
Calcium (Ca2+) is an essential element of signal transduction. Ca2+ signaling is involved in various cellular processes, such as proliferation and apoptosis and thus is also crucial in cancer. In particular, modulation of Ca2+ signaling can change cells’ sensitivity to apoptotic signals, such as chemotherapeutic agents.Citation1
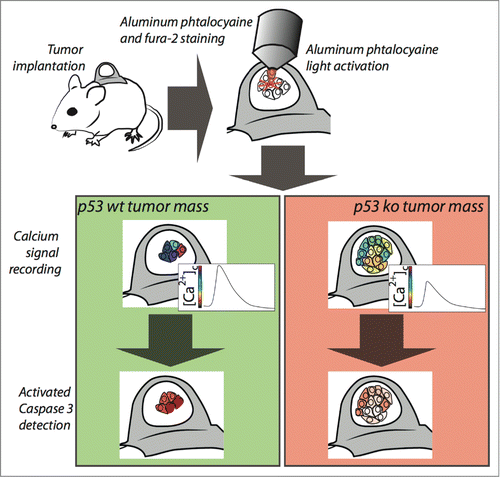
Figure 1. Imaging of calcium signaling and apoptosis into tumor mass. Tumors grown within the skinfold chamber are loaded with aluminum phtalocyanine and the Ca2+ indicator Fura-2. Phtalocyanine activation and Ca2+ signal recording are performed in the same situ using the microscope optics. After Ca2+ live imaging, apoptosis is measured by intravenous administration of a fluorescent marker measuring caspase activity.
Previous studies indicate that both tumor-suppressors (PML, PTEN, Bax) and oncogenes (Bcl-2, Ras, Akt) are able to regulate Ca2+ dependent apoptosis by modulating Ca2+ release from the endoplasmic reticulum (ER) store.Citation2 In turn, these factors can mediate the opening of the mitochondrial permeability transition pore.Citation3
In contrast, little information was available on the transformation-related protein 53 (TRP53), better known as p53 in the regulation of Ca2-dependent apoptosis. This oncosuppressor becomes activated in response to a myriad of stressors, and its activity is crucial for the regulation of cell death pathways. Since the p53s prominent pro-apoptotic role was demonstrated, the mechanism through which p53 mediates apoptosis has been a matter of intense study. Numerous publications have described the importance of p53 in both extrinsic and intrinsic apoptotic pathways.Citation4
In a recent study we demonstrated that p53 is present at the ER and mitochondria-associated membranes (MAMs), where it becomes enriched upon activation. At these sites, p53 interacts with the C-terminal portion of the sarco/ER Ca2+−ATPase (SERCA) pump, changing its oxidative state and, in turn, increasing ER Ca2+ loading. Furthermore, p53 activation enhances ER Ca2+ transfer to mitochondria, thus triggering pro-apoptotic mitochondrial Ca2+ overload and mitochondrial morphological alterations, leading to release of pro-apoptotic factors. Consistently, pharmacological p53 inactivation, as well as naturally occurring loss-of-function mutations of p53, inhibit p53s ability to increase Ca2+ signaling to the mitochondria, and allow for its oncogenic function.Citation5
Whereas most of the mechanisms concerning intracellular Ca2+ handling have been successfully elucidated in vitro, we still know very little about the actual physiological role of these processes in the context of the tumor environment. Recent advancements in intravital imaging and genetically-encoded sensor technologies have allowed us to visualize Ca2+ transient changes in live mice, overcoming previous technical limitations.
In order to elucidate chemoresistance signaling and, thus, to study the effect of novel drugs on the induction of apoptosis, we investigated the role of intra-tumor Ca2+ signaling and apoptosis in a 3D tumor mass, by intravital fluorescence microscopy of mouse tumor models (Fig. 1).Citation6
To that end, we generated a clone deriving from p53−/− mouse embryo fibroblasts (MEFs) transduced with H-RASV12 and a clone from p53−/− MEFs transduced with H-RASV12 with re-introduced wild-type p53. These clones were then transplanted to a dorsal skinfold chamber, in athymic mice to allow tumor growth. We treated the mice with aluminum phthalocyanine chloride a photosensitizer commonly used in the photodynamic therapy (PDT) of cancer. Aluminum phthalocyanine chloride accumulates in intracellular organelles, including mitochondria and ER, where it initiates the apoptotic pathway by increasing cytosolic and mitochondrial Ca2+ upon photoactivation.Citation7
We found that p53 is necessary and sufficient for the control of Ca2+-dependent apoptosis. In particular, we were able to demonstrate that Ca2 response induced by p53 is correlated with the ability of PDT to initiate apoptosis.
To investigate whether the observed Ca2+ modulation was a side effect of our genetic manipulation of p53, or whether it was a result of p53s mechanism of action, we used various pharmacological and molecular approaches to mimic p53s effects on intracellular Ca2+ homeostasis. Independently of the mechanism, all conditions that increased Ca2+ responses rescue the sensitivity to apoptosis in p53-/- cells, while treatments that blunted the Ca2+ responses in p53 wild-type background were associated with the inhibition of apoptosis as in p53−/− cells.Citation6
Finally, the methodology used in this study is compatible with all the fluorescent probes currently available to follow intracellular parameters.
It is of the utmost importance to understand all the signaling pathways deregulated in cancer cells. Understanding the molecular mechanisms underlying the regulation of tumor cell fate is a great challenge in cancer research and will provide further insights for the development of new approaches for the treatment and cure of malignancy.
The development of novel in vivo imaging techniques will facilitate an even greater understanding of the Ca2+ signal-function relationship in live organism and in tumors, and will be an invaluable tool in the identification and classification of new pharmacological targets, as well as in the optimization of current treatments.
References
- Roderick HL, Cook SJ. Nat Rev Cancer 2008; 8:361-75; PMID:18432251; http://dx.doi.org/10.1038/nrc2374
- Giorgi C, et al. Antioxid Redox Signal 2015 PMID:25557408; http://dx.doi.org/10.1089/ars.2014.6223
- Bonora M, et al. Cell Cycle 2013; 12:674-83; PMID:23343770; http://dx.doi.org/10.4161/cc.23599
- Vousden KH, Prives C. Cell 2009; 137:413-31; PMID:19410540; http://dx.doi.org/10.1016/j.cell.2009.04.037
- Giorgi C, et al. Proc Natl Acad Sci USA 2015; 112(6):1779-84; http://dx.doi.org/10.1073/pnas.1410723112
- Giorgi C, et al. Oncotarget 2015; 6(3):1435-45; PMID:25544762.
- Shahzidi S, et al. Photochem Photobiol Sci 2011; 10:1773-82; PMID:21881674; http://dx.doi.org/10.1039/c1pp05169e