is calculated based on gray scale of the blots examined by ImageJ.
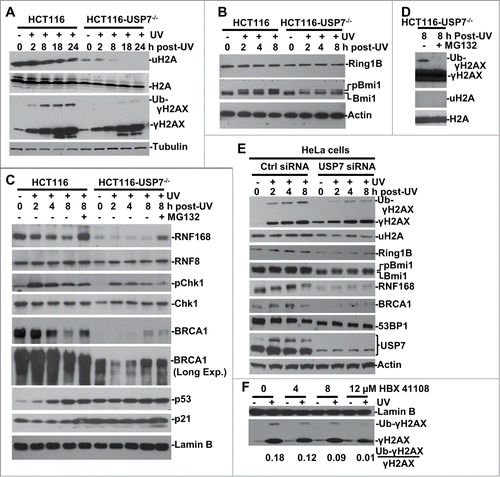
While global reduction of uH2A may be due to compromised Bmi1/Ring1B E3 activity, it is uncertain how USP7 deficiency could contribute to the decrease of γH2AX monoubiquitination. So, we closely examined the cellular status of RNF168, RNF8, BRCA1 and other DDR specific factors (). In HCT116, UVR led to a decreased RNF168 level that was restored by proteasome inhibitor MG132 treatment. Surprisingly, in HCT116-USP7−/− cells, RNF168 level was very low and unresponsive to UVR exposure. MG132 treatment partially restored the RNF168 level. Nevertheless, due to Ub pool depletion caused by proteasome inhibition, MG132 treatment further diminished the remaining traces of γH2AX monoubiquitination in HCT116-USP7−/− cells, which could only be detected upon film overexposures (), and confirming that γH2AX ubiquitination is sensitive to Ub stress. USP7 disruption, however, caused a slight decrease of cellular RNF8 levels. As expected, USP7 disruption stabilized p53, and led to a consequent increase in p53-downstream target proteins p21, which in turn affected the levels of pChk1 in HCT116-USP7−/− cells. These results are in accord with the known regulatory circuit of p53-mdm2 by USP7.Citation32-34 Interestingly, the results further showed that BRCA1 levels were reduced over time following UVR and the reduction was prevented upon proteasome inhibition (). Like RNF168, BRCA1 levels were very low in HCT116-USP7−/− cells but were to some extent stabilized by UVR and MG132. These results suggested that UVR induces RNF168 and BRCA1 degradation in USP7-proficient cells, whereas USP7 disruption destabilizes and diminishes RNF168 and BRCA1 in HCT116-USP7−/− cells.
Next, we utilized RNAi approach to knockdown USP7 in HeLa cells to test whether USP7 depletion has similar effect on RNF168, BRCA1 and other DDR factors in a different cell type and also rule out the possibility that chronic USP7 deficiency may lead to uncharacteristic cellular changes. The USP7 depletion was achieved by 2 rounds of siRNA transfection (). Consistent with the observations in HCT116-USP7−/− cells, the USP7 depletion decreased UVR-induced γH2AX monoubiquitination without any significant impact on the UV-induced γH2AX levels. However, the USP7 depletion reduced the levels of uH2A, BRCA1, RNF168, Ring1B and Bmi1. Notably, 53BP1 levels fairly decreased over time following UVR in parental HCT116 cells. The USP7 depletion had moderately reduced 53BP1 levels. In conclusion, siRNA-mediated USP7 depletion, recapitulated the effects of chronic USP7 deficiency on the regulation of H2A and H2AX monoubiquitination. Next, we checked the effect of USP7-specific inhibitor HBX 41108 on γH2AX ubiquitination (). As expected, the inhibitor treatment of HCT116 cells led to a dose-dependent decrease in γH2AX ubiquitination. Based on the combined results of 3 different approaches, we concluded that USP7 regulates the cellular H2A and γH2AX monoubiquitination upon UVR.
USP7 disruption compromises focus formation of uH2A and FK2, thereby affecting recruitment of BRCA1 and 53BP1
Upon invoking DDR, accumulation of H2A/X Ub-conjugates and DDR factors form characteristic UVRIF or IRIF at the sites of DNA damage. For example, micropore UV irradiation induced UVRIF of uH2A are formed due to H2A ubiquitination.Citation24 Whereas, IRIF of H2A/X polyubiquititination (as detected by anti-Ub FK2 antibody, also called FK2 foci) are formed at the sites of DNA breaks.Citation7-10 It is noteworthy that although FK2 antibody recognizes both mono- and poly-Ub, detection of FK2 foci depends on γH2A/X polyubiquititination, MDC1, RNF8 and RNF168.Citation7,8,10,11 Therefore, we examined UVRIF and IRIF of uH2A and FK2 to further characterize the monoubiquitination of H2A and polyubiquititination of γH2A/X in HCT116 vs. HCT116-USP7−/− cells. Upon micropore UV irradiation, which generates localized DNA damage in distinct sub-nuclear areas, γH2AX foci were distinctly formed and colocalized with uH2A foci in HCT116 cells at 0.5 and 2 h. In HCT116-USP7−/− cells, formation of γH2AX foci was delayed as seen at 0.5 h but appeared as distinct foci at 2 h. Nevertheless, uH2A foci were almost undetectable at any time, although UV-induced cyclobutane pyrimidine dimers (CPD), marking the sites of damage, were present at all the time points (). Similarly, HCT116-USP7−/− cells also failed to form FK2 foci (), indicating that polyubiquititination of γH2A/X are compromised in the absence of USP7. Interestingly, the results showed that the formation of UVRIF of BRCA1 was severely compromised in HCT116-USP7−/− cells. For example, at 2 h, UVRIF of BRCA1 appeared in ∼20% of HCT116-USP7−/− cells but showed in ∼45% of parental cells ().
Figure 2. USP7 disruption compromises the formation of UVRIF of uH2A, FK2 and BRCA1 (A) HCT116 and HCT116-USP7−/− cells were exposed to micropore UV irradiation at 100 J/m2. Sub-nuclear spot accumulations of indicated DDR factors were visualized by immunofluorescence using specific antibodies. Calibration bar is 10 μm. Left panel: UVRIF of uH2A and γH2AX; right panel: UVRIF of CPD and γH2AX. (B) UVRIF of FK2 and γH2AX. (C) UVRIF of BRCA1 and γH2AX. (D) The quantitative data of γH2AX, uH2A, FK2 and BRCA1 foci from HCT116 and HCT116-USP7−/− cells. Mean ± SD were calculated from 4–6 microscopic fields of 3 independent experiments.
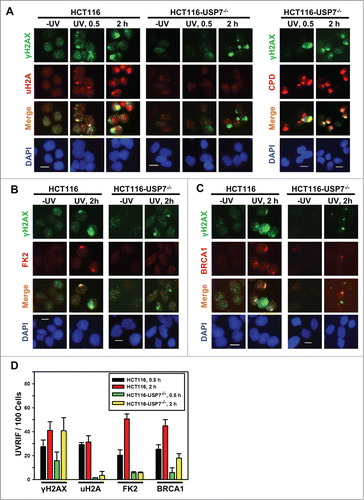
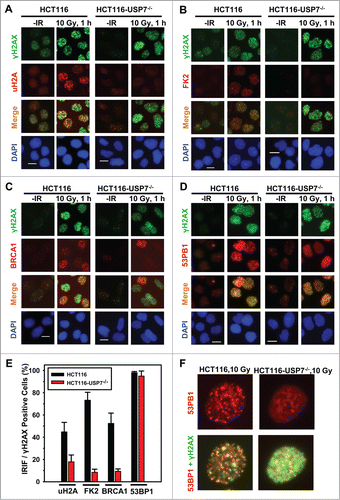
Since both BRCA1 and 53BP1 are attracted to the sites of DNA strand breaks by polyubiquitinated γH2AX/A, and are involved in strand break repair, we further confirmed the effect of USP7 deficiency on IRIF upon IR exposures. The results showed that IR at 10 Gy induced the formation of uH2A, FK2 and BRCA1 foci in ∼42%, 75%, and 50% of γH2AX positive HCT116 cells, respectively (). In comparison, the formation of uH2A, FK2, and BRCA1 in HCT116-USP7−/− cells was reduced to ∼20%, 10% and 10%, respectively (). The formation of 53BP1 foci occurred at a high efficiency of 97% in parental HCT116 cells. Moreover, the overall 53BP1 focus formation rate was not significantly affected by USP7 disruption. However, a closer look at 53BP1 IRIF indicated that the 53BP1 foci in most cells were smaller, diffuse and often indistinguishable from the strong background signals (). We further examined IRIF at 2 Gy IR. Again, 53BP1 foci were seen to be smaller in size with diffuse background signals in ∼30% HCT116-USP7−/− cells (). It was recently demonstrated that RNF168 ubiquitinates 53BP1 with K63 linkage and controls 53BP1 recruitment.Citation41 Thus, USP7 disruption might affect 53BP1 recruitment through both down regulation of RNF168 and RNF168-mediated H2A/X ubiquitination. Taken together, we concluded that USP7 function is required for the efficient monoubiquitination of H2A and polyubiquititination of γH2A/X, The insufficient γH2A/X polyubiquititination resulting from USP7 deficiency affects the recruitment of BRCA1 and, to a lesser extent, the recruitment of 53BP1.
Figure 3. USP7 disruption compromises the formation of IRIF of uH2A and FK2, thereby affecting the BRCA1 and to a lesser extent the 53BP1 recruitment to DNA strand breaks. HCT116 and HCT116-USP7−/− cells were exposed to IR at 10 Gy. Nuclear accumulations of uH2A, γH2AX, FK2, BRCA1 and 53BP1 were visualized by immunofluorescence using specific antibodies. Calibration bar is 10 μm. (A) IRIF of uH2A and γH2AX. (B) IRIF of FK2 and γH2AX. (C) IRIF of BRCA1 and γH2AX. (D) IRIF of 53BP1 and γH2AX. (E) The quantitative data of IRIF of γH2AX, uH2A, FK2, BRCA1 and 53BP1 from HCT116 and HCT116-USP7−/− cells. Only the cells with 10 or more distinctive IRIF were considered IRIF positive and scored for quantitation. Mean ± SD of IRIF vs. γH2AX positive cell ratio was calculated from 4–6 microscopic fields of 3 independent experiments. (F) Magnified views of IRIF of 53BP1 in HCT116 and HCT116-USP7−/− cells exposed to IR at 10 Gy.
USP7 deubiquitinates RNF168
Decrease in the steady-state level of RNF168 by USP7 disruption and siRNA-mediated depletion suggests that USP7 might possibly regulate cellular level of RNF168, whose function is required for polyubiquititination of γH2A/X. To follow this thinking, we first examined the physical interaction between USP7 and RNF168. We performed an in vitro GST pull-down assay using GST fusion proteins containing various USP7 structural domains (). Consistent with the presence of MIUs (motif interacting with Ub, MIU) in RNF168,Citation42 GST fusion protein containing a single UBL domain (UBL1, spanning from 560 to 664 amino acid) was able to bind RNF168 (, L1–3). Moreover, GST fusion proteins containing UBL1 and catalytic domains, or UBL2 to UBL5 domains, bind RNF168 with much higher efficiency (, L1’-6). To discern the contribution of UBL domains for RNF168 binding in vivo, we used FLAG-tagged wild-type (WT) and M1 mutant USP7.Citation43 The M1 mutant contains the tryptophan (W) and phenylalanine (F) to serine (S) substitutions in UBL1 domain at amino acids 623 and 661 (W623S/F661S), respectively. Co-immunoprecipitation assay showed that WT USP7 interacted with RNF168. However, M1 mutation abrogated the USP7 interaction with RNF168 (). Together, these experiments demonstrated that USP7 directly interacts with RNF168.
Figure 4. USP7 binds to RNF168, deubiquitinates RNF168 ubiquitin (Ub)-conjugates and protects RNF168 from UV-induced degradation. (A) Diagram of structural domains of USP7. TRAF represents tumor necrosis factor-receptor associated factor (TRAF) domain. UBL represents Ub-like domain. (B) USP7 interacts with RNF168 in vitro. GST pull-down assay were conducted using whole cell lysates of HCT 116 in RIPA buffer. The GST fusion proteins loaded on beads were verified as described in materials and methods. (C) Interaction between RNF168 and USP7 in vivo. FLAG-tagged USP7 and M1 mutant were transiently expressed in HCT116 cells. Immunoprecipitation was performed using anti-FLAG agarose gels and cells lysates made in E1A buffer, followed by Western blot analysis for presence of RNF168. Non-transfected HCT116 cells were used as control. (D) Overexpression of WT-USP7 protects RNF168 from UV-induced degradation. Construct for expressing FLAG-tagged USP7 was transfected into HCT116 cells for 48 h. The transfected cells were UV irradiated at 20 J/m2 and allowed to repair DNA for 2 h. RNF168 and USP7 were detected by Western blotting. Anti-Lamin B blots served as loading control. (E) Overexpression of M1 mutant USP7. (G) USP7 deubiquitinates RNF168 Ub-conjugates in vivo. HCT116 cells were transfected with expressing constructs for RNF168, HA-tagged Ub and FLAG-tagged USP7 in combination as illustrated in the figure. The transfected cells were treated with proteasome inhibitor MG132 or vehicle DMSO for 8 h. Expression of transfected RNF168 and USP7 was examined by Western blotting (Input); RNF168 Ub-conjugates were examined by immunoprecipitation followed by Western blotting.
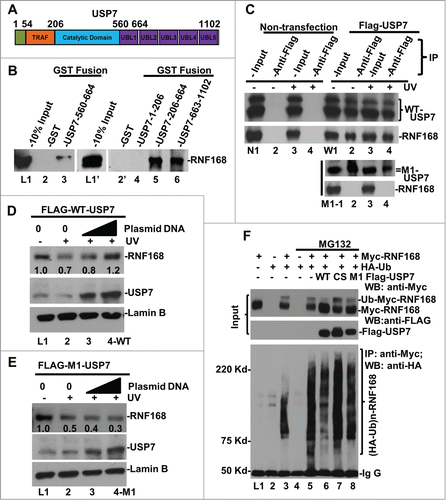
In HCT116 cells, UVR induced the proteasome-mediated degradation of RNF168 (). Based on this distinctive response, we explored whether ectopic expression of USP7 protects RNF168 from UV-induced proteolysis. The results showed that ectopic expression of WT USP7 indeed prevented the endogenous RNF168 from UVR-induced proteolysis (). In contrast, endogenous RNF168 was not protected by M1 mutant (). We do not yet understand how the M1 (UBL1 domain) mutation affects in vivo USP7-RNF168 interaction, when other UBL domains are still intact. It is known that catalytic domain of USP7 alone is maintained in a nonfunctional state.Citation44 The binding of UBL4–5 to catalytic domain activates USP7 catalytic activity toward p53, while binding of UBL1–3 domains to GMP-synthetase allows allosteric regulation (activation) of USP7 in vivo.Citation45 Perhaps, the UBL1 domain is more important for in vivo interaction, when UBL4–5 domains are occupied by catalytic domain for catalytic activation.
Since RNF168 level was dramatically lowered by USP7 disruption as well as siRNA-mediated depletion without any exposure to UVR (), we next investigated whether USP7 deubiquitinates Ub-conjugates of RNF168 in the absence of cellular DNA damage. Thus, epitope-tagged RNF168, Ub and USP7 were ectopically expressed through various combinations of transfections of HCT116 cells with and without the proteasome inhibition. The RNF168 and USP7 were then detected by Western blotting, while RNF168 Ub-conjugates were examined by immunoprecipitation followed by Western blotting (). In detecting Myc-tagged RNF168, an extra band migrating slower than RNF168 appeared only in transfection combinations of HA-Ub plus Myc-tagged RNF168 (, L3, 5–8), indicating the occurrence of ubiquitinated-RNF168 forms. Interestingly, these RNF168 species decreased significantly upon ectopic expression of USP7 but not CS or M1 mutant (, L5 vs. L6–8), suggesting that USP7 specifically deubiquitinates these RNF168 forms. Indeed, immunoprecipitation experiments showed that RNF168 Ub-conjugates in higher molecular sizes were clearly detectable after immunoprecipitation and higher levels of RNF168 Ub-conjugates accumulated upon proteasome inhibition (, L3 and 5). Ectopically expressed WT USP7 decreased RNF168 Ub-conjugates, but CS and M1 mutant lost this function (, L5 vs. L6–8). This data is consistent with the inability of M1 mutant proteins to protect RNF168 from UVR-induced proteolysis (). Taken together, these results indicate that USP7 directly binds RNF168 and protects it from proteolytic turnover via deubiquitination.
Enforced expression of RNF8 and RNF168 bypasses USP7 deficiency
As mentioned above, E3 Ub ligases RNF8, RNF168, Ring1B and Bmi1 all function in the ubiquitination of H2A and γH2AX. The complexities and inter-relationship between monoubiquitination and polyubiquitination of H2A/X make it challenging to dissect the contributions of USP7-mediated RNF168 regulation to Ub-dependent signaling. Here, we undertook a functional bypass approach to experimentally address this quandary. We utilized an adenoviral expression system to deliver RNF8 and RNF168 to HCT116-USP7−/− cells, which are resistant to transfection presumably due to their slow growth from elevated cellular p53 and p21 protein levels. Upon adenoviral infection at the similar MOIs, HA-tagged RNF8 and RNF168 were readily detected in HCT116-USP7−/− cells () and, RNF8 and RNF168 appeared to stabilize each other when expressed together. In comparison to GFP-expressing control, expression of RNF8, RNF168 or RNF8 plus RNF168 increasingly enhanced the UV-induced γH2AX monoubiquitination (). Also, RNF8 and RNF168 caused the stabilization of BRCA1 and elevated its levels upon UVR exposure.
Figure 5. Adenovirus-mediated expression of RNF168 and RNF8 or168 partially rescues the formation of UVRIF of uH2A, FK2 and BRCA1 in HCT116-USP7−/− cells. (A) HCT116-USP7−/− cells were infected with the indicated adenoviral vectors expressing HA-tagged RNF8 and RNF168. Expression of RNF8 and RNF168 was examined by anti-HA Western blotting. (B) HCT116-USP7−/− cells were infected with indicated adenoviral vector or vector combination. The infected cells were harvested 2 h after UV exposure. The cell lysates from infected cells were examined by Western blotting for γH2AX and BRCA1 with anti-Actin blot as loading control. (C) The adenoviral vector infected cells were exposed to micropore UV irradiation at 100 J/m2. Two hour after UV irradiation, UVRIF of uH2A and γH2AX were visualized by immunofluorescence using specific antibodies. (D) UVRIF of FK2 and γH2AX. (E) UVRIF of BRCA1 and γH2AX. (F) Bar graph illustrates quantitative data of UVRIF. Mean ± SD of UVRIF vs. γH2AX positive cell ratio was calculated from 4–6 microscopic fields of 3 independent experiments. The p values were results from Student's t-test. Symbol * indicates P ≤ 0.05; Symbol ** indicates P ≤ 0.01. Calibration bar is 10 μm.
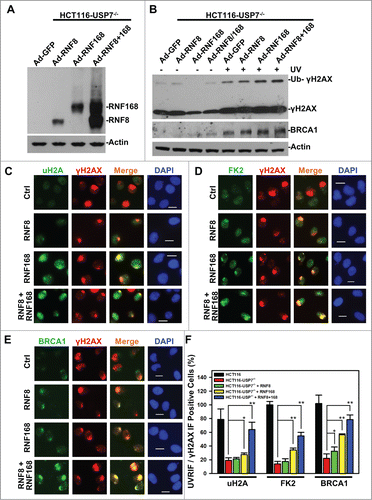
We next explored whether delivery of RNF8 or RNF168 rescues the formation of UVRIF of uH2A, FK2 and BRCA1 (). The results showed that RNF168 alone moderately increased the formation of UVRIF of uH2A. Whereas, RNF8 and RNF168 E3 ligases together significantly increased the formation of UVRIF of uH2A from ∼20 to 60%. More importantly, RNF168 alone significantly increased the formation of UVRIF of FK2 and BRCA1, which is further enhanced by joint expression of RNF8 and RNF168. It is noteworthy that the rescue of BRCA1 UVRIF by RNF168 alone or RNF8 and RNF168 together is in concurrence with stabilization of BRCA1 by these E3 ligases upon UVR exposure. Perhaps, the BRCA1 is stabilized due to its recruitment/binding to DNA strand breaks in chromatin; while non-chromatin bound BRCA1 is degraded in nucleoplasm. Similar phenomenon seems to exist for 53BP1, where 53BP1 is degraded upon IR except when bound to damage sites.Citation46
We noticed that formation of UVRIF of uH2A and FK2 was not fully restored to the levels in parental HCT116 cells, even when RNF8 and RNF168 were co-expressed. Given the function of Ring1B/Bmi1 in H2A/X monoubiquitination, we speculated that this is partially due to the regulatory effect of USP7 on Ring1B/Bmi1. Nonetheless, we cannot rule out the possibility that other regulatory components may also be involved. Thus, we concluded that in the absence of USP7 function, RNF168 becomes a limiting factor for the formation of γH2A/X polyubiquitination and subsequent BRCA1 recruitment.
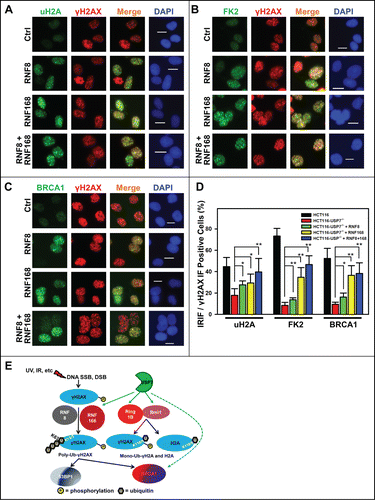
We further addressed whether delivery of RNF8 or RNF168 would also rescue the formation of IRIF. The results showed that expression of RNF8, RNF168 or RNF8 and RNF168 together increasingly promoted the formation of IRIF of uH2A, with a more significant boost in IRIF seen after co-expression of RNF8 and RNF168. Similarly, IRIF of FK2 and BRCA1 were also significantly increased by expression of RNF168 (). RNF168 expression alone elevated the IRIF of FK2 and BRCA1 to the levels comparable to that of joint expression of RNF8 and RNF168. Again, we observed that formation of the IRIF of uH2A, FK2 and BRCA1 was not fully restored to the levels in parental HCT116 cells (). Taken together, we concluded that expression of RNF168 or RNF8 and RNF168 partially bypass the USP7 deficiency in generating Ub-dependent signaling.
Figure 6. Adenovirus-mediated expression of RNF168 and RNF8 or168 partially rescues the formation of IRIF of uH2A, FK2 and BRCA1 in HCT116-USP7−/− cells. (A) HCT116-USP7−/− cells were infected with the indicated adenoviral vectors as in . The infected cells were exposed to IR at 10 Gy. One hour after IR, sub-nuclear foci formations as IRIF of uH2A, γH2AX, FK2 and BRCA1 were visualized by immunofluorescence using specific antibodies. (A) IRIF of uH2A and γH2AX. (B) IRIF of FK2 and γH2AX. (C) IRIF of BRCA1 and γH2AX. (D) Bar graph illustrates quantitative data of IRIF of γH2AX, uH2A, FK2 and BRCA1. Mean ± SD of IRIF vs. γH2AX positive cell ratio was calculated from 4–6 microscopic fields of 3 independent experiments. Symbol * indicates P ≤ 0.05; Symbol ** indicates P ≤ 0.01. Calibration bar is 10 μm. (E) Graphic illustration of regulation of DDR by USP7. Upon DNA damage, phosphorylation of H2AX is triggered by DNA strand-breaks and accumulated at damage sites. γH2AX is subsequently ubiquitinated by the concerted action of RNF168 and RNF8. The ubiquitinated γH2AX in turn facilitates the accumulation of repair factor BRCA1 and 53BP1. USP7 (green hue) regulates polyubiquitination of γH2AX, mono-ubiquitination of γH2AX and H2A through modulating the stability of RNF168, Ring1B and Bmi1 E3 ligases (red hue). USP7 may also directly regulate stability of BRCA1.
Discussion
In this report, we have uncovered a novel role of USP7 in regulation of H2A/X ubiquitination in DDR. Our data reveal that USP7 is involved in the regulation of H2A monoubiquitination and H2A/X polyubiquitination via its action on RNF168 in addition to Ring1B/Bmi1 Ub ligases. Mechanistically, USP7 directly interacts with RNF168 and deubiquitinates RNF168 Ub-conjugates. Although we do not rule out the possibility that other regulatory components in DDR may be subjected to USP7-mediated regulation, our functional bypass experiments support the hypothesis that USP7 plays an important role in generating H2A/X polyubiquitination signal. Previously, we and others demonstrated that deubiquitinating enzymes USP3 and USP44 counteract RNF168 via deubiquitinating H2A and γH2AX.Citation47,48 Thus, different deubiquitinating enzyme can regulate Ub-dependent signaling along divergent regulatory axis.
The regulation of Ring1B and Bmi1 Ub ligases by USP7 has been documented by previous studies.Citation35,36 USP7 directly deubiquitinates Ring1B Ub-conjugates, but it does not discriminate between the activating and proteolysis-target modes of ubiquitination.Citation35 Similarly, USP7 has been shown to regulate the ubiquitination status of Bmi1.Citation36 Knockdown of USP7 resulted in a reduction of total Bmi1 and caused a substantial decrease in the chromatin-associated Bmi1. Interestingly, ablation of USP7 resulted in de-repression of the INK4a tumor suppressor accompanied by loss of PRC1 occupancy at the INK4a locus, suggesting that USP7 plays a role in regulation of PRC1-mediated gene silencing. In this study, we found that this regulatory pathway affects global H2A ubiquitination and damage-induced H2A monoubiquitination as indicated by uH2A level as well as the formation of UVRIF and IRIF of uH2A. We like to suggest that USP7-mediated regulation of Ring1B/Bmi1 has a broader impact on cellular processes than its documented impact on gene silencing.
Here, we have identified RNF168 as a new target of USP7-mediated regulation. The defect in γH2AX monoubiquitination and H2A/X polyubiquitination in HCT116-USP7−/− cells may be readily explained by the reduction of RNF168. Nevertheless, the tangled relationship between Ring1B/Bmi1-mediated H2A/X ubiquitination and RNF8/RNF168-mediated H2A/X ubiquitination prohibits us to consider RNF168 as a single contributing factor. In several recent studies, it has been demonstrated that Bmi1-mediated histone ubiquitination promotes DSB repair. Yet, whether Bmi1-mediated histone ubiquitination contributes to γH2A/X polyubiquitination is a matter of debate. It was reported that Bmi1 knockout severely affected the formation of uH2A but not FK2 stripes when Bmi1 knockout MEFs were treated with laser scissors.Citation31 However, it was also reported that Bmi1 knockout MEF not only decreased the H2AX monoubiquitination but also reduced the FK2 IRIF.Citation30 In this study, both UVRIF and IRIF of uH2A and FK2 were severely compromised by USP7 deficiency. Adenovirus-mediated delivery of RNF168 or joint delivery of RNF8 and RNF168 into HCT116-USP7−/− cells partially restored the formation of uH2A and FK2 foci. Thus, the results support the hypothesis that Ring1B/Bmi1 contributes to H2A/X polyubiquitination.
Although H2A/X polyubiquitination drives the recruitment of both 53BP1 and BRCA1, we found that the recruitment of BRCA1 was more severely affected by USP7 deficiency than that of 53BP1. It was recently reported that 53BP1 contains a methyl-lysine-binding Tudor domain and ubiquitination-dependent recruitment (UDR) motif.Citation49 These structural features enable 53BP1 to recognize both dimethylated H4K20 and Ub mark in chromatin. It may be speculated that 53BP1 reads H2A/X polyubiquitination in chromatin with high efficiency. In support of this notion, we observed that formation of UVRIF and IRIF of 53BP1 was consistently high in HCT116 cells. In the case of BRCA1, besides the recruitment defect caused by compromised H2A/X polyubiquitination, we also found that BRCA1 abundance was dramatically reduced in HCT116-USP7−/− cells. Consistent with this, siRNA-mediated ablation of USP7 also decreased BRCA1 level. Although adenovirus-mediated expression of RNF8 or RNF168 or joint expression of RNF8 and RNF168 stabilizes the endogenous BRCA1 in the presence of DNA damage, but once again, expression of RNF8 and RNF168 only partially restored the UVRIF and IRIF of BRCA1. Given that UVR () and IR both induce BRCA1 degradationCitation50 and USP7 disrupted or depleted cells have low level of BRCA1, it is possible that USP7 also regulates BRCA1 stability through a direct mechanism (). The dual regulation of BRCA1 stability and recruitment by USP7 is a very likely the reason for the more severely defective foci formation of UVRIF and IRIF of BRCA1 in HCT116-USP7−/− cells.
In summary, our results describe a unique role of USP7 in regulation of Ub-dependent signaling in DDR by regulating Ring1B/Bmi1 and by monitoring RNF168 via deubiquitination (). These molecular mechanisms offer another regulatory layer in the control of Ub-dependent signaling besides the canonical function of disassembling the H2A/X polyubiquitination chains.
Materials and Methods
DNA constructs and antibodies
USP7 and its mutant constructs for both eukaryotic and bacterial expression were obtained from Dr. Yanhui Xu, Department of Biochemistry, Fudan University Medical School (Shanghai, China). Expression constructs for RNF168 were provided by Dr Junjie Chen in Department of Experimental Oncology, the University of Texas MD Anderson Cancer Center (Houston, Texas 77030). Rabbit anti-CPD antibodies were as previously described.Citation51,52 Anti-γH2A, BRCA1, 53BP1, Chk1, pChk1, p53, p21 and Bmi1 antibodies were purchased from Cell Signaling (Danvers, MA 01923). Anti-FLAG M2 agarose beads, anti-FLAG M2 and Anti-Myc antibodies were purchased from Sigma-Aldrich (St. Louis, MO 63103). Monoclonal FK2, anti-uH2A clone E6C5, RNF168 antibodies were from EMB Millipore (Billerica, MA 01821). RNF8 antibody and anti-USP7 antibodies were purchased from Epitomics/Abcam (http://www.abcam.com/). Ring1B antibody was from MBL international (Woburn, MA 01801).
Cell culture and transfection
HCT116 and HCT116-USP7−/− cells were obtained from Bert Vogelstein's laboratory, grown in McCoy's 5A medium supplemented with 10% FCS and antibiotics at 37°C in a humidified atmosphere of 5% CO2. HeLa cells were maintained in DMEM with 10% FCS. For transfection, exponentially growing cells were plated at a desired seeding density. Plasmid DNAs were transfected into HeLa or HCT116 cells lines using Fugene 6 transfection reagents (Promega Corporation, Madison, WI 53711).
RNA interference
USP7 siRNA (5′-ACCCUUGGACAAUAUUCCUdTdT-3′) and control siRNA (5′-UUCUCCGAACGUGUCACGUdT-3′) were synthesized by Thermo Scientific (Lafayette, CO 80026). The siRNA was transfected at final concentration of 100 nM into cells with Lipofectamine 2000 transfection reagent from Invitrogen (Life Technologies, Grand Island, NY 14072), according to manufacturer's instructions. HeLa cells were seeded at a proper density and grown overnight. For cell transfection, Lipofectamine2000 reagent was diluted with proper amount of Opti-MEM medium. The gene specific or control siRNA was added to the diluted reagent, incubated at room temperature (RT) for 20 min and then the Lipofectamine–small interfering RNA (siRNA) complex mix was added to the cells and incubation continued for 48 h. Second round siRNA transfection was carried out for 24-h.
GST pull-down assays
The GST and its fusion proteins were expressed in E. coli BL21 strain. The bacterial extracts were made in lysis buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1 mM EDTA, 1 mM DTT, 1% Triton X-100) with or without 1% sarkosyl. Equal amount of GST fusion proteins was immobilized on glutathione sepharose 4B beads in binding buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 0.1% (v/v) Triton X-100) and examined by Coomassie Blue staining.Citation38 The loaded beads were incubated with whole cell extracts containing ∼1.0 mg proteins made from control or 20 J/m2 UV-treated HCT116 cells in RIPA buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1% NP40, 0.5% deoxycholate and protease inhibitors). After incubation at 4 °C for 16 h, the beads were washed with RIPA and boiled in SDS sample buffer. The bound proteins were analyzed by Western blotting.
Immunoprecipitation
The FLAG-tagged WT-USP7 and M1 mutants were transiently expressed in HCT116 cells by 48 h-transfection using Fugene 6 reagents. The transfected cells were UV-irradiated at 20 J/m2 or left untreated and maintained for indicated time period. The cells were lysed in RIPA buffer in presence of protease inhibitors. The cell lysates were incubated with the anti-FLAG-M2 beads at 4°C overnight. The beads were washed 4 times with RIPA buffer and the bound proteins were released by boiling in SDS sample buffer and examined by Western blotting. For detecting Ub-conjugates of RNF168, the FLAG-tagged WT and mutant USP7 constructs were transfected into HCT116 cells in various combinations with expression vectors for Myc-tagged RNF168 and/or HA-tagged Ub. The transfected cells were treated with MG132 or its vehicle DMSO for additional 8 h and then lysed in RIPA buffer. The immunoprecipitation was done with anti-Myc antibody and the immuno-complexes were captured by protein A Plus G agarose beads and analyzed by Western blotting.
Adenovirus-mediated gene expression
Recombinant adenoviruses expression GFP, HA-tagged RNF8 or RNF168 were purchased from Applied Biological Materials (ABM) (Richmond, BC Canada). The adenoviruses were propagated and the multiplicity of infection (MOI) was assayed on HEK 293 cell line. To infect target cells, adenoviruses at desired MOI were incubated with the cells for 4 h to allow infection. The adenoviruses were then removed and the cell culture continued for 48 h. The infected cells were further processed for western blotting or immunofluorescence analysis.
Immunofluorescence
Immunofluorescent staining was conducted according to the method described previously.Citation27,51,53,54 For micropore UV irradiation, the cells or adenoviruses-infected cells were washed once with PBS and a 5-μm isopore polycarbonate filter was placed on top of cell monolayer, followed by 100-J/m2 UV irradiation. For IR, the cells or adenoviruses-infected cells were exposed to gamma ray source at 2 or 10 Gy. The UV or IR irradiated cells were maintained in a suitable medium for indicated time periods. The cells were then washed twice with cold PBS, permeabilized with 0.5% Triton X-100/PBS for about 8 min on ice as needed, and/or fixed with 2% paraformaldehyde in 0.5% Triton X-100 at 4°C for 30 min. The fixed cells were rinsed with twice with cold PBS and blocked with 20% normal goat serum in 0.1% Triton X-100/PBS, and stained with an appropriate primary antibody as well as fluorescein isothiocyanate (FITC) or Alexa Fluor 488 or Texas Red-conjugated secondary antibodies. The coverslips were mounted in Vectashield mounting medium with DAPI. The fluorescence images were obtained with a Nikon fluorescence microscope E80i (Tokyo, Japan) and processed with SPOT software and ImageJ software.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
1007785_Supplementary_Materials.zip
Download Zip (319.8 KB)Acknowledgments
The authors thank Dr. Bert Vogelstein for providing HCT116 and HCT116-USP7−/− cell lines. Authors also thank Drs. Yanhui Xu at Department of Biochemistry, Fudan University Medical School, and Dr. Yang Shi at Department of Cell Biology, Harvard Medical School, for providing expression constructs of USP7. Authors are grateful to Dr Junjie Chen at Department of Experimental Oncology, The University of Texas MD Anderson Cancer Center, for providing RNF168 expression constructs.
Funding
This work was supported by Public Health service Grants (ES2388, ES12991) from National Institute of Health.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Related Research Data
References
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 2009; 461:1071-8; PMID:19847258; http://dx.doi.org/10.1038/nature08467
- Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell 2007; 28:739-45; PMID:18082599; http://dx.doi.org/10.1016/j.molcel.2007.11.015
- Celeste A, Fernandez-Capetillo O, Kruhlak MJ, Pilch DR, Staudt DW, Lee A, Bonner RF, Bonner WM, Nussenzweig A. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat Cell Biol 2003; 5:675-9; PMID:12792649; http://dx.doi.org/10.1038/ncb1004
- Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ and Jackson SP. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 2005; 123:1213-26; PMID:16377563; http://dx.doi.org/10.1016/j.cell.2005.09.038
- Lukas C, Falck J, Bartkova J, Bartek J, Lukas J. Distinct spatiotemporal dynamics of mammalian checkpoint regulators induced by DNA damage. Nat Cell Biol 2003; 5:255-60; PMID:12598907; http://dx.doi.org/10.1038/ncb945
- Doil C, Mailand N, Bekker-Jensen S, Menard P, Larsen DH, Pepperkok R, Ellenberg J, Panier S, Durocher D, Bartek J, et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell 2009; 136:435-46; PMID:19203579; http://dx.doi.org/10.1016/j.cell.2008.12.041
- Huen MS, Grant R, Manke I, Minn K, Yu X, Yaffe MB, Chen J. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 2007; 131:901-14; PMID:18001825; http://dx.doi.org/10.1016/j.cell.2007.09.041
- Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, Lukas J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 2007; 131:887-900; PMID:18001824; http://dx.doi.org/10.1016/j.cell.2007.09.040
- Wang B, Elledge SJ. Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proc Natl Acad Sci U S A 2007; 104:20759-63; PMID:18077395; http://dx.doi.org/10.1073/pnas.0710061104
- Stewart GS, Panier S, Townsend K, Al-Hakim AK, Kolas NK, Miller ES, Nakada S, Ylanko J, Olivarius S, Mendez M, et al. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell 2009; 136:420-34; PMID:19203578; http://dx.doi.org/10.1016/j.cell.2008.12.042
- Mattiroli F, Vissers JH, Van Dijk WJ, Ikpa P, Citterio E, Vermeulen W, Marteijn JA, Sixma TK. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell 2012; 150:1182-95; PMID:22980979; http://dx.doi.org/10.1016/j.cell.2012.08.005
- Bergink S, Jentsch S. Principles of ubiquitin and SUMO modifications in DNA repair. Nature 2009; 458:461-7; PMID:19325626; http://dx.doi.org/10.1038/nature07963
- Lukas J, Bartek J. DNA repair: new tales of an old tail. Nature 2009; 458:581-3; PMID:19340068; http://dx.doi.org/10.1038/458581a
- Messick TE, Greenberg RA. The ubiquitin landscape at DNA double-strand breaks. J Cell Biol 2009; 187:319-26; PMID:19948475; http://dx.doi.org/10.1083/jcb.200908074
- Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010; 141:243-54; PMID:20362325; http://dx.doi.org/10.1016/j.cell.2010.03.012
- Callen E, Di VM, Kruhlak MJ, Nieto-Soler M, Wong N, Chen HT, Faryabi RB, Polato F, Santos M, Starnes LM, et al. 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell 2013; 153(6):1266-80; PMID:23727112
- Cao L, Xu X, Bunting SF, Liu J, Wang RH, Cao LL, Wu JJ, Peng TN, Chen J, Nussenzweig A, et al. A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol Cell 2009; 35:534-41; PMID:19716796; http://dx.doi.org/10.1016/j.molcel.2009.06.037
- Munoz MC, Laulier C, Gunn A, Cheng A, Robbiani DF, Nussenzweig A, Stark JM. RING finger nuclear factor RNF168 is important for defects in homologous recombination caused by loss of the breast cancer susceptibility factor BRCA1. J Biol Chem 2012; 287:40618-28; PMID:23055523; http://dx.doi.org/10.1074/jbc.M112.410951
- Sobhian B, Shao G, Lilli DR, Culhane AC, Moreau LA, Xia B, Livingston DM, Greenberg RA. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science 2007; 316:1198-202; PMID:17525341; http://dx.doi.org/10.1126/science.1139516
- Goldknopf IL, Taylor CW, Baum RM, Yeoman LC, Olson MO, Prestayko AW, Busch H. Isolation and characterization of protein A24, a "histone-like" non-histone chromosomal protein. J Biol Chem 1975; 250:7182-7; PMID:1165239
- Buchwald G, van der Stoop P, Weichenrieder O, Perrakis A, van LM, Sixma TK. Structure and E3-ligase activity of the Ring-Ring complex of polycomb proteins Bmi1 and Ring1b. EMBO J 2006; 25:2465-74; PMID:16710298; http://dx.doi.org/10.1038/sj.emboj.7601144
- Cao R, Tsukada Y, Zhang Y. Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol Cell 2005; 20:845-54; PMID:16359901; http://dx.doi.org/10.1016/j.molcel.2005.12.002
- Wang H, Wang L, Erdjument-Bromage H, Vidal M, Tempst P, Jones RS, Zhang Y. Role of histone H2A ubiquitination in Polycomb silencing. Nature 2004; 431:873-8; PMID:15386022; http://dx.doi.org/10.1038/nature02985
- Bergink S, Salomons FA, Hoogstraten D, Groothuis TA, de WH, Wu J, Yuan L, Citterio E, Houtsmuller AB, Neefjes J, et al. DNA damage triggers nucleotide excision repair-dependent monoubiquitylation of histone H2A. Genes Dev 2006; 20:1343-52; PMID:16702407; http://dx.doi.org/10.1101/gad.373706
- Marteijn JA, Bekker-Jensen S, Mailand N, Lans H, Schwertman P, Gourdin AM, Dantuma NP, Lukas J, Vermeulen W. Nucleotide excision repair-induced H2A ubiquitination is dependent on MDC1 and RNF8 and reveals a universal DNA damage response. J Cell Biol 2009; 186:835-47; PMID:19797077; http://dx.doi.org/10.1083/jcb.200902150
- Wu J, Huen MS, Lu LY, Ye L, Dou Y, Ljungman M, Chen J, Yu X. Histone ubiquitination associates with BRCA1-dependent DNA damage response. Mol Cell Biol 2009; 29:849-60; PMID:19015238; http://dx.doi.org/10.1128/MCB.01302-08
- Zhu Q, Wani G, Arab HH, El-Mahdy MA, Ray A, Wani AA. Chromatin restoration following nucleotide excision repair involves the incorporation of ubiquitinated H2A at damaged genomic sites. DNA Repair (Amst) 2009; 8:262-73; PMID:19059499; http://dx.doi.org/10.1016/j.dnarep.2008.11.007
- Pan MR, Peng G, Hung WC, Lin SY. Monoubiquitination of H2AX protein regulates DNA damage response signaling. J Biol Chem 2011; 286:28599-607; PMID:21676867; http://dx.doi.org/10.1074/jbc.M111.256297
- Ismail IH, McDonald D, Strickfaden H, Xu Z, Hendzel MJ. A small molecule inhibitor of polycomb repressive complex 1 inhibits ubiquitin signaling at DNA double-strand breaks. J Biol Chem 2013; 288:26944-54; PMID:23902761; http://dx.doi.org/10.1074/jbc.M113.461699
- Ismail IH, Andrin C, McDonald D, Hendzel MJ. BMI1-mediated histone ubiquitylation promotes DNA double-strand break repair. J Cell Biol 2010; 191:45-60; PMID:20921134; http://dx.doi.org/10.1083/jcb.201003034
- Ginjala V, Nacerddine K, Kulkarni A, Oza J, Hill SJ, Yao M, Citterio E, van LM, Ganesan S. BMI1 is recruited to DNA breaks and contributes to DNA damage-induced H2A ubiquitination and repair. Mol Cell Biol 2011; 31:1972-82; PMID:21383063; http://dx.doi.org/10.1128/MCB.00981-10
- Meulmeester E, Maurice MM, Boutell C, Teunisse AF, Ovaa H, Abraham TE, Dirks RW, Jochemsen AG. Loss of HAUSP-mediated deubiquitination contributes to DNA damage-induced destabilization of Hdmx and Hdm2. Mol Cell 2005; 18:565-76; PMID:15916963
- Li M, Chen D, Shiloh A, Luo J, Nikolaev AY, Qin J, Gu W. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 2002; 416:648-53; PMID:11923872; http://dx.doi.org/10.1038/nature737
- Cummins JM, Rago C, Kohli M, Kinzler KW, Lengauer C, Vogelstein B. Tumour suppression: disruption of HAUSP gene stabilizes p53. Nature 2004; 428:1; PMID:15058298; http://dx.doi.org/10.1038/nature02501
- de BP, Zaaroor-Regev D, Ciechanover A. Regulation of the Polycomb protein RING1B ubiquitination by USP7. Biochem Biophys Res Commun 2010; 400:389-95; PMID:20800574; http://dx.doi.org/10.1016/j.bbrc.2010.08.082
- Maertens GN, El Messaoudi-Aubert S, Elderkin S, Hiom K, Peters G. Ubiquitin-specific proteases 7 and 11 modulate Polycomb regulation of the INK4a tumour suppressor. EMBO J 2010; 29:2553-65; PMID:20601937; http://dx.doi.org/10.1038/emboj.2010.129
- Nguyen LK, Munoz-Garcia J, Maccario H, Ciechanover A, Kolch W, Kholodenko BN. Switches, excitable responses and oscillations in the Ring1B/Bmi1 ubiquitination system. PLoS Comput Biol 2011; 7:e1002317; PMID:22194680; http://dx.doi.org/10.1371/journal.pcbi.1002317
- He J, Zhu Q, Wani G, Sharma N, Han C, Qian J, Pentz K, Wang QE, Wani AA. Ubiquitin-specific protease 7 regulates nucleotide excision repair through deubiquitinating XPC protein and preventing XPC protein from undergoing ultraviolet light-induced and VCP/p97 protein-regulated proteolysis. J Biol Chem 2014; 289:27278-89; PMID:25118285; http://dx.doi.org/10.1074/jbc.M114.589812
- de Napoles M, Mermoud JE, Wakao R, Tang YA, Endoh M, Appanah R, Nesterova TB, Silva J, Otte AP, Vidal M, et al. Polycomb group proteins Ring1A/B link ubiquitylation of histone H2A to heritable gene silencing and X inactivation. Dev Cell 2004; 7:663-76; PMID:15525528; http://dx.doi.org/10.1016/j.devcel.2004.10.005
- Nacerddine K, Beaudry JB, Ginjala V, Westerman B, Mattiroli F, Song JY, van der Poel H, Ponz OB, Pritchard C, Cornelissen-Steijger P, et al. Akt-mediated phosphorylation of Bmi1 modulates its oncogenic potential, E3 ligase activity, and DNA damage repair activity in mouse prostate cancer. J Clin Invest 2012; 122:1920-32; PMID:22505453; http://dx.doi.org/10.1172/JCI57477
- Bohgaki M, Bohgaki T, El GS, Srikumar T, Maire G, Panier S, Fradet-Turcotte A, Stewart GS, Raught B, Hakem A, et al. RNF168 ubiquitylates 53BP1 and controls its response to DNA double-strand breaks. Proc Natl Acad Sci U S A 2013; 110:20982-7; PMID:24324146; http://dx.doi.org/10.1073/pnas.1320302111
- Pinato S, Scandiuzzi C, Arnaudo N, Citterio E, Gaudino G, Penengo L. RNF168, a new RING finger, MIU-containing protein that modifies chromatin by ubiquitination of histones H2A and H2AX. BMC Mol Biol 2009; 10:55; PMID:19500350; http://dx.doi.org/10.1186/1471-2199-10-55
- Ma H, Chen H, Guo X, Wang Z, Sowa ME, Zheng L, Hu S, Zeng P, Guo R, Diao J, et al. M phase phosphorylation of the epigenetic regulator UHRF1 regulates its physical association with the deubiquitylase USP7 and stability. Proc Natl Acad Sci U S A 2012; 109:4828-33; PMID:22411829; http://dx.doi.org/10.1073/pnas.1116349109
- Hu M, Li P, Li M, Li W, Yao T, Wu JW, Gu W, Cohen RE, Shi Y. Crystal structure of a UBP-family deubiquitinating enzyme in isolation and in complex with ubiquitin aldehyde. Cell 2002; 111:1041-54; PMID:12507430; http://dx.doi.org/10.1016/S0092-8674(02)01199-6
- Faesen AC, Dirac AM, Shanmugham A, Ovaa H, Perrakis A, Sixma TK. Mechanism of USP7/HAUSP activation by its C-terminal ubiquitin-like domain and allosteric regulation by GMP-synthetase. Mol Cell 2011; 44:147-59; PMID:21981925; http://dx.doi.org/10.1016/j.molcel.2011.06.034
- Hu Y, Wang C, Huang K, Xia F, Parvin JD, Mondal N. Regulation of 53BP1 protein stability by RNF8 and RNF168 is important for efficient DNA double-strand break repair. PLoS One 2014; 9:e110522; PMID:25337968; http://dx.doi.org/10.1371/journal.pone.0110522
- Sharma N, Zhu Q, Wani G, He J, Wang QE, Wani AA. USP3 counteracts RNF168 via deubiquitinating H2A and gammaH2AX at lysine 13 and 15. Cell Cycle 2014; 13:106-14; PMID:24196443; http://dx.doi.org/10.4161/cc.26814
- Mosbech A, Lukas C, Bekker-Jensen S, Mailand N. The deubiquitylating enzyme USP44 counteracts the DNA double-strand break response mediated by the RNF8 and RNF168 ubiquitin ligases. J Biol Chem 2013; 288:16579-87; PMID:23615962; http://dx.doi.org/10.1074/jbc.M113.459917
- Fradet-Turcotte A, Canny MD, Escribano-Diaz C, Orthwein A, Leung CC, Huang H, Landry MC, Kitevski-LeBlanc J, Noordermeer SM, Sicheri F, et al. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 2013; 499:50-4; PMID:23760478; http://dx.doi.org/10.1038/nature12318
- Liu W, Zong W, Wu G, Fujita T, Li W, Wu J, Wan Y. Turnover of BRCA1 involves in radiation-induced apoptosis. PLoS One 2010; 5:e14484; PMID:21217819; http://dx.doi.org/10.1371/journal.pone.0014484
- Wang QE, Zhu Q, Wani G, El-Mahdy MA, Li J, Wani AA. DNA repair factor XPC is modified by SUMO-1 and ubiquitin following UV irradiation. Nucleic Acids Res 2005; 33:4023-34; PMID:16030353; http://dx.doi.org/10.1093/nar/gki684
- Wani AA, D'Ambrosio SM, Alvi NK. Quantitation of pyrimidine dimers by immunoslot blot following sublethal UV-irradiation of human cells. Photochem Photobiol 1987; 46:477-82; PMID:3423120; http://dx.doi.org/10.1111/j.1751-1097.1987.tb04798.x
- Arab HH, Wani G, Ray A, Shah ZI, Zhu Q, Wani AA. Dissociation of CAK from core TFIIH reveals a functional link between XP-G/CS and the TFIIH disassembly state. PLoS One 2010; 5:e11007; PMID:20543986; http://dx.doi.org/10.1371/journal.pone.0011007
- Zhu Q, Wani G, Sharma N, Wani A. Lack of CAK complex accumulation at DNA damage sites in XP-B and XP-B/CS fibroblasts reveals differential regulation of CAK anchoring to core TFIIH by XPB and XPD helicases during nucleotide excision repair. DNA Repair (Amst) 2012; 11:942-50; PMID:23083890; http://dx.doi.org/10.1016/j.dnarep.2012.09.003