
In mammalian cells there are 5 NF-κB subunits, RelA(p65), RelB, c-Rel, p50/p105 (NF-κB1) and p52/p100 (NF-κB2). These combine, through formation of a wide variety of homo and heterodimers, to control the cellular responses to a diverse array of environmental challenges, including bacterial or viral infection and physiological stresses such as hypoxia or DNA damage. Of the NF-κB subunits, RelA, RelB and c-Rel contain C-terminal transactivation domains not found in p50 and p52, the nuclear and proteolytically processed forms of NF-κB1 and NF-κB2. Consequently the majority of research investigating transcriptional regulation of NF-κB target genes has focused on the 'Rel' subunits, with RelA receiving the majority of attention. p50 and p52 are frequently (although wrongly) thought to fulfil an accessory function, contributing to high affinity DNA binding of heterodimers with Rel subunits or functioning as competitive, negative regulators when present as homodimers. There is a comparative absence of research demonstrating that they participate in dynamic regulation of NF-κB function. For example, while a large number of RelA post-translational modifications have been demonstrated, associated with regulation of nuclear localization, transcriptional activity, protein stability and DNA-binding, very few have been described for p50. Furthermore, while RelA knockout mice die as embryos due to liver apoptosis, NF-κB1 knockouts survive with few immediately obvious problems. Therefore p50 is often seen as the boring 'other subunit' of the ubiquitous p50/RelA heterodimer. Importantly, this assumption is now being challenged by a number of groups and increasingly p50 is being demonstrated as an important regulator in its own right.
The physiological significance of dynamic p50 regulation is shown by Crawley et al. previously in Cell Cycle.Citation1 Although NF-κB is best known as a mediator of inflammatory signaling it also responds to DNA damage. However, DNA damage comes in many forms and the NF-κB response can differ depending on the nature of the genotoxic stress. The clearest example of this is the NF-κB response to DNA replication stress, where rather than functioning as an anti-apoptotic regulator, NF-κB can promote cell death.Citation2 Here, Crawley and co-workers demonstrate the vital role regulation of p50 has in this switch of NF-κB function. DNA replication stress results in the activation of the checkpoint kinases ATR and Chk1. Building on previous work from the Yamini group demonstrating that Chk1 can phosphorylate p50 at serine 329,Citation3 here it is shown that this modification is required for NF-κB dependent cell death following DNA replication stress.Citation1 This effect, which is associated with down regulation of anti-apoptotic genes such as Bcl-xL, is lost in NF-κB1 null cells reconstituted with S329 mutant forms of p50. Moreover, p50 is shown to undergo Chk1 dependent S329 phosphorylation during normal S-phase and mutation of this site leads to genome instability. It is tempting to speculate that this effect may be linked to the accelerated aging phenotype recently reported for NF-κB1 null mice.Citation4,5
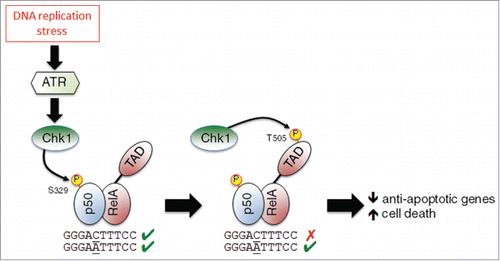
Crawley et al. report that p50‘s effect on cell survival correlates with inhibition of NF-κB DNA binding. However, the same authors have recently demonstrated that p50 S329 phosphorylation has a more selective effect on DNA binding, leading to a change in κB element specificity rather than total inhibition.Citation6 This change, dependent on whether κB-sites have a central cytosine or adenine, with the former inhibiting binding, implies a more modulatory function of ATR/Chk1 activity on NF-κB, with loss of DNA binding occurring only at some genomic locations (). This is also consistent with the location of S329 in p50, where it is a surface exposed residue well away from any direct DNA contacts, implying that these effects arise from a conformational change. Previous data from my group also demonstrated that Chk1 phosphorylates the T505 residue in the RelA subunit, an effect also resulting in pro-apoptotic NF-κB activity following DNA damage.Citation7 Therefore, Chk1 activation resulting from DNA replication stress simultaneously modulates NF-κB activity through at least 2 pathways, targeting different components of the p50/RelA heterodimer. Phosphorylation of p50 at S329 changes the genomic binding of NF-κB, while phosphorylation of RelA at T505 regulates NF-κB transcriptional activity. In combination these result in changes in NF-кB dependent gene expression leading to increased cell death ().
Figure 1. A model depicting regulation of the p50/RelA(p65) NF-κB complex as a result of DNA replication stress. Replication stress activates the checkpoint kinases ATR and Chk1. Chk1 then phosphorylates p50 at S329, inhibiting its ability to bind some κB elements. Chk1 also phosphorylates RelA at T505 in its transactivation domain (TAD). The combined effect of these modifications is the down regulation of anti-apoptotic genes and an increase in cell death.
References
- Crawley CD, et al. Cell Cycle 2015; 14(4):566-76; PMID:25590437; http://dx.doi.org/10.4161/15384101.2014.991166
- Wu ZH, Miyamoto S. EMBO L 2008; 27:1963–73; PMID:18583959; http://dx.doi.org/10.1038/emboj.2008.127
- Schmitt AM, et al. Mol Cell 2011; 44:785-96; PMID:22152481; http://dx.doi.org/10.1016/j.molcel.2011.09.026
- Bernal GM, et al. Aging 2014; 6:931-43; PMID:25553648
- Jurk D, et al. Nat Commun 2014; 2:4172; PMID:24960204; http://dx.doi.org/10.1038/ncomms5172
- Crawley CD, et al. Nucl Acids Res 2013; 41:764-74; PMID:23180782; http://dx.doi.org/10.1093/nar/gks1120
- Msaki A, et al. Mol Biol Cell 2011; 22:3032-40; PMID:21737676; http://dx.doi.org/10.1091/mbc.E11-04-0280