
Abstract
Human corneal endothelial cells (HCECs) responsible for corneal transparency have limited proliferative capacity in vivo because of “contact-inhibition." This feature has hampered the ability to engineer HCECs for transplantation. Previously we have reported an in vitro model of HCECs in which contact inhibition was re-established at Day 21, even though cell junction and cell matrix interaction were not perturbed during isolation. Herein, we observe that such HCEC monolayers continue to expand and retain a normal phenotype for 2 more weeks if cultured in a leukemia inhibitory factor (LIF)-containing serum-free medium. Such expansion is accompanied initially by upregulation of Cyclin E2 colocalized with nuclear translocation of phosphorylated retinoblastoma tumor suppressor (p-Rb) at Day 21 followed by a delay in contact inhibition through activation of LIF-Janus kinase1 (JAK1)-signal transducer and activator of transcription 3 (STAT3) signaling at Day 35. The LIF-JAK1-STAT3 signaling is coupled with upregulation of E2F2 colocalized with nuclear p-Rb and with concomitant downregulation of p16INK4a, of which upregulation is linked to senescence. Hence, activation of LIF-JAK1-STAT3 signaling to delay contact inhibition can be used as another strategy to facilitate engineering of HCEC grafts to solve the unmet global shortage of corneal grafts.
Abbreviations
| HCEC | = | human corneal endothelial cell |
| LIF | = | leukemia inhibitory factor |
| JAK | = | Janus kinase |
| STAT3 | = | signal transducer and activator of transcription 3 |
| NC | = | neural crest |
| ESC | = | embryonic stem cell |
| Rb | = | retinoblastoma tumor suppressor |
| CDK | = | cyclin-dependent kinase |
| CKI | = | cyclin-dependent kinase inhibitors |
| bFGF | = | basic fibroblast growth factor |
| EGF | = | epidermal growth factor |
| EMT | = | endothelial mesenchymal transition |
| SHEM | = | supplemental hormonal epithelial medium |
| p120 | = | p120 catenin |
| RPE | = | retinal pigment epithelial cells |
| MESCM | = | modified embryonic stem cell medium |
| BMP | = | bone morphogenetic protein |
| BrdU | = | bromodeoxyuridine |
| EDTA | = | ethylenediaminetetraacetic acid |
| ID | = | inhibitor of differentiation |
| iPSCs | = | induced pluripotent stem cells |
| LEF1 | = | lymphoid enhancer-binding factor 1 |
| NFkB | = | nuclear factor kappa-light-chain-enhancer of activated B cells |
| scRNA | = | scramble RNA |
| siRNA | = | small interfering ribonucleic acid |
| ZO-1 | = | Zona occludens protein 1 |
| DMEM | = | Dulbecco's modified Eagle's medium |
| HBSS | = | Hanks’ balanced salt solution |
| PBS | = | phosphate-buffered saline |
| FBS | = | fetal bovine serum |
| ITS | = | insulin-transferrin-sodium selenite |
| GAPDH | = | glyceraldehyde-3- phosphate dehydrogenase |
Introduction
The corneal endothelium is a delicate monolayer of neural crest (NC)-derived cells that play a pivotal role in regulating corneal stromal hydration and hence transparency.Citation1 Unlike the corneal endothelial cells of other species, such as rabbit and bovine, human corneal endothelial cells (HCECs) are notorious for their limited proliferative capacity in vivo Citation2 because of the mitotic block at the G1 phase of the cell cycle due to “contact-inhibition."Citation3 In mammalian cells, the positive control of the G1/S phase transition is mainly regulated by a transcription factor, E2F, of which the activity is inhibited by non-phosphorylated retinoblastoma tumor suppressor, Rb.Citation4 Release of such inhibition is mediated by phosphorylation of Rb, which is positively controlled by cyclin D1/cyclin-dependent kinase-4 (CDK4), cyclin E/CDK2 complex and negatively regulated by cyclin-dependent kinase inhibitors (CKIs) such as p16INKCitation4a, p15INKCitation4b, p18INKCitation4c, p19INK4d, p21CIP1, p27KIP1, and p57KIP2.Citation5
The conventional approach of expanding HCECs in vitro is to disrupt cell junctions by EDTA with or without trypsin, followed by culturing in a medium supplemented with mitogens, such as basic fibroblast growth factor (bFGF).Citation6 However, these approaches run a high risk of endothelial-mesenchymal transition (EMT).Citation7 To circumvent this undesirable outcome, we have discovered a different isolation method of HCECs using collagenase instead of trypsin-EDTA and dispase to avoid disruption of intercellular junctions and cell-matrix (basement membrane) interaction.Citation8 Using this isolation method, HCECs cultured in epidermal growth factor (EGF) and serum-containing supplemented hormonal epithelial medium (SHEM) exhibit contact inhibition without EMT when adherent junctions mature to an in vivo pattern in a course of 21 d.Citation9
Using the above in vitro model system, we have discovered that contact inhibition of HCECs can safely be perturbed by transient knockdown with p120 catenin (hereafter termed p120) siRNA with or without Kaiso siRNA, resulting in notable expansion of HCEC monolayers without EMT.Citation10 A similar result has also been confirmed in confluent retinal pigment epithelial cells (ARPE-19).Citation11 Such expansion of HCEC monolayer is mediated by activation of non-canonical bone morphogenetic protein (BMP) signaling when cultured in SHEM.Citation10,12 Interestingly, a switch of the culture medium from SHEM to a serum-free medium termed modified embryonic stem cell medium (MESCM) containing leukemia inhibitory factor (LIF) and bFGF, which is routinely used in our laboratory to expand limbal stromal niche cells,Citation13–15 further expanded HCEC monolayers to a transplantable size of approximately 11 mm in diameter after 6 weeks.Citation16 Such further expansion of HCEC monolayers is mediated by canonical BMP signaling and is coupled with reprogramming into NC progenitors.Citation16 However, it is unclear how MESCM, a LIF- and bFGF-containing medium, promotes dramatic expansion of HCECs. As a first step to dissect the mechanism, we herein report that activation of LIF-JAK1-STAT3 by LIF, but not bFGF, delays contact inhibition by upregulation of E2F2 colocalized with nuclear phosphorylated Rb (p-Rb, Ser807/811) with concomitant downregulation of p16INK4a. The significance of this finding in senescence and future tissue engineering by reprogramming of NC progenitors is further discussed.
Results
LIF in MESCM plays a major role in delaying contact inhibition of HCECs
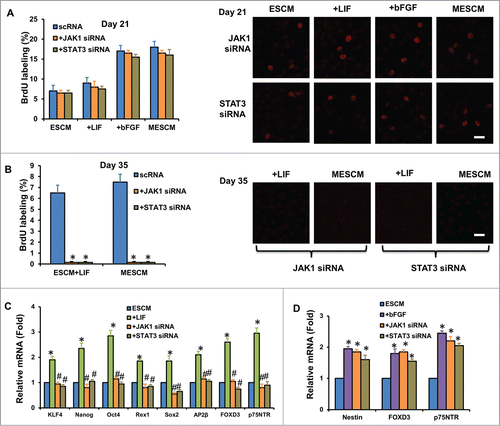
Similar to what we have previously reported,Citation16 the average size of HCEC monolayers was promoted from 1.5 ± 0.3 mm to 4.3 ± 0.2 mm in diameter in 6 weeks after switching the medium from SHEM to MESCM at Day 3 (, #,*P < 0.05, n = 3). Expansion of HCEC monolayers ceased in SHEM but continued in MESCM after Day 21 (). This finding was confirmed by double immunostaining of BrdU (red) and p120 (green) performed at Day 7, 21, 35 and 42. At Day 7, a similar positive nuclear BrdU labeling index, approximately 10%, was noted among cultures in SHEM, ESCM, ESCM+LIF, ESCM+bFGF or MESCM (, P > 0.05, n = 3). At Day 21, the BrdU labeling vanished in SHEM as reportedCitation9 but was maintained at 7 ± 0.8% in ESCM, and further elevated to 9 ± 0.5%, 16 ± 1.2%, and 18 ± 1.1% by adding LIF, bFGF, and LIF+bFGF in ESCM, respectively (, #P < 0.05, n = 3), suggesting that contact inhibition was not re-established in the latter three conditions. At Day 35, the BrdU labeling was not only nil in SHEM but also diminished in ESCM (). Under this scenario, positive nuclear BrdU labeling was still observed in ESCM+LIF or MESCM, but not in ESCM+bFGF (, *P < 0.05, n = 3), suggesting that contact inhibition was significantly delayed by LIF, but not bFGF, in MESCM. No BrdU labeling was observed in any groups at Day 42 (, P > 0.05, n = 3), suggesting eventual re-establishment of contact inhibition.
Figure 1. LIF in delays contact inhibition. The size of HCECs in diameter was compared between cultures in SHEM and MESCM for 6 weeks (A) *P < 0.05, when compared to the size a week before in the same medium; #P < 0.05, when compared to that in SHEM at the same time point, n = 3). BrdU labeling of HCECs in different culture media at Day 7, 21, 35 and 42 was compared (B) *P < 0.05, **P < 0.01 when BrdU labeling in SHEM at the same time point was used as control; #P < 0.05, when BrdU labeling in ESCM at the same time point was used as control, n = 3). At Day 21, p120 was located at intercellular junctions in the cells cultured in SHEM (C). At Day 42, BrdU labeling was compared in all cultures (C). After 6 weeks of culture, the cell morphology, the junctional staining of p120, N-cadherin, β-catenin, ZO-1, F-actin, and Na-K-ATPase, and negative staining patterns of LEF1 and S100A4 were compared (D). Scale bar: 25 μm.
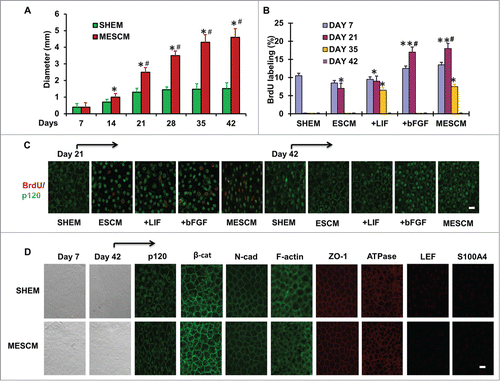
At Day 21, the intercellular junction of p120 was noted in HCECs cultured in SHEM when contact inhibition was re-established but remained lacking in those cultured in ESCM-based media (). By Day 42, HCECs maintained a hexagonal shape and expressed junctional or cytoplasmic expression of p120, β-catenin, N-cadherin, ZO-1, Na/K-ATPase and F-actin in both SHEM and MESCM (), suggesting that HCECs retained a normal phenotype as reported.Citation10,12 Negative immunostaining to LEF1 and S100A4 () confirmed the lack of EMT in SHEM or MESCM, consistent with our previous reports.Citation10,12,16 Collectively, these results indicate that LIF, but not bFGF, in MESCM plays a major role in delaying re-establishment of contact inhibition, explaining how a switch of SHEM to MESCM at Day 3 further expands the size of HCEC monolayers as previously reported.
LIF, but not bFGF, upregulates expression of embryonic stem cell (ESC) and NC markers without reprogramming into NC progenitors
We have reported that if a switch of the culture medium from SHEM to MESCM at Day 3–7 is accompanied by weekly knockdown with p120-Kaiso siRNAs, HCEC monolayers are notably expanded to a transplantable size.Citation16 Such a successful expansion is accompanied by reprogramming of HCECs to NC progenitors highlighted by marked upregulation of a number of markers such as Nanog, Oct 4, Sox2, Nestin, SSEA4, SOX9 and FOXD3.Citation16 We thus wondered whether the aforementioned delay of contact inhibition might also be associated with such reprogramming. We chose to study the expression of these markers at Day 21 when there was a dramatic difference of the BrdU labeling index between SHEM and MESCM (). Compared to SHEM, MESCM indeed upregulated expression of KLF4, Nanog, Nestin, Sox2, TFAP2β, FOXD3 and p75NTR transcripts in HCECs even in the absence of p120-Kaiso siRNA knockdown (, **P < 0.01 and ***P < 0.001, n = 3). In the ESCM control where there was upregulation of Nestin and TFAP2β transcripts, addition of bFGF further upregulated FOXD3 and p75NTR transcripts, while addition of LIF not only retained upregulation of all markers that were elevated by MESCM, but also further upregulated expression of Oct4, Rex1 and SSEA4 transcripts (, *P < 0.05 and **P < 0.01, n = 3). Immunostaining confirmed stronger cytoplasmic expression of Sox2, Nanog, Nestin, KLF4, FOXD3 and p75NTR in MESCM when compared to SHEM (). Compared to ESCM, addition of LIF, but not bFGF, enhanced cytoplasmic staining of Sox2, Oct4, Nanog, Nestin, KLF4, FOXD3 and p75NTR (). Collectively, the results suggest that LIF, but not bFGF, in MESCM upregulates expression of ESC and NC markers.
Figure 2. LIF promotes over-expression of ESC and NC markers. Transcript expression of ESC markers (A) and neural crest cell markers (B) was measured by qRT-PCR after 21 days of culturing of HCECs in different media (*P < 0.05, **P < 0.01 and ***P < 0.001, compare with that of SHEM group as the control, n = 3). The immunostaining was performed at Day 21 for expression of Sox2, Oct4, Nanog, KLF4, Nestin, p75NTR and FOXD3 (C). Scale bar: 25 μm.
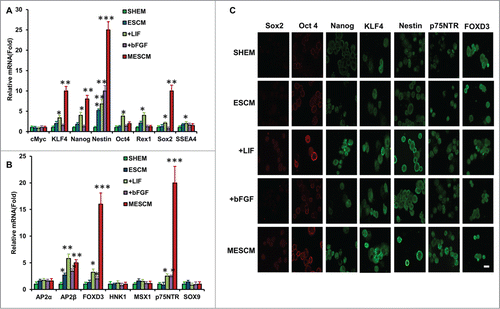
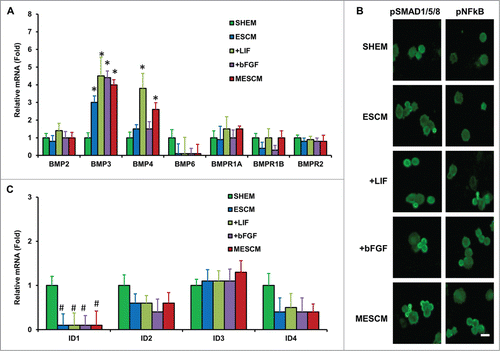
However, the aforementioned upregulation was not accompanied by nuclear translocation of Oct4, Sox2 and Nanog observed during reprogramming changed by additional knockdown with p120-Kaiso siRNAs.Citation16 We strongly suspected that the said reprogramming under the latter scenario might be absent. To substantiate this notion, we studied whether there was activation of non-canonical BMP-NFkB signaling as reported in SHEMCitation12 or activation of canonical BMP signaling as reported in MESCMCitation16 when p120-Kaiso knockdown was deployed. Quantitative RT-PCR analysis at Day 21 revealed upregulation of BMP3 and BMP4 transcripts in MESCM when compared to SHEM (, *P < 0.05, n = 3). Compared to ESCM alone, which promoted expression of BMP3 transcript, addition of LIF promoted expression of both BMP3 and BMP4 while addition of bFGF only promoted that of BMP3 (, *P < 0.05, n = 3). Importantly, the aforementioned upregulation was accompanied by neither nuclear staining of pSMAD1/5/8 and pNFkB (S276) () nor upregulation of ID1, ID2, ID3, or ID4, i.e., SMAD-targeted downstream genes (, P > 0.05).
Figure 3. LIF or bFGF or combination does not activate BMP signaling. Expression of BMP ligands and receptors (A), and BMP downstream genes, ID1-4 (C), was measured by qRT-PCR (*P < 0.05 when compared to the corresponding SHEM group, n = 3) in HCECs at Day 21 cultured in different media. Immunofluorescence staining of pSMAD1/5/8 and pNF-κB was performed to examine the canonical BMP signaling and non-canonical BMP-NFκB signaling in HCECs (B). Scale bars: 25 μm.
LIF activates LIF-JAK1-STAT3 signaling
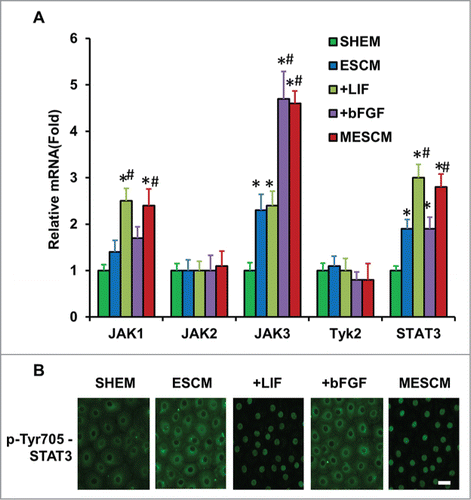
LIF, a member of the IL-6 family, is a key cytokine for sustaining self-renewal and pluripotency of mouse ESCsCitation17 and induced pluripotent stem cells (iPSCs).Citation18 Upon binding to the LIF receptor, LIF activates JAK, which phosphorylates latent STAT3 to elicit nuclear translocation.Citation19 Because phosphorylation of STAT3 is maximized between 5 and 30 min in mouse ESCsCitation20 and nuclear pSTAT3 occurs 2 h in distal neurites after addition of LIF,Citation21 we performed immunostaining of Tyr705-pSTAT3 at 1, 3, 5 and 24 h after addition of LIF or bFGF or both in ESCM at Day 21. Compared to SHEM, transcript expression of JAK3 and STAT3 was significantly higher in ESCM (, *P < 0.05 n = 3). Compared to ESCM, addition of bFGF upregulated transcript expression of JAK3 and addition of LIF upregulated that of JAK1 and STAT3, while addition both LIF and bFGF did not exhibit any additional change in upregulation of JAK1, 3, or STAT3 transcript (, #P < 0.05 when compare to that of ESCM). Immunostaining indicated that pSTAT3 (Tyr705) was only in the nuclei of HCECs cultured in ESCM+LIF or MESCM as early as 3 h (). Collectively, these findings suggest that JAK1-STAT3 signaling is indeed activated by LIF in HCECs when added to ESCM.
Figure 4. LIF-JAK1-STAT3 signaling in HCECs. The expression of mRNA of JAKs and STAT3 was measured at Day 21 in different cultures by qRT-PCR (A) *P < 0.05 when the corresponding SHEM group as the control; #P < 0.05 when the corresponding ESCM group as the control, n = 3). The immunostaining of Tyr705-phosphorylated form of STAT3 in HCECs cultured in ESCM+LIF and MESCM for 3 h was obtained from the confocal microscope (B). Scale bars: 25 μm.
LIF-JAK1-STAT3 signaling is correlated with delay of contact inhibition and overexpression of ESC and NC markers
We then examined whether activation of LIF-JAK1-STAT3 signaling was responsible for the delay of contact inhibition and the upregulation of gene expression of ESC and NC markers caused by LIF through knockdown with JAK1 or STAT3 siRNA. At Day 21, the BrdU labeling index in HCECs cultured in ESCM, ESCM+LIF, ESCM+bFGF or MESCM decreased without any statistical significance by knockdown with JAK1 or STAT3 siRNA for 48 h when compared to their respective control cultures (, all P > 0.05, n = 3). In contrast, the BrdU labeling was completely abolished in HCECs cultured in ESCM+LIF and MESCM by knockdown with JAK1 or STAT3 siRNA for 48 h after Day 35 (, *P < 0.05, n = 3). At Day 21, JAK1 or STAT3 knockdown also suppressed overexpression of all ESC and NC markers in ESCM+LIF (, #P < 0.05, n = 3) but not that of Nestin, FOXD3 and p75NTR in ESCM+bFGF (, P > 0.05, n = 3) when compared to those cultured in ESCM as the control. These results collectively indicate that activation of LIF-JAK1-STAT3 signaling is involved in delaying contact inhibition in the late, but not early, phase of HCEC cultures, and also responsible for the up-regulation of ESC and NC markers.
Figure 5. LIF delays contact inhibition and overexpression of ESC and NC markers. At Day 21, scRNA, JAK1 or STAT3 siRNA was added to the cultures for 48h and BrdU labeling was measured by immunostaining (A). At Day 35, JAK1 or STAT3 siRNA was added to the cultures for 48 h and BrdU labeling was measured by immunostaining (B) *P < 0.05 when compared to the scRNA group). The expression of ESC and NC markers was measured 48 h after JAK1 and STAT3 siRNA knockdown in HCECs cultured in ESCM+LIF (C) *P < 0.05 using ESCM group as the control, #P < 0.05 using ESCM+LIF group as the control) and ESCM+bFGF (D) *P < 0.05 using ESCM group as the control, #P < 0.05 using ESCM+bFGF group as the control) at Day 21 by qRT-PCR. Scale bars: 25 μm.
Upregulation of E2F2 is associated with the delay of contact inhibition
Because contact inhibition of HCECs is caused by an arrest of the G1/S phase transition of the cell cycle,Citation3 we wondered if the LIF-JAK1-STAT3 signaling delayed contact inhibition by modulating expression of cell cycle related genes. At Day 21, compared to SHEM, the expression of positive cell cycle regulation genes such as CDK2, CDK4, CyclinD1, CyclinE2, E2F1 as well as CKIs such as p16INK4a and p27KIP1 was significantly increased in ESCM (, *P < 0.05), among which upregulation of Cyclin E2 was most notable. Immunostaining confirmed nuclear translocation of p-Rb (Ser807/811) only in ESCM-based media but not in SHEM (). Double immunostaining between Cyclin E2 and p-Rb also confirmed colocalization of p-Rb with Cyclin E2 () but not Cyclin D1 (Fig. S1A) in ESCM but not in SHEM. In ESCM, addition of LIF did not significantly influence the expression of these cell cycle related genes, while addition of bFGF upregulated mRNA expression of CDK2, Cyclin E2 and E2F1 but downregulated that of CKIs such as p27KIP1 (, #P < 0.05, n = 3). However, immunostaining showed similar nuclear staining of p27KIP1 in HCECs cultured in ESCM and ESCM+ bFGF, suggesting that promote proliferation of HCECs by bFGF in MESCM at this stage might not be through selective downregulation of p27KIP1. Compared to ESCM+LIF, addition of STAT3 siRNA affected neither expression of cell cycle related genes () nor nuclear translocation of p-Rb in ESCM+LIF (), suggesting the lack of involvement of LIF-STAT3 signaling in this early phase.
Figure 6. Association of expression of cell cycle related genes and contact inhibition. The mRNA expression of cell cycle related genes at Day 21 (A) and Day 35 (B) were measured by qRT-PCR (*P < 0.05 when the corresponding SHEM group as the control; #P < 0.05 when the corresponding ESCM group as the control, n = 3). The immunostaining of p-Rb (Ser807/811) at Day 21 and Day 35 in HCECs cultured in different media (C). The double immunostaining of p-Rb (Ser807/811) and Cyclin E2 (D), E2F2 (E) and p16INK4a (F) at Day 21 or Day 35 was obtained from the confocal microscope. Scale bars: 25 μm.
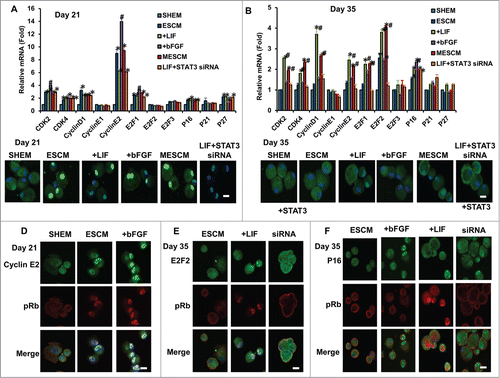
At Day 35, compared to SHEM, ESCM upregulated the mRNA expression of E2F2 and p16INK4a (, *P < 0.05, n = 3). However, double immunostaining did not detect nuclear translocation of E2F2, p16INK4a or p-Rb in ESCM (), suggesting that contact inhibition already ensued in ESCM. When compared to ESCM, addition of bFGF further upregulated the expression of p16INK4a (, #P < 0.05, n = 3). The double immunostaining between p-Rb and p16INK4a or p27KIP1 confirmed nuclear translocation of p16INK4a () but not p27KIP1 (Fig. S1B) only in ESCM+bFGF but not in ESCM+LIF, supporting the expression of p16INK4a, but not p27KIP1, led to contact inhibition by bFGF at this late phase. In contrast, addition of LIF upregulated expression of CDK2, CDK4, Cyclin D1, Cyclin E2, E2F1 and E2F2, but not p16INK4a (, *P < 0.05, n = 3). Addition of STAT3 siRNA in ESCM+LIF not only abolished the aforementioned upregulation of CDK2, CDK4, Cyclin D1, Cyclin E2, E2F1, E2F2 () and nuclear translocation of p-Rb (), but also promoted nuclear translocation of p16INK4a (). Immunostaining confirmed nuclear translocation of p-Rb only in LIF-containing media at Day 35 (). Double immunostaining also demonstrated colocalization of E2F2 with p-Rb only in ESCM+LIF, but not ESCM, and disappearance of such colocalization by STAT3 siRNA (). Collectively, these results indicate that LIF-JAK1-STAT3 signaling promotes G1/S phase transition to delay contact inhibition of HCECs in the late phase (Day 35) by upregulating E2F2 with concomitant downregulation of p16INK4a.
Discussion
Presumably because of the preservation of cell-cell contact and cell matrix interaction by collagenase isolation,Citation8,9 HCECs cultured in SHEM on coated collagen IV withstands the risk of undergoing EMT (). Nonetheless, in a course of 21 d the proliferative potential eventually succumbs to re-establishment of contact inhibition when adherent junctions matured to an in vivo pattern as evidenced by cessation of BrdU nuclear labeling and emergence of junctional expression of p120 ().Citation9 Consistent with our earlier observation,Citation16 a switch of the medium from SHEM to MESCM at Day 3 could expand the size of HCEC monolayers with a normal phenotype from 1.5 ± 0.3 mm to 4.3 ± 0.2 mm in diameter in 6 weeks (). Herein, we gathered strong evidence supporting the notion that such outcome was attributed to LIF, but not bFGF, added in the serum-free ESC medium by delaying contact inhibition. This conclusion was supported by continuous BrdU nuclear labeling, a delay of junctional expression of p120 () and upregulation of a number of positive cell cycle regulation genes (). Using knockdown by JAK1 and STAT3 siRNAs, we conclude that the addition of LIF indeed activates the JAK1-STAT3 signaling pathway, which is responsible for the delay in contact inhibition.
Unlike EGF and FBS-containing SHEM, ESCM alone could delay contact inhibition at Day 21 by upregulation of such transcripts as CDK2, CDK4, Cyclin D1, Cyclin E2, E2F1 as well as p16INK4a and p27KIP1, among which we noted colocalization of Cyclin E2 with nuclear translocation of p-Rb (Ser807/811) (). Addition of bFGF in ESCM could also further promote HCEC growth at this time presumably through upregulation of CDK2, Cyclin E2 and E2F1 but downregulation of CKIs such as p27KIP1 transcript (). Because nuclear staining of p27KIP1 was similarly noted in HCECs cultured in ESCM and ESCM+ bFGF, we believed that the mutagenic effect of bFGF is not through selective downregulation of p27KIP1, a finding resembling what has been reported in rat glomerular epithelial cells.Citation22 Nonetheless, continuous exposure to bFGF up to Day 35 re-established contact inhibition because of upregulation of p16INK4a (). This finding resembles what has been reported in human mesenchymal stem cells in which the mitogenic effect of bFGF is eventually mitigated by concomitant induction of p16INK4a, resulting in premature senescence.23 Because the contact-inhibition (high cell density) and the conditioning of the medium (serum-free) can protect cells from aging and inhibit the cells from senescence, we believe HCECs under such culture condition are quiescent without aging, but not senescent, probably though deactivation of the mammalian target of rapamycin pathway, thus avoiding geroconversion.Citation24,25
In contrast, addition of LIF in ESCM at Day 21 with or without addition of STAT3 siRNA did not significantly influence the expression of the aforementioned cell cycle related genes and nuclear translocation of p-Rb (). This finding explained why LIF did not play a significant role in delaying the contact inhibition in an early phase. Unlike bFGF, continuous exposure of LIF to a late phase, i.e., Day 35, actually upregulated transcription expression of CDK2, CDK4, Cyclin D1, Cyclin E2, E2F1 and E2F2 without concomitant upregulation of p16INK4a transcript (). We noted positive BrdU labeling and colocalization of nuclear E2F2 and p-Rb (Ser807/811) (). Because overexpression of E2F2 induces proliferation and increases the cell density of in ex vivo organ cultures of HCECs,Citation26 our finding suggests that LIF overcame contact inhibition via overexpression and nuclear translocation of E2F2 during this late phase. Previous studies have disclosed an important role of STAT3 in gp130-mediated cell survival and G1-S transition in B cells and myeloma cellsCitation27,28 as well as promoting cell proliferation of neurula-stage embryonic cells in Xenopus.Citation29 Our study, for the first time, suggests that E2F2 may be a target of STAT3 in mediating these actions. It is equally important to point out that addition of STAT3 siRNA not only prevented the nuclear translocation of E2F2 and p-Rb but also promoted nuclear translocation of p16INK4a (). This finding strongly suggests that the effect of LIF-JAK1-STAT3 signaling in delaying contact inhibition is also mediated by concomitant downregulation of p16INK4a. Although in a rat corneal endothelial model the gradual decrease in proliferation after birth is correlated with an increased expression of p27KIP1,Citation30,31 the reduction of p27 levels is only sufficient to promote proliferation in HCEC cultured from young but not old donors.Citation3 Knockdown with p21CIP1 and p16 INK4a siRNAs facilitates HCECs to enter the cell cycle,Citation32 indicating that both p21CIP1 and p16INK4a are involved in negative regulation of the cell cycle in HCECs. The phenomenon of age-related senescence of HCECs is correlated with an increase in p16 INK4a expression,Citation33 and cell density is co-related with p21CIP1-induced senescence.Citation24,25 Hence, future studies are needed to determine whether LIF-JAK1-STAT3 signaling delays contact inhibition by controlling expression of p16 INK4a and p21CIP1 to counteract senescence.
LIF -JAK1-STAT3 signaling is also linked to sustain self-renewal and pluripotency of mouse ESCsCitation17 and iPSCs,Citation18 suggesting its potential role in reprogramming of adult somatic cells. In MESCM, we have recently reported that reprogramming of HCECs to NC progenitors can be induced with additional p120/Kaiso knockdown.Citation16 Herein, although we did not observe conclusive evidence of such reprogramming in the absence of p120/Kaiso knockdown (), for example, we did not observe nuclear translocation of Oct4, Sox2 and Nanog, which is one salient feature of such reprogramming,Citation16 we have noted significant upregulation of ESC markers such as KLF4, Nanog, Nestin and Sox2 as well as NC markers such as TFAP2β, FOXD3 and p75NTR () and extended growth of HCECs when LIF -JAK1-STAT3 signaling is activated. Indeed, if SHEM is replaced by MESCM+5% FBS at the beginning of culture to facilitate the cell attachment, we can generate 4 transplantable size (8 mm in diameter) HCEC grafts from one remaining corneoscleral rim after 6 weeks of culture (not shown). Since delay of contact inhibition occurs only in HCECs cultured in LIF-containing MESCM, and p120-Kaiso knockdown promotes reprogramming only in HCECs cultured in MESCM but not SHEM, we envision that delay of contact inhibition caused by MESCM is prerequisite for HCEC reprogramming to NC progenitors. Because reprogramming of HCECs to NC progenitors requires MESCM containing LIF and additional p120/Kaiso knockdown to trigger activation of Rho-ROCK- canonical BMP signaling,Citation16 future studies are also needed to determine how LIF-JAK1-STAT3 signaling interacts with Rho-ROCK- canonical BMP signaling in achieving the said reprogramming. These studies will help unravel how to safely perturb "contact inhibition" and allow efficient expansion of HCEC monolayers in vitro as a new technology of engineering HCEC surgical grafts to solve the unmet global shortage of corneal grafts.
Materials and Methods
Reagents
Dulbecco's modified Eagle's medium (DMEM), Ham's/F12 medium, human EGF, human bFGF, HEPES buffer, Hanks’ balanced salt solution (HBSS), phosphate-buffered saline (PBS), gentamicin, fetal bovine serum (FBS), knockout serum, Texas-Red-X phalloidin, Alexa-Fluor-conjugated secondary IgG and all real-time PCR primers and probes, JAK1 (catalog number 4390824, ID: s7646) and STAT3 siRNAs (catalog number 4390824, ID: s745) and the monoclonal antibody against ZO-1 were purchased from Life Technologies (Carlsbad, CA). Collagenase A and insulin-transferrin-sodium selenite (ITS) medial supplement were obtained from Roche Applied Science (Indianapolis, IN). Human LIF, hydrocortisone, dimethyl sulfoxide, cholera toxin, bovine serum albumin, paraformaldehyde, methanol, Triton X-100 and Hoechst 33342 dye were purchased from Sigma-Aldrich (St Louis, MO). Collagen IV coated 24-well dishes and a monoclonal antibody against β-catenin were obtained from BD Biosciences (Franklin Lakes, NJ). Monoclonal antibodies against BrdU, Oct4, and Na+/K+-ATPase were purchased from Millipore (Billerica, MA). Monoclonal antibodies against FOXD3, Nestin, Cyclin D1, Cyclin E2, p16 INK4a, and p27KIP1 and polyclonal antibodies against LEF1, Nanog, KLF4, p75NTR, Sox2, N-cadherin (type I), and S100A4 were obtained from Abcam (La Jolla, CA). The polyclonal antibodies against p120, E2F2 and p-Rb (Ser807/811) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The monocolonal antibody against pSTAT3 (pTy705) and the polyclonal antibodies against p-Rb (Ser807/811), pNFκB (pSer276) and pSMAD1/5/8 were obtained from Cell Signaling (Danvers, MA). RNeasy Mini Kit and HiPerFect siRNA transfection reagent were obtained from Qiagen (Valencia, CA).
HCEC Isolation and Culture
The human corneas from 46 individuals aged 18–78 y maintained at 4°C in Optisol (Chiron Vision, Irvine, CA) for less than 7 d after death were obtained from the Florida Lions Eye Bank (Miami, FL) and handled according to the Declaration of Helsinki. HCEC were isolated and cultured as previously reported.Citation8,9 In short, after central corneal buttons had been used for corneal transplantation, the remaining corneoscleral rims were rinsed 3 times with DMEM containing 50 μg/ml gentamicin and 1.25 μg/ml amphotericin B. Under a dissecting microscope, the trabecular meshwork was cleaned up to the Schwalbe's line, and Descemet membranes were stripped from different sizes of the rim. After digestion at 37°C for 16 h with 2 mg/ml collagenase A in SHEM made of an equal volume of HEPES-buffered DMEM and Ham's F12 supplemented with 5% FBS, 0.5% dimethyl sulfoxide, 2 ng/ml hEGF, 5 μg/ml insulin, 5 μg/ml transferrin, 5 ng/ml selenium, 0.5 μg/ml hydrocortisone, 1 nM cholera toxin, 50 μg/ml gentamicin, and 1.25 μg/ ml amphotericin B, HCEC aggregates were collected by centrifugation at 2000 r.p.m. for 3 min to remove the digestion solution, and cultured in 24-well dishes coated with collagen IV in SHEM or ESCM made of DMEM/F-12 (1:1) supplemented with 10% knockout serum, 5 μg/ml insulin, 5 μg/ml transferrin, 5 ng/ml sodium selenite, 50 μg/ml gentamicin, and 1.25 μg/ml amphotericin B or ESCM+LIF (10 ng/ml) or ESCM+bFGF (4 ng/ml) or ESCM+LIF+bFGF (MESCM), depending on the experimental purpose. Cultures were continuously monitored by phase contrast microscopy, and the size of HCECs was determined by digitizing the surface area using Image J and the cell counting was analyzed using Axio Vision software (Carl Zeiss, Thornhood, NY). The cultures from the same corneoscleral rims were used for control and experimental groups, respectively, in each experiment.
siRNA Transfection and other treatments
For the siRNA knockdown, parallel HCECs were subjected to transfection by mixing 50 μl of serum-free, antibiotic-free DMEM with 1 μl of HiPerFect siRNA transfection reagent (final dilution, 1:300) and 1.5 μl of 20 μM of scrambled (sc) RNA, JAK1 siRNA, or STAT3 siRNA, adding the mix (the final concentration of 100 nM) drop-wise to a culture with 250 μl of fresh ESCM or ESCM+LIF or ESCM+bFGF or MESCM at 37°C. BrdU was added at a final concentration of 10 μM to a culture 24 h before termination. For each culture, at least 2000 total nuclei were counted for the BrdU labeling index, defined as the number of BrdU-labeled nuclei divided by the total number of labeled and unlabeled nuclei.
RNA extraction, reverse transcription and real-time PCR
Total RNAs were extracted using RNeasy Mini Kit (Qiagen, Valencia, CA) and reverse transcribed into cDNA by High Capacity Reverse Transcription Kit (Applied Biosystems, Foster City, CA). cDNAs were then amplified by qRT-PCR using specific primer-probe mixtures and Taq DNA polymerase in a 7300 Real-Time PCR System (Applied Biosystems, Foster City, CA). The amplification program consisted of an initial activation for 10 min at 95°C followed by 40 cycles of denaturation for 15 sec at 95°C and an annealing and extension cycle for 1 min at 60°C. The relative gene expression data were analyzed by the comparative CT method (ΔΔCT). All assays were performed in triplicate. The results were normalized by glyceraldehyde-3- phosphate dehydrogenase (GAPDH) as an internal control.
Immunostaining
HCEC cultures were air-dried and fixed in 4% formaldehyde, pH 7.0, for 15 min at room temperature, rehydrated in PBS, incubated with 0.2% Triton X-100 for 15 min, and rinsed 3 times with PBS for 5 min each. For double immunostaining to both BrdU and p120, samples were fixed with 75% methanol plus 25% acetic acid for 15 min, denatured with 2 M HCl for 30 min at 37°C and neutralized by 0.1 M borate buffer, pH 8.5 for 5 min 3 times. After incubation with 2% BSA to block non-specific staining for 30 min, the samples were incubated with the desired first antibody (all at 1:50 dilution) for 16 h at 4°C. After three washes with PBS, they were incubated with corresponding Alexa-Fluor-conjugated secondary IgG (all at 1:100 dilution) for 60 min. The samples were then counterstained with Hoechst 33342 and analyzed with Zeiss LSM 700 confocal microscope (Thornhood, NY). Corresponding mouse and rabbit sera were used as negative controls for primary monoclonal and polyclonal antibodies, respectively.
Statistical analysis
All summary data were reported as means ± SD calculated for each group and compared using ANOVA and the Student's paired and unpaired t-test by Microsoft Excel (Microsoft, Redmont, WA). Test results were reported as 2-tailed p values, where P < 0.05 was considered statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The innovations, methods for promoting cellular proliferation limited by contact inhibition because of adherent junctions in cells including HCECs, and for generating surgical grafts and tissues, were filed in an International PCT Patent Application (PCT/US07/79757) on September 27, 2007.
Funding
This study has been supported by the National Eye Institute, National Institutes of Health [grant numbers R43 EY 022502-01 and R44 EY 022502-02 to Y.-T.Z. and S.C.G.T.]; and in part by TissueTech, Miami, FL.
References
- Bonanno JA. Identity and regulation of ion transport mechanisms in the corneal endothelium. Prog Retin Eye Res 2003; 22:69–94; PMID:12597924; http://dx.doi.org/10.1016/S1350-9462(02)00059-9
- Laing RA, Neubauer L, Oak SS, Kayne HL, Leibowitz HM. Evidence for mitosis in the adult corneal endothelium. Ophthalmology 1984; 91:1129–34; PMID:6392976; http://dx.doi.org/10.1016/S0161-6420(84)34176-8
- Joyce NC. Cell cycle status in human corneal endothelium. Exp Eye Res 2005; 81:629–38; PMID:16054624; http://dx.doi.org/10.1016/j.exer.2005.06.012
- DeGregori J, Leone G, Miron A, Jakoi L, Nevins JR. Distinct roles for E2F proteins in cell growth control and apoptosis. Proc Natl Acad Sci U S A 1997; 94:7245–50; PMID:9207076; http://dx.doi.org/10.1073/pnas.94.14.7245
- Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev 1995; 9:1149–63; PMID:7758941; http://dx.doi.org/10.1101/gad.9.10.1149
- Engelmann K, Bohnke M, Friedl P. Isolation and long-term cultivation of human corneal endothelial cells. Invest Ophthalmol Vis Sci 1988; 29:1656–62; PMID:3182201
- Lee JG, Kay EP. FGF-2-mediated signal transduction during endothelial mesenchymal transformation in corneal endothelial cells. Exp Eye Res 2006; 83:1309–16; PMID:16769055; http://dx.doi.org/10.1016/j.exer.2006.04.007
- Li W, Sabater AL, Chen YT, Hayashida Y, Chen SY, He H, Tseng SC. A novel method of isolation, preservation, and expansion of human corneal endothelial cells. Invest Ophthalmol Vis Sci 2007; 48:614–20; PMID:17251457; http://dx.doi.org/10.1167/iovs.06-1126
- Zhu YT, Hayashida Y, Kheirkhah A, He H, Chen SY, Tseng SC. Characterization and comparison of intercellular adherent junctions expressed by human corneal endothelial cells in vivo and in vitro. Invest Ophthalmol Vis Sci 2008; 49:3879–86; PMID:18502989; http://dx.doi.org/10.1167/iovs.08-1693
- Zhu YT, Chen HC, Chen SY, Tseng SC. Nuclear p120 catenin unlocks mitotic block of contact-inhibited human corneal endothelial monolayers without disrupting adherent junctions. J Cell Sci 2012; 125:3636–48; PMID:22505615; http://dx.doi.org/10.1242/jcs.103267
- Chen HC, Zhu YT, Chen SY, Tseng SC. Selective activation of p120ctn-Kaiso signaling to unlock contact inhibition of ARPE-19 cells without epithelial-mesenchymal transition. PLoS One 2012; 7:e36864; PMID:22590627; http://dx.doi.org/10.1371/journal.pone.0036864
- Zhu YT, Han B, Li F, Chen SY, Tighe S, Zhang S, Tseng SC. Knockdown of both p120 catenin and Kaiso promotes expansion of human corneal endothelial monolayers via RhoA-ROCK-noncanonical BMP-NFkappaB pathway. Invest Ophthalmol Vis Sci 2014; 55:1509–18; PMID:24474278; http://dx.doi.org/10.1167/iovs.13-13633
- Li GG, Chen SY, Xie HT, Zhu YT, Tseng SC. Angiogenesis potential of human limbal stromal niche cells. Invest Ophthalmol Vis Sci 2012; 53:3357–67; PMID:22538425; http://dx.doi.org/10.1167/iovs.11-9414
- Li GG, Zhu YT, Xie HT, Chen SY, Tseng SC. Mesenchymal stem cells derived from human limbal niche cells. Invest Ophthalmol Vis Sci 2012; 53:5686–97; PMID:22836771; http://dx.doi.org/10.1167/iovs.12-10300
- Xie HT, Chen SY, Li GG, Tseng SC. Isolation and expansion of human limbal stromal niche cells. Invest Ophthalmol Vis Sci 2012; 53:279–86; PMID:22167096; http://dx.doi.org/10.1167/iovs.11-8441
- Zhu YT, Li F, Han B, Tighe S, Zhang S, Chen SY, Liu X, Tseng SC. Activation of RhoA-ROCK-BMP signaling reprograms adult human corneal endothelial cells. J Cell Biol 2014; 206:799–811; PMID:25202030; http://dx.doi.org/10.1083/jcb.201404032
- Hirai H, Karian P, Kikyo N. Regulation of embryonic stem cell self-renewal and pluripotency by leukaemia inhibitory factor. Biochem J 2011; 438:11–23; PMID:21793804; http://dx.doi.org/10.1042/BJ20102152
- Tang Y, Tian XC. JAK-STAT3 and somatic cell reprogramming. JAKSTAT 2013; 2:e24935; PMID:24470976
- Matsuda T, Nakamura T, Nakao K, Arai T, Katsuki M, Heike T, Yokota T. STAT3 activation is sufficient to maintain an undifferentiated state of mouse embryonic stem cells. EMBO J 1999; 18:4261–9; PMID:10428964; http://dx.doi.org/10.1093/emboj/18.15.4261
- Forrai A, Boyle K, Hart AH, Hartley L, Rakar S, Willson TA, Simpson KM, Roberts AW, Alexander WS, Voss AK, et al. Absence of suppressor of cytokine signalling 3 reduces self-renewal and promotes differentiation in murine embryonic stem cells. Stem Cells 2006; 24:604–14; PMID:16123385; http://dx.doi.org/10.1634/stemcells.2005-0323
- O'Brien JJ, Nathanson NM. Retrograde activation of STAT3 by leukemia inhibitory factor in sympathetic neurons. J Neurochem 2007; 103:288–302; PMID:17608645
- Shankland SJ, Floege J, Thomas SE, Nangaku M, Hugo C, Pippin J, Henne K, Hockenberry DM, Johnson RJ, Couser WG. Cyclin kinase inhibitors are increased during experimental membranous nephropathy: potential role in limiting glomerular epithelial cell proliferation in vivo. Kidney Int 1997; 52:404–13; PMID:9263996; http://dx.doi.org/10.1038/ki.1997.347
- Takahashi H, Toyoda M, Birumachi J, Horie A, Uyama T, Miyado K, Matsumoto K, Saito H, Umezawa A. Shortening of human cell life span by induction of p16ink4a through the platelet-derived growth factor receptor beta. J Cell Physiol 2009; 221:335–42; PMID:19582773; http://dx.doi.org/10.1002/jcp.21860
- Sun P. Contact inhibition against senescence. Oncotarget 2014; 5:7212–3; PMID:25277173
- Leontieva OV, Demidenko ZN, Blagosklonny MV. Contact inhibition and high cell density deactivate the mammalian target of rapamycin pathway, thus suppressing the senescence program. Proc Natl Acad Sci U S A 2014; 111:8832–7; PMID:24889617; http://dx.doi.org/10.1073/pnas.1405723111
- McAlister JC, Joyce NC, Harris DL, Ali RR, Larkin DF. Induction of replication in human corneal endothelial cells by E2F2 transcription factor cDNA transfer. Invest Ophthalmol Vis Sci 2005; 46:3597–603; PMID:16186339; http://dx.doi.org/10.1167/iovs.04-0551
- Fukada T, Hibi M, Yamanaka Y, Takahashi-Tezuka M, Fujitani Y, Yamaguchi T, Nakajima K, Hirano T. Two signals are necessary for cell proliferation induced by a cytokine receptor gp130: involvement of STAT3 in anti-apoptosis. Immunity 1996; 5:449–60; PMID:8934572; http://dx.doi.org/10.1016/S1074-7613(00)80501-4
- Catlett-Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R, Ciliberto G, Moscinski L, Fernandez-Luna JL, Nunez G, et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity 1999; 10:105–15; PMID:10023775; http://dx.doi.org/10.1016/S1074-7613(00)80011-4
- Nichane M, Ren X, Bellefroid EJ. Self-regulation of Stat3 activity coordinates cell-cycle progression and neural crest specification. EMBO J 2010; 29:55–67; PMID:19851287; http://dx.doi.org/10.1038/emboj.2009.313
- Joyce NC, Harris DL, Zieske JD. Mitotic inhibition of corneal endothelium in neonatal rats. Invest Ophthalmol Vis Sci 1998; 39:2572–83; PMID:9856767
- Joyce NC, Harris DL, Mello DM. Mechanisms of mitotic inhibition in corneal endothelium: contact inhibition and TGF-beta2. Invest Ophthalmol Vis Sci 2002; 43:2152–9; PMID:12091410
- Joyce NC, Harris DL. Decreasing expression of the G1-phase inhibitors, p21Cip1 and p16INK4a, promotes division of corneal endothelial cells from older donors. Mol Vis 2010; 16:897–906; PMID:20508865
- Song Z, Wang Y, Xie L, Zang X, Yin H. Expression of senescence-related genes in human corneal endothelial cells. Mol Vis 2008; 14:161–70; PMID:18334933