
Abstract
The retinoblastoma tumor suppressor (RB) was the first identified tumor suppressor based on germline predisposition to the pediatric eye tumor. Since these early studies, it has become apparent that the functional inactivation of RB is a common event in nearly all human malignancy. A great deal of research has gone into understanding how the loss of RB promotes tumor etiology and progression. Since malignant tumors are characterized by aberrant cell division, much of this research has focused upon the ability of RB to regulate the cell cycle by repression of proliferation-related genes. However, it is progressively understood that RB is an important mediator of multiple functions. One area that is gaining progressive interest is the emerging role for RB in regulating diverse features of immune function. These findings suggest that RB is more than simply a regulator of cellular proliferation; it is at the crossroads of proliferation and the immune response. Here we review the data related to the functional roles of RB on the immune system, relevance to immune evasion, and potential significance to the response to immune-therapy.
Canonical Function for RB
Multiple studies have demonstrated that RB can associate with numerous proteins across a disparate range of biological functions. However, given that RB has no catalytic activity, much of the attention has focused on how associations between RB and critical transcription factors impact transcriptional activity. These studies have taken on a particular importance given that RB is inactivated in a majority of human malignancies.Citation1,2 Although RB is capable of acting as a transcriptional activatorCitation3,4 as well as a repressor, the tumor suppressive functions of RB have generally been attributed to its ability to repress transcription or otherwise modulate cell cycle progression. In this regard, the interaction between RB and the E2F family of transcription factors serves as the prototypical example of RB function. The genes involved in DNA replication, DNA repair, and G2/M progression are largely modulated by the E2F/DP heterodimer.Citation1 During quiescence hypophosphorylated RB masks the transcriptional activation domains of E2F/DP both directly through interaction with these proteins as well as indirectly through the recruitment of additional co-repressors. Mitogenic signals lead to the induction of CDK4/6 and CDK2 activities that promote the hyperphosphorylation of RB. This event limits RB binding to E2F proteins and co-repressors, allowing for increased transcription of genes responsible for cell cycle progression.Citation1 Thus the role of RB in cell cycle control is relatively well established. However, additional roles for RB exist in the regulation of immune system development and the immune response.
Contribution to Immune Progenitor Fate Determination
The most obvious way in which RB impacts immune function is by acting as a critical regulator of transcriptional pathways at multiple checkpoints during progenitor differentiation. Haematopoietic stem cells (HSC) are multipotent progenitor cells with the capacity to differentiate into any of the haematopoietic lineages. The first decision in the process of HSC differentiation is whether the cell will become a lymphoid-lineage cell or a myeloid-lineage cell (). One of the determinants at this checkpoint in stem cell differentiation is the expression of the transcription factor PU.1.Citation5 PU.1 is a member of the ets family of transcription factors that is highly expressed in early myeloid lineage cells as well as specific mature myeloid populations. Thus, increased PU.1 transcriptional activity in multipotential progenitors directs these cells toward the myeloid lineage, whereas lower PU.1 activity leads these cells toward the lymphoid lineage (). PU.1 expression is still a factor in lineage determination in lymphoid cells, where cells with low levels of PU.1 will ultimately become B cells as opposed to T cells which do not seem to rely on PU.1 expression beyond very early stages. During early lineage commitment decisions, Id2, a member of the inhibitor of DNA binding family, binds PU.1 and keeps transcriptional activity in check, but ultimately this balance is maintained by mitogenic signals. In slowly proliferating cells, hypophosphorylated RB competitively binds Id2, allowing transcription of PU.1 target genes and commitment to the myeloid lineage, whereas in more rapidly dividing cells, hyperphosphorylated RB is unable to bind Id2, resulting in commitment to the lymphoid lineage.Citation6
Figure 1. RB is a critical regulator of haematopoietic differentiation and immune cell development. Haematopoietic lineage fate is determined by the activity of a handful of transcription factors including PU.1, GATA-1, SP1, and the C/EBP family. RB influences the transcriptional activity of all of these factors through various interactions. Beginning at the earliest stages of haematopoietic development, (A) RB hypophosphorylation allows RB to competitively bind Id2, allowing for increased transcription of PU.1 target genes that favor myeloid differentiation over lymphoid differentiation. RB plays an important role in determining common myeloid progenitor (CMP) fate as well. (B) While RB and E2F2 promote GATA1-mediated transcription, RB also cooperates with PU.1 to repress the GATA1 program and further enhances PU.1 driven gene expression by enhancing PU.1-promoting C/EBP family members. In myeloid lineage cells, (C) RB contributes to myeloid cell differentiation and activation by binding MDM2, enhancing the transcriptional availability of Sp1. Meanwhile, epigenetic silencing of RB by HDAC2 skews myeloid derived suppressor cell populations toward PMN-MDSCs, which are predominantly expressed in cancer.
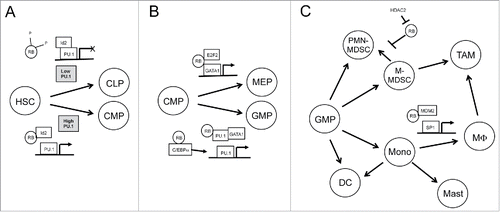
After the initial commitment to the myeloid lineage, RB remains an important factor in determining the ultimate fate of cells (). High expression of the transcription factor GATA-1 is required for maturation of common myeloid progenitors (CMPs) into megakaryocytes or erythroid cells and this is opposed by PU.1, leading to further myeloid differentiation. In either case RB is involved in the process as GATA-1 binding to a RB-E2F2 is required for erythropoiesis,Citation7 but cooperation between RB and PU.1 acts to repress GATA-1 expression, blocking erythroid differentiation.Citation8 In addition to PU.1, further maturation of myeloid cells into macrophages or granulocytes, and the proper expression of key immunoregulatory receptors and genes in these cells, also relies on the C/EBP family transcription factors as well as Sp1. The transcriptional activity of the C/EBP family is activated through complexes with RB and, similar to PU.1, RB acts as a transcriptional activator for Sp1 by releasing the factor from its inhibitor, MDM2.Citation9,10 Thus, RB manages to play a critical role in the development of lymphoid, myeloid, and erythroid lineage cells at multiple points and these lineage decisions have obvious ramifications for immune function ().
Given the numerous roles RB plays in myeloid cell development, it stands to reason that alterations in the RB family may play a role in myeloid-derived cancers. Indeed, a significant number of patients with acute myeloid leukemia (AML) have either truncated RB or a total loss of RB at the protein level.Citation11 Similarly, RB-family deficiency has myeloproliferative effects in mouse models and inhibition of the RB-suppressor CDK4/6 impedes proliferation in vitro and increases survival in an in vivo model of Flt3-associated AML.Citation12,13 On the other hand, it seems reasonable that loss or RB in early haematopoietic progenitors may also skew these developing cells toward the lymphocytic lineage. This possibility seems to be supported by findings in low-hypodiploid acute lymphoblastic leukemia, where more than 40% of cases have alteration in RB.Citation14
Functional Implications of RB Loss on Immune Responsiveness and Evasion
Loss of RB impacts the development of maturing leukocytes. However, this is not particularly representative of the events surrounding malignancy, where RB is functionally inactivated within the tumor but is still expressed in surrounding cell types. In this context, RB loss appears to have dual functions one of which is to influence the action of myeloid suppressor cells and influence the overall immunogenicity of the tumor.
One of the crucial functions of the immune system is detecting cellular abnormalities and clearing the aberrant cells before malignancy can develop. Thus, tumors that arise despite the normal immune response are likely to be the result of some form of immune evasion or localized immune suppression.Citation15 To this end, while inflammatory monocytes mature into macrophages under normal circumstances, as regulated by interactions between RB/PU.1/C/EBPs and other factors (Fig. 1), myelopoiesis is skewed in cancer resulting in the increased presence of immature, activated myeloid cells known as myeloid-derived suppressor cells (MDSCs).Citation5,9,10,16 Expansion of these cells results in the negative regulation of immune processes including the suppression of much of the T cell response and the production of cytokines and chemokines responsible for increased rates of angiogenesis and metastasis.Citation17,18 As opposed to cells undergoing physiologic myelopoiesis, MDSCs in mice have been shown to not upregulate RB expression after entering the periphery from the bone marrow.Citation19 As a result, these cells express less RB than equivalent monocyte populations and restoration of RB expression in these cells leads to increases in traditional myeloid populations at the expense of MDSCs. This alternative pathway of myeloid development is enforced by histone deacetylase-mediated silencing of RB in the presence of tumors (Fig. 1).Citation19
Studies with genetic deletion of RB have demonstrated the suppression of a broad range of genes associated with the immune response; including surface receptors, complement, lymphocyte factors, and cytokines.Citation20,21 Interestingly, this response appears to be highly evolutionarily conserved, wherein even in Drosophila models there is evidence for the suppression of genes associated with immune function in concert with RB loss.Citation22 The complex nature of the immune system makes it difficult to infer specifically how these alterations will affect immune recognition of resultant tumors. However, several key elements of RB loss impact Major Histocompatibility Complex (MHC), Interferon gamma response, and IL-6 signaling.
When activated, immune cells secrete cytokines that act as cell autonomous signaling mediators. These signals are generally considered to be either type 1, which stimulate cellular immune responses (e.g. IFNγ, TNFα) or type 2, which stimulate antibody responses (e.g., TGFβ, IL-6, IL-10) and proper immune function relies on the production, secretion, and recognition of these mediators to aid in recognition, uptake, presentation, and killing of pathogens and deleterious cells, such as those that would develop into tumors.Citation23-25 RB inhibits IL-6 gene expression and, conversely, IL-6 expression in multiple myeloma inhibits RB, leading to increased proliferation.Citation26,27 Similarly, in the context of RB-deficient acute myeloid leukemia, IL-6 mediates unchecked myeloid blast proliferation in an autocrine fashion.Citation28 Meanwhile, in mouse embryonic fibroblasts lacking RB, IFNγ is unable to stimulate MHC class II expression and this function is restored upon the addition of RB.Citation29 This is similar to the effect of RB loss in human tumor cell lines where it was found that RB-deficiency increases the stability of an Oct-1 containing repressosome on the HLA-DRA promoter.Citation30-38 The fact that this relationship could be manipulated by small molecule inhibition of RB suggests that this is an important gene regulatory network underlying tumor immunity and, presumably, the cell cycle.Citation39,40 Taken together, RB functions to limit type 1 cytokine signaling (IL-6) while also serving as a critical mediator of type 2 cytokine signaling (IFNγ) and MHC class II-mediated antigen presentation.
Anti-Viral Response—Is RB a Target for Immune Evasion by Oncogenic Viruses?
It has long been appreciated that RB protein is a target of various viral oncoproteins including human papilloma virus protein E7, SV40 large T antigen, and components of hepatitis B virus and hepatitis C virus.Citation41-45 That multiple viruses target RB suggests that RB is playing an important role in reducing the ability of these viruses to replicate efficiently. While this has previously been attributed to the loss of RB yielding enhanced capacity for DNA replication, and thus disseminating viral products more rapidly, there is mounting evidence to suggest that RB is also a key regulator of the anti-viral immune response ().
Figure 2. Anti-viral immunity requires RB. Recognition of viral motifs by mediators of the innate immune response triggers an anti-viral immune response through a variety of signaling pathways. Expression of NF-κB and IFN-mediated gene targets require RB to elicit an efficient immune response. Various viral products including E1A, E7, and Large T antigen disrupt these essential processes through their interactions with RB.
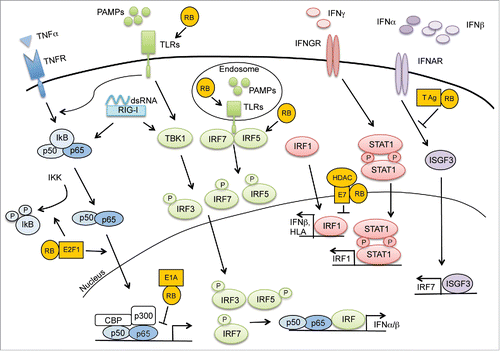
The innate immune system is designed to respond rapidly once specific molecular patterns are detected.Citation46 One example of this is the expression of toll-like receptors (TLRs) on antigen presenting cells. Each TLR has a defined ligand that corresponds to a particular pathogen (e.g. LPS-TLR4, ssRNA-TLR7, dsRNA-TLR3). Once a pathogenic ligand engages its cognate TLR, a series of signaling pathways are activated including those related to type I IFNs as well as NF-κB.Citation46 Thus, proper regulation of TLRs and their signaling pathways are crucial to recognition and clearance of pathogens and aberrant-self cells.Citation47 Examination of RB-deficient MEFs or cells transduced with RB-specific shRNA revealed that RB was necessary for TLR3 expression and efficient downstream signaling, including cytokine production. Meanwhile, antithetical to its typical role as a transactivator, E2F1 downregulated TLR3 expression in epithelial cells by binding to the TLR3 promoter and repressing transcription. This inhibition was initially independent of RB, but treatment with the TLR3-ligand poly(I:C) stimulated RB expression, leading to increased levels of TLR3 as a result of RB-mediated sequestration of E2F1 away from the TLR3 promoter. This positive feedback loop created by poly(I:C) and RB demonstrates the importance of RB as a regulator of immune signaling.Citation48
Initial recognition of viral motifs leads to increased type I IFN production through pathways mediated by interferon regulatory transcription factors (IRFs) and NF-κB. Subsequent binding of type I IFN to its receptor results in the expression of numerous immune function genes in anti-viral gene networks. Thus, IRF/NF-κB-mediated signaling provides the first line of defense against infection by viruses. In RB-deficient cells, viral production is greater than in RB-sufficient cells, suggesting that RB serves to limit viral infectivity. This enhanced rate of viral infection is associated with decreased activation of NF-κB and reintroduction of RB results in measurable IκB degradation, allowing for the restoration of some NF-κB activity.Citation49 These data suggest that RB acts as a modulator of IκB stability and is required for NF-κB activity. Although loss of RB does not impact IRF3 expression, IRF5 is decreased in RB-deficient cells, suggesting that RB plays other roles in the IFN response aside from NF-κB.Citation21,49 Given that type I IFNs and NF-κB have numerous transcriptional targets, this implies a mechanism by which RB may greatly impact expression of numerous immune-related genes ().
Polyoma viruses have long been recognized as having oncogenic potential at least in part because the polyoma large T antigen (T Ag) is capable of driving DNA synthesis and inhibiting cell cycle suppressors (including the RB family).Citation50-52 Additionally, T Ag can negatively impact the host immune response. As has previously been discussed, viral immunity relies on a robust IFN response stimulated by the recognition of viral components. Merkel cell polyoma virus T Ag promotes immune evasion via the downregulation of TLR9 expression, the receptor that mediates immune responsiveness to unmethylated CpG DNA dinucleotides.Citation53 Similarly, infection with murine polyoma virus elicits an inefficient immune response characterized by resistance to IFNβ and ultimately results in the development of tumors in the host. While wild-type virus is able to limit the activation of the STAT1/STAT2/IRF9 complex (ISGF3), a mutant virus lacking RB-binding capacity is incapable of mediating the same response (). This is a function of the RB-binding deficient virus being unable to bind and inhibit the STAT`1-activating tyrosine kinase JAK1.Citation54 Although the connection between T Ag, RB, and JAK1 remains poorly understood, this report demonstrates an immunoregulatory role for the T Ag-RB complex that would seemingly complement the proliferative impact of this association in promoting viral replication.
Although type I IFN plays an immediate role in establishing the anti-viral host response, this initial response can also result in the production of IFNγ.Citation55 IFNγ production can lead to the chemoattraction of numerous immune cells through the production of CXCL10 in addition to inducing increased antigen presentation on macrophages.Citation56 Limiting these responses is vital to the ability of viruses to evade the immune response. Given that RB plays a role in antigen presentation in response to IFNγ stimulation, it is not surprising that RB acts as a viral target in dampening the IFNγ response. Following IFNγ binding, STAT1 is phosphorylated and translocates to the nucleus where it binds interferon gamma activated sequence (GAS) domain, resulting in increased transcription of genes such as IRF1. Upon further activation of IRF1, numerous genes are transcribed including several genes associated with antigen presentation (e.g., HLA, TAP1), chemokines (e.g. CCL5), and IFNβ.Citation57 To counteract this process, the human papilloma virus (HPV) oncoprotein E7 transrepresses IRF-1 targets by recruiting HDAC ().Citation58 HPV-E7 is strongly associated with the shift from latency to cellular transformation in HPV, largely due to its affinity for host RB and the resultant increase in cellular proliferation caused by HPV-E7-mediated RB-loss. Although E7 binds HDAC independently of RB, the RB-binding domain is necessary for the repression, suggesting that RB plays a role in this process.Citation58 HPV-E7 has been shown to interfere with this pathway by blocking STAT1 activation as well, although RB was neither implicated nor excluded in this association.Citation59
Following the initial interferon responses to viral infection, other mediators, including tumor necrosis factor α (TNFα), provide critical support to the immune response in its efforts to eliminate viruses. There are 2 distinct outcomes following the recognition of TNFα by its receptor. One results in caspase-mediated apoptosis while the other promotes NF-κB activation resulting in increased expression of pro-inflammatory and anti-apoptotic proteins. Adenoviral infection leads to the suppression of the pro-inflammatory TNFα pathway, presumably to protect against the initiation of host immunity that would threaten viral survival and propagation even at the cost of promoting cellular death by apoptosis. To this end, it has been determined that this skewing toward TNFα-induced cellular damage and apoptosis is dependent on the formation of a complex between the adenoviral E1A protein, RB, and p300 that directly disrupts NF-κB activation at late stages in the nucleus ().Citation60,61 Given the functional and structural similarities with E1A, it is worth noting that E7 plays a similar role in NF-kB inhibition but does not form a ternary structure with RB and p300 and thus there is currently no evidence that RB plays a role in this association.Citation61
These roles in regulating the innate immune response to viral infection have important implications in light of the paradigmatic role of RB in cell cycle regulation. Prolonged tumor progression depends on multiple factors including aberrantly increased cellular proliferation and the ability to evade immune recognition. Given that these ends coincide with the viral imperative to replicate, it is of little surprise that some viral infections ultimately result in the rise of malignancies. Indeed, it was recently shown that the pattern of decreased immune gene expression demonstrated by acute RB-loss is similar to that found in patients with advanced hepatocellular carcinoma (HCC), a disease characterized by both RB-silencing and decreased immune responsiveness at later stages. This gene signature also was associated with poor prognosis, suggesting a potential therapeutic approach for a subset of patients with advanced HCC.Citation21
Impact on Immunotherapies
Given the important role for RB in limiting proliferation and the prevalence of RB disruption in malignancies, it is not surprising that RB has been an attractive target for cancer therapies. Citation62 As additional roles for RB in the immune response are reported, it is similarly tempting to consider how RB modulation may impact cancer immunotherapies. Recent reports offer some evidence that RB contributes in immunotherapy efficacy. Since a number of cancers result primarily from viral infections, vaccination against these viruses is perhaps the most basic form of immunotherapy. Cervical cancer, where HPV accounts for virtually all cases, is a prime example of where vaccination can reduce cancer incidence.Citation63 Continued development of more effective vaccines has led to the usage of DNA-based vaccines. While these have advantages over typical protein-based vaccine approaches, they tend to be less immunogenic than traditional vaccines and they also run the risk of inserting viral DNA into the host genome and, in the case of RB-associating viruses like HPV, causing unchecked proliferation. To avoid this, a DNA vaccine for HPV was designed in which the E7 protein was altered to abrogate RB binding. Not only was this vaccine stable but it was also more immunogenic than a DNA virus produced from full E7, directly implicating RB in the immunogenic response against HPV.Citation64
Baccilus Calmette-Guérin (BCG) therapy was initially designed as a vaccine against tuberculosis. More recently, BCG has become a promising immunotherapy adjuvant in several malignancies including melanoma, colorectal cancer, and cervical cancer. Usage of BCG is particularly prevalent in superficial bladder cancer where instillation of BCG after tumor resection has become a first-line treatment. While the exact mechanism by which BCG triggers an immune response against bladder tumors remains unclear, RB expression predicts response to BCG.Citation65,66 Although these data certainly include the effects of RB alteration on proliferation, given that RB-underexpression specifically predicts BCG nonresponse when BCG is given in concert with IFNα, these reports provide evidence that RB is playing a role in the immunological response mediated by BCG instillation.Citation65
These reports suggest that tumors deficient in RB could represent a particular challenge for immune-therapies. Perhaps the most well known such therapies involve immune checkpoint blockade as a result of inhibiting the function of CTLA-4 or PD-L1.Citation67 While expression of PD-L1 in tumor cells or tumor-associated stroma directly inhibits the cytotoxic function of CD8+ T cells by blocking co-receptor binding, CTLA-4 expression on T cells reduces the capacity of antigen presenting cells to stimulate antigen-specific responses in other T cells.Citation68 Although these 2 immune checkpoints have differing mechanisms, blockade of either checkpoint is intended to jumpstart an anti-tumor immune response through the restoration of T cell function. In this regard, proper function of either immune checkpoint blockade relies on TCR-MHC interaction. Similarly, a number of MHC class II-based tumor vaccines have demonstrated some efficacy in various solid tumor models based on their ability to initiate a tumor-specific CD4+ T cell response.Citation69-72 Given that RB is required for proper MHC expression, checkpoint blockade or other MHC class II dependent immunotherapies may be less effective in RB-deficient tumors as a result of a potentially inadequate T cell response.Citation21,29-39,59 Further study in the context of ongoing clinical studies would be necessary to fully understand how RB loss impacts checkpoint inhibitors and other immune-based therapies.
Summary
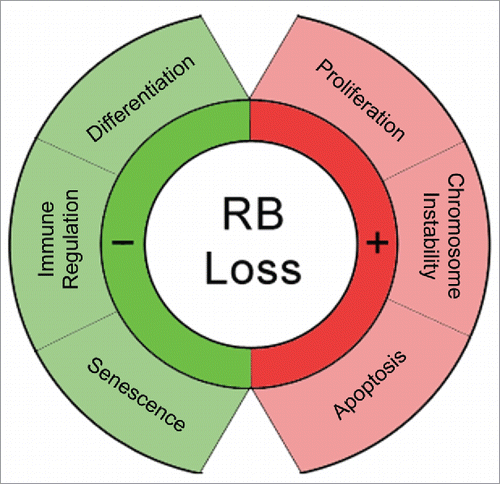
The retinoblastoma tumor suppressor has been extensively studied, yet we still do not fully appreciate the diverse roles it plays in maintaining homeostasis by regulating proliferation, apoptosis, immunity, and development (). Although RB has been associated with the immune response for many years now, the role of RB in immunity has expanded significantly over the last few years to suggest that RB is an important mediator of innate immunity and that at least some of this functionality may be separate from the role of RB in regulating proliferation. Taken in concert, RB appears to be positioned at a key crossroad between proliferation and immunity and this can potentially be exploited in future cancer immunotherapies to improve therapeutic efficacy. Given that RB function is lost or inactivated in a majority of all human malignancies, further study is warranted to explore whether the molecular interactions that mediate the cell cycle functionality of RB and those that mediate the immune functionality of RB overlap or if it is possible to target individual aspects of RB.
Figure 3. Loss of RB impacts multiple key functions. RB is a multi-functional protein with diverse roles in regulating cellular processes. Therapeutic interventions targeting RB should keep in mind the variety of functions that are potentially being altered.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer 2008; 8:671-82; PMID:18650841; http://dx.doi.org/10.1038/nrc2399
- Knudsen ES, Knudsen KE. Tailoring to RB: tumour suppressor status and therapeutic response. Nat Rev Cancer 2008; 8:714-24; PMID:19143056; http://dx.doi.org/10.1038/nrc2401
- Miccadei S, Provenzano C, Mojzisek M, Natali PG, Civitareale D. Retinoblastoma protein acts as Pax 8 transcriptional coactivator. Oncogene 2005; 24:6993-7001; PMID:16007137; http://dx.doi.org/10.1038/sj.onc.1208861
- Thomas DM, Carty SA, Piscopo DM, Lee JS, Wang WF, Forrester WC, Hinds PW. The retinoblastoma protein acts as a transcriptional coactivator required for osteogenic differentiation. Mol Cell 2001; 8:303-16; PMID:11545733; http://dx.doi.org/10.1016/S1097-2765(01)00327-6
- Scott EW, Fisher RC, Olson MC, Kehrli EW, Simon MC, Singh H. PU.1 functions in a cell-autonomous manner to control the differentiation of multipotential lymphoid-myeloid progenitors. Immunity 1997; 6:437-47; PMID:9133423; http://dx.doi.org/10.1016/S1074-7613(00)80287-3
- Kueh HY, Champhekar A, Nutt SL, Elowitz MB, Rothenberg EV. Positive feedback between PU.1 and the cell cycle controls myeloid differentiation. Science 2013; 341:670-3; PMID:23868921; http://dx.doi.org/10.1126/science.1240831
- Kadri Z, Shimizu R, Ohneda O, Maouche-Chretien L, Gisselbrecht S, Yamamoto M, Romeo PH, Leboulch P, Chretien S. Direct binding of pRb/E2F-2 to GATA-1 regulates maturation and terminal cell division during erythropoiesis. PLoS Biol 2009; 7:e1000123; PMID:19513100; http://dx.doi.org/10.1371/journal.pbio.1000123
- Rekhtman N, Choe KS, Matushansky I, Murray S, Stopka T, Skoultchi AI. PU.1 and pRB interact and cooperate to repress GATA-1 and block erythroid differentiation. Mol Cell Biol 2003; 23:7460-74; PMID:14559995; http://dx.doi.org/10.1128/MCB.23.21.7460-7474.2003
- Charles A, Tang X, Crouch E, Brody JS, Xiao ZX. Retinoblastoma protein complexes with C/EBP proteins and activates C/EBP-mediated transcription. J Cell Biochem 2001; 83:414-25; PMID:11596110; http://dx.doi.org/10.1002/jcb.1239
- Johnson-Pais T, Degnin C, Thayer MJ. pRB induces Sp1 activity by relieving inhibition mediated by MDM2. Proc Natl Acad Sci U S A 2001; 98:2211-6; PMID:11226218; http://dx.doi.org/10.1073/pnas.051415898
- Jamal R, Gale RE, Shaun N, Thomas B, Wheatley K, Linch DC. The retinoblastoma gene (rb1) in acute myeloid leukaemia: analysis of gene rearrangements, protein expression and comparison of disease outcome. Br J Haematol 1996; 94:342-51; PMID:8759895; http://dx.doi.org/10.1046/j.1365-2141.1996.d01-1804.x
- Walkley CR, Shea JM, Sims NA, Purton LE, Orkin SH. Rb regulates interactions between hematopoietic stem cells and their bone marrow microenvironment. Cell 2007; 129:1081-95; PMID:17574022; http://dx.doi.org/10.1016/j.cell.2007.03.055
- Viatour P, Somervaille TC, Venkatasubrahmanyam S, Kogan S, McLaughlin ME, Weissman IL, Butte AJ, Passegue E, Sage J. Hematopoietic stem cell quiescence is maintained by compound contributions of the retinoblastoma gene family. Cell Stem Cell 2008; 3:416-28; PMID:18940733; http://dx.doi.org/10.1016/j.stem.2008.07.009
- Holmfeldt L, Wei L, Diaz-Flores E, Walsh M, Zhang J, Ding L, Payne-Turner D, Churchman M, Andersson A, Chen SC, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet 2013; 45:242-52; PMID:23334668; http://dx.doi.org/10.1038/ng.2532
- Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol 2002; 3:991-8; PMID:12407406; http://dx.doi.org/10.1038/ni1102-991
- Solito S, Marigo I, Pinton L, Damuzzo V, Mandruzzato S, Bronte V. Myeloid-derived suppressor cell heterogeneity in human cancers. Ann N Y Acad Sci 2014; 1319:47-65; PMID:24965257; http://dx.doi.org/10.1111/nyas.12469
- Ostrand-Rosenberg S, Sinha P, Beury DW, Clements VK. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin Cancer Biol 2012; 22:275-81; PMID:22313874; http://dx.doi.org/10.1016/j.semcancer.2012.01.011
- Dai J, El Gazzar M, Li GY, Moorman JP, Yao ZQ. Myeloid-Derived Suppressor Cells: Paradoxical Roles in Infection and Immunity. J Innate Immun 2014; PMID:25401944
- Youn JI, Kumar V, Collazo M, Nefedova Y, Condamine T, Cheng P, Villagra A, Antonia S, McCaffrey JC, Fishman M, et al. Epigenetic silencing of retinoblastoma gene regulates pathologic differentiation of myeloid cells in cancer. Nat Immunol 2013; 14:211-20; PMID:23354483; http://dx.doi.org/10.1038/ni.2526
- Markey MP, Bergseid J, Bosco EE, Stengel K, Xu H, Mayhew CN, Schwemberger SJ, Braden WA, Jiang Y, Babcock GF, et al. Loss of the retinoblastoma tumor suppressor: differential action on transcriptional programs related to cell cycle control and immune function. Oncogene 2007; 26:6307-18; PMID:17452985; http://dx.doi.org/10.1038/sj.onc.1210450
- Hutcheson J, Bourgo RJ, Balaji U, Ertel A, Witkiewicz AK, Knudsen ES. Retinoblastoma protein potentiates the innate immune response in hepatocytes: significance to hepatocellular carcinoma. Hepatology 2014; PMID:24824777
- Longworth MS, Walker JA, Anderssen E, Moon NS, Gladden A, Heck MM, Ramaswamy S, Dyson NJ. A shared role for RBF1 and dCAP-D3 in the regulation of transcription with consequences for innate immunity. PLoS Genet 2012; 8:e1002618; PMID:22496667; http://dx.doi.org/10.1371/journal.pgen.1002618
- Ikeda H, Chamoto K, Tsuji T, Suzuki Y, Wakita D, Takeshima T, Nishimura T. The critical role of type-1 innate and acquired immunity in tumor immunotherapy. Cancer science 2004; 95:697-703; PMID:15471553; http://dx.doi.org/10.1111/j.1349-7006.2004.tb03248.x
- Pulendran B, Artis D. New paradigms in type 2 immunity. Science 2012; 337:431-5; PMID:22837519; http://dx.doi.org/10.1126/science.1221064
- Dalessandri T, Strid J. Beneficial autoimmunity at body surfaces - immune surveillance and rapid type 2 immunity regulate tissue homeostasis and cancer. Front Immunol 2014; 5:347; PMID:25101088; http://dx.doi.org/10.3389/fimmu.2014.00347
- Urashima M, Ogata A, Chauhan D, Vidriales MB, Teoh G, Hoshi Y, Schlossman RL, DeCaprio JA, Anderson KC. Interleukin-6 promotes multiple myeloma cell growth via phosphorylation of retinoblastoma protein. Blood 1996; 88:2219-27; PMID:8822942
- Santhanam U, Ray A, Sehgal PB. Repression of the interleukin 6 gene promoter by p53 and the retinoblastoma susceptibility gene product. Proc Natl Acad Sci U S A 1991; 88:7605-9; PMID:1652755; http://dx.doi.org/10.1073/pnas.88.17.7605
- Zhu YM, Bradbury DA, Keith FJ, Russell N. Absence of retinoblastoma protein expression results in autocrine production of interleukin-6 and promotes the autonomous growth of acute myeloid leukemia blast cells. Leukemia 1994; 8:1982-8; PMID:7967742
- Zhu X, Pattenden S, Bremner R. pRB is required for interferon-gamma-induction of the MHC class II abeta gene. Oncogene 1999; 18:4940-7; PMID:10490828; http://dx.doi.org/10.1038/sj.onc.1202876
- Lu Y, Tschickardt ME, Schmidt BJ, Blanck G. IFN-gamma inducibility of class II transactivator is specifically lacking in human tumour lines: relevance to retinoblastoma protein rescue of IFN-gamma inducibility of the HLA class II genes. Immunol Cell Biol 1997; 75:325-32; PMID:9315472; http://dx.doi.org/10.1038/icb.1997.50
- Lu Y, Boss JM, Hu SX, Xu HJ, Blanck G. Apoptosis-independent retinoblastoma protein rescue of HLA class II messenger RNA IFN-gamma inducibility in non-small cell lung carcinoma cells. Lack of surface class II expression associated with a specific defect in HLA-DRA induction. J Immunol 1996; 156:2495-502; PMID:8786310
- Lu Y, Ussery GD, Jacim M, Tschickardt M, Boss JM, Blanck G. Retinoblastoma protein regulation of surface CD74 (invariant chain) expression in breast carcinoma cells. Mol Immunol 1994; 31:1365-8; PMID:7997248; http://dx.doi.org/10.1016/0161-5890(94)90055-8
- Lu Y, Ussery GD, Muncaster MM, Gallie BL, Blanck G. Evidence for retinoblastoma protein (RB) dependent and independent IFN-gamma responses: RB coordinately rescues IFN-gamma induction of MHC class II gene transcription in noninducible breast carcinoma cells. Oncogene 1994; 9:1015-9; PMID:8134104
- Osborne A, Tschickardt M, Blanck G. Retinoblastoma protein expression facilitates chromatin remodeling at the HLA-DRA promoter. Nucleic Acids Res 1997; 25:5095-102; PMID:9396821; http://dx.doi.org/10.1093/nar/25.24.5095
- Osborne AR, Zhang H, Blanck G. Oct-1 DNA binding activity unresponsive to retinoblastoma protein expression prevents MHC class II induction in a non-small cell lung carcinoma cell line. Mol Immunol 2006; 43:710-6; PMID:16360016; http://dx.doi.org/10.1016/j.molimm.2005.03.013
- Osborne A, Zhang H, Yang WM, Seto E, Blanck G. Histone deacetylase activity represses gamma interferon-inducible HLA-DR gene expression following the establishment of a DNase I-hypersensitive chromatin conformation. Mol Cell Biol 2001; 21:6495-506; PMID:11533238; http://dx.doi.org/10.1128/MCB.21.19.6495-6506.2001
- Zhang H, Shepherd AT, Eason DD, Wei S, Diaz JI, Djeu JY, Wu GD, Blanck G. Retinoblastoma protein expression leads to reduced Oct-1 DNA binding activity and enhances interleukin-8 expression. Cell Growth Differ 1999; 10:457-65; PMID:10437913
- Osborne AR, Zhang H, Fejer G, Palubin KM, Niesen MI, Blanck G. Oct-1 maintains an intermediate, stable state of HLA-DRA promoter repression in Rb-defective cells: an Oct-1-containing repressosome that prevents NF-Y binding to the HLA-DRA promoter. J Biol Chem 2004; 279:28911-9; PMID:15105429; http://dx.doi.org/10.1074/jbc.M403118200
- Pillai S, Szekeres K, Lawrence NJ, Chellappan SP, Blanck G. Regulation of interlocking gene regulatory network subcircuits by a small molecule inhibitor of retinoblastoma protein (RB) phosphorylation: cancer cell expression of HLA-DR. Gene 2013; 512:403-7; PMID:23041127; http://dx.doi.org/10.1016/j.gene.2012.09.092
- Xu L, Niesen MI, Blanck G. Linkage of a tumor immune function and cell cycle de-regulation via a gene regulatory network subcircuit. Mol Immunol 2009; 46:569-75; PMID:18952290; http://dx.doi.org/10.1016/j.molimm.2008.07.035
- Gage JR, Meyers C, Wettstein FO. The E7 proteins of the nononcogenic human papillomavirus type 6b (HPV-6b) and of the oncogenic HPV-16 differ in retinoblastoma protein binding and other properties. J Virol 1990; 64:723-30; PMID:2153238
- Zwerschke W, Jansen-Durr P. Cell transformation by the E7 oncoprotein of human papillomavirus type 16: interactions with nuclear and cytoplasmic target proteins. Adv Cancer Res 2000; 78:1-29; PMID:10547667; http://dx.doi.org/10.1016/S0065-230X(08)61022-2
- Croul S, Otte J, Khalili K. Brain tumors and polyomaviruses. J Neurovirol 2003; 9:173-82; PMID:12707848; http://dx.doi.org/10.1080/13550280390194055
- Helt AM, Galloway DA. Mechanisms by which DNA tumor virus oncoproteins target the Rb family of pocket proteins. Carcinogenesis 2003; 24:159-69; PMID:12584163; http://dx.doi.org/10.1093/carcin/24.2.159
- Knudsen ES, Gopal P, Singal AG. The changing landscape of hepatocellular carcinoma: etiology, genetics, and therapy. Am J Pathol 2014; 184:574-83; PMID:24388934; http://dx.doi.org/10.1016/j.ajpath.2013.10.028
- Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol 2004; 4:499-511; PMID:15229469; http://dx.doi.org/10.1038/nri1391
- Rakoff-Nahoum S, Medzhitov R. Toll-like receptors and cancer. Nat Rev Cancer 2009; 9:57-63; PMID:19052556; http://dx.doi.org/10.1038/nrc2541
- Taura M, Suico MA, Koyama K, Komatsu K, Miyakita R, Matsumoto C, Kudo E, Kariya R, Goto H, Kitajima S, et al. Rb/E2F1 regulates the innate immune receptor Toll-like receptor 3 in epithelial cells. Mol Cell Biol 2012; 32:1581-90; PMID:22310660; http://dx.doi.org/10.1128/MCB.06454-11
- Garcia MA, Gallego P, Campagna M, Gonzalez-Santamaria J, Martinez G, Marcos-Villar L, Vidal A, Esteban M, Rivas C. Activation of NF-kB pathway by virus infection requires Rb expression. PLoS One 2009; 4:e6422; PMID:19649275; http://dx.doi.org/10.1371/journal.pone.0006422
- DeCaprio JA, Ludlow JW, Figge J, Shew JY, Huang CM, Lee WH, Marsilio E, Paucha E, Livingston DM. SV40 large tumor antigen forms a specific complex with the product of the retinoblastoma susceptibility gene. Cell 1988; 54:275-83; PMID:2839300; http://dx.doi.org/10.1016/0092-8674(88)90559-4
- Harris KF, Christensen JB, Imperiale MJ. BK virus large T antigen: interactions with the retinoblastoma family of tumor suppressor proteins and effects on cellular growth control. J Virol 1996; 70:2378-86; PMID:8642665
- Krynska B, Gordon J, Otte J, Franks R, Knobler R, DeLuca A, Giordano A, Khalili K. Role of cell cycle regulators in tumor formation in transgenic mice expressing the human neurotropic virus, JCV, early protein. J Cell Biochem 1997; 67:223-30; PMID:9328827; http://dx.doi.org/10.1002/(SICI)1097-4644(19971101)67:2%3c223::AID-JCB7%3e3.0.CO;2-Z
- Shahzad N, Shuda M, Gheit T, Kwun HJ, Cornet I, Saidj D, Zannetti C, Hasan U, Chang Y, Moore PS, et al. The T antigen locus of Merkel cell polyomavirus downregulates human Toll-like receptor 9 expression. J Virol 2013; 87:13009-19; PMID:24067965; http://dx.doi.org/10.1128/JVI.01786-13
- Weihua X, Ramanujam S, Lindner DJ, Kudaravalli RD, Freund R, Kalvakolanu DV. The polyoma virus T antigen interferes with interferon-inducible gene expression. Proc Natl Acad Sci U S A 1998; 95:1085-90; PMID:9448289; http://dx.doi.org/10.1073/pnas.95.3.1085
- Nguyen KB, Cousens LP, Doughty LA, Pien GC, Durbin JE, Biron CA. Interferon alpha/beta-mediated inhibition and promotion of interferon gamma: STAT1 resolves a paradox. Nat Immunol 2000; 1:70-6; PMID:10881178; http://dx.doi.org/10.1038/76940
- Liu M, Guo S, Stiles JK. The emerging role of CXCL10 in cancer (Review). Oncol Lett 2011; 2:583-9; PMID:22848232
- Schoenborn JR, Wilson CB. Regulation of interferon-gamma during innate and adaptive immune responses. Adv Immunol 2007; 96:41-101; PMID:17981204; http://dx.doi.org/10.1016/S0065-2776(07)96002-2
- Park JS, Kim EJ, Kwon HJ, Hwang ES, Namkoong SE, Um SJ. Inactivation of interferon regulatory factor-1 tumor suppressor protein by HPV E7 oncoprotein. Implication for the E7-mediated immune evasion mechanism in cervical carcinogenesis. J Biol Chem 2000; 275:6764-9; PMID:10702232; http://dx.doi.org/10.1074/jbc.275.10.6764
- Zhou F, Chen J, Zhao KN. Human papillomavirus 16-encoded E7 protein inhibits IFN-gamma-mediated MHC class I antigen presentation and CTL-induced lysis by blocking IRF-1 expression in mouse keratinocytes. J General Virol 2013; 94:2504-14; PMID:23956301; http://dx.doi.org/10.1099/vir.0.054486-0
- Cook JL, Walker TA, Worthen GS, Radke JR. Role of the E1A Rb-binding domain in repression of the NF-kappa B-dependent defense against tumor necrosis factor-alpha. Proc Natl Acad Sci U S A 2002; 99:9966-71; PMID:12119420; http://dx.doi.org/10.1073/pnas.162082999
- Fera D, Marmorstein R. Different regions of the HPV-E7 and Ad-E1A viral oncoproteins bind competitively but through distinct mechanisms to the CH1 transactivation domain of p300. Biochemistry 2012; 51:9524-34; PMID:23121466; http://dx.doi.org/10.1021/bi3011863
- Knudsen ES, Wang JY. Targeting the RB-pathway in cancer therapy. Clin Cancer Res 2010; 16:1094-9; PMID:20145169; http://dx.doi.org/10.1158/1078-0432.CCR-09-0787
- Elfstrom KM, Herweijer E, Sundstrom K, Arnheim-Dahlstrom L. Current cervical cancer prevention strategies including cervical screening and prophylactic human papillomavirus vaccination: a review. Curr Opin Oncol 2014; 26:120-9; PMID:24248011; http://dx.doi.org/10.1097/CCO.0000000000000034
- Bahrami AA, Ghaemi A, Tabarraei A, Sajadian A, Gorji A, Soleimanjahi H. DNA vaccine encoding HPV-16 E7 with mutation in L-Y-C-Y-E pRb-binding motif induces potent anti-tumor responses in mice. J Virol Methods 2014; 206:12-8; PMID:24880067; http://dx.doi.org/10.1016/j.jviromet.2014.05.013
- Esuvaranathan K, Chiong E, Thamboo TP, Chan YH, Kamaraj R, Mahendran R, Teh M. Predictive value of p53 and pRb expression in superficial bladder cancer patients treated with BCG and interferon-alpha. Cancer 2007; 109:1097-105; PMID:17311305; http://dx.doi.org/10.1002/cncr.22503
- Cormio L, Tolve I, Annese P, Saracino A, Zamparese R, Sanguedolce F, Bufo P, Battaglia M, Selvaggi FP, Carrieri G. Retinoblastoma protein expression predicts response to bacillus Calmette-Guerin immunotherapy in patients with T1G3 bladder cancer. Urol Oncol 2010; 28:285-9; PMID:18976938; http://dx.doi.org/10.1016/j.urolonc.2008.08.003
- Page DB, Postow MA, Callahan MK, Allison JP, Wolchok JD. Immune modulation in cancer with antibodies. Annu Rev Med 2014; 65:185-202; PMID:24188664; http://dx.doi.org/10.1146/annurev-med-092012-112807
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012; 12:252-64; PMID:22437870; http://dx.doi.org/10.1038/nrc3239
- Dissanayake SK, Thompson JA, Bosch JJ, Clements VK, Chen PW, Ksander BR, Ostrand-Rosenberg S. Activation of tumor-specific CD4(+) T lymphocytes by major histocompatibility complex class II tumor cell vaccines: a novel cell-based immunotherapy. Cancer Res 2004; 64:1867-74; PMID:14996751; http://dx.doi.org/10.1158/0008-5472.CAN-03-2634
- Bosch JJ, Thompson JA, Srivastava MK, Iheagwara UK, Murray TG, Lotem M, Ksander BR, Ostrand-Rosenberg S. MHC class II-transduced tumor cells originating in the immune-privileged eye prime and boost CD4(+) T lymphocytes that cross-react with primary and metastatic uveal melanoma cells. Cancer Res 2007; 67:4499-506; PMID:17483366; http://dx.doi.org/10.1158/0008-5472.CAN-06-3770
- Thompson JA, Srivastava MK, Bosch JJ, Clements VK, Ksander BR, Ostrand-Rosenberg S. The absence of invariant chain in MHC II cancer vaccines enhances the activation of tumor-reactive type 1 CD4+ T lymphocytes. Cancer Immunol 2008; 57:389-98; PMID:17724589; http://dx.doi.org/10.1007/s00262-007-0381-5
- Srivastava MK, Bosch JJ, Wilson AL, Edelman MJ, Ostrand-Rosenberg S. MHC II lung cancer vaccines prime and boost tumor-specific CD4+ T cells that cross-react with multiple histologic subtypes of nonsmall cell lung cancer cells. Int J Cancer 2010; 127:2612-21; PMID:20473949; http://dx.doi.org/10.1002/ijc.25462