
Abstract
The Nobel prized discovery of nuclear reprogramming is swiftly providing mechanistic evidence of a role for metabolism in the generation of cancer stem cells (CSC). Traditionally, the metabolic demands of tumors have been viewed as drivers of the genetic programming detected in cancer tissues. Beyond the energetic requirements of specific cancer cell states, it is increasingly recognized that metabolism per se controls epi-transcriptional networks to dictate cancer cell fate, i.e., metabolism can define CSC. Here I review the CSC-related metabolic features found in induced pluripotent stem (iPS) cells to provide an easily understandable framework in which the infrastructure and functioning of cellular metabolism might control the efficiency and kinetics of reprogramming in the re-routing of non-CSC to CSC-like cellular states. I suggest exploring how metabolism-dependent regulation of epigenetics can play a role in directing CSC states beyond conventional energetic demands of stage-specific cancer cell states, opening a new dimension of cancer in which the “physiological state” of CSC might be governed not only by cell-autonomous cues but also by local micro-environmental and systemic metabolo-epigenetic interactions. Forthcoming studies should decipher how specific metabolites integrate and mediate the overlap between the CSC-intrinsic “micro-epigenetics” and the “upstream” local and systemic “macro-epigenetics," thus paving the way for targeted epigenetic regulation of CSCs through metabolic modulation including "smart foods" or systemic "metabolic nichotherapies."
The 2012 Nobel prized discovery of induced pluripotent stem (iPS) cellsCitation1 mechanistically supports one of the most contemporary hypotheses in cancer pathophysiology, which states that the molecularly distinct subpopulation of so-called cancer stem cells (CSC) are responsible for many if not all aspects of tumorigenesis including the existence of deadly metastases, and also the clinical failure of the majority of available cancer therapies. The question of whether CSC ultimately originate via mutations that occur in normal stem cells, or from differentiated cells which reacquire stem cell attributes i.e., the acquisition of capacities to self-renew and to maintain multipotency or pluripotency through dedifferentiation, remains to be answered unequivocally.Citation2-11 Nonetheless, the striking similarity of the molecular features shared between iPS cell generation and tumorigenesis is providing key mechanistic insights on how CSC could actually arise, in some cases, from differentiated cells through a process of “pathological nuclear reprogram-ming."Citation12-21
A proof-of-concept demonstration of the close association between acquisition of stem cell properties by induced pluripotency and CSC-driven tumorigenesis has been recently carried out in a landmark study, showing that transient in vivo expression of reprogramming factors generates tumors with altered epigenetic states which cause abnormal growth of incompletely reprogrammed cells.Citation22 Though these findings are the first to confirm that premature termination of induced pluripotency can result in cancer development, it should be noted that oncogenic-transformed cells and iPS cells generated from common parental fibroblasts have been found to represent highly related, yet distinct, cell types based on expression profiling,Citation15 thus suggesting that they should share common “cellular ancestors” that develop along an equivalent molecular pathway(s) before they diverge. Indeed, a model comparing malignant transformation and (non-malignant) nuclear cell reprogramming demonstrated that differentiated cells should first acquire epigenetic changes that lead to a downregulation of the differentiation machinery, which is paralleled by an activation of glycolysis and other metabolic pathways.Citation15 Crucially, only then are the oncogenic or the pluripotent phenotypes fully acquired, depending on other stimuli such as stemness factors. Moreover, whereas reprogrammed pluripotent stem cells can acquire oncogenic traits, the converse is not true because oncogenic cells cannot acquire the bona fide pluripotent state possessed by stem cells.Citation15
If the acquisition of stem cell properties in induced pluripotency is closely associated with CSC-driven tumorigenesis, it then follows that determining the mechanisms that positively regulate the efficiency and kinetics of somatic reprogramming to iPS cellular states may provide a proof-of-concept validation for the novel self-renewing tumor-initiating mechanisms that regulate both the number and aberrant functionality of CSC.Citation23 Following this line of reasoning, Tung and KnoepflerCitation24 have recently reviewed the shared epigenetic machinery by which pluripotency and oncogenicity are established and regulated. Interestingly, while the close similarity between iPS cell generation and the acquisition of CSC is shedding new light on the roles of bona fide oncogenes, tumor suppressor genes, transcription factors and chromatin regulators, in mediating the transition from differentiated-to-stem cell states in cancer tissues, an increasing number of experimental studies have consistently revealed that, similar to embryonic and adult stem cells, iPS cells are metabolically distinct from their differentiated counterparts.Citation25-32 Moreover, the precise metabolic properties of stem cells appear to be functionally relevant for stem cell identity and specification regardless of their cellular sizes or cell duplication dynamics, implicating a metabolism-centric regulation of stemness and cell fate.
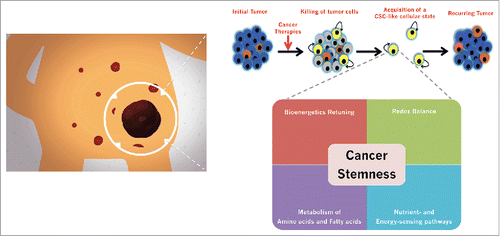
Here I briefly review the CSC-related metabolic features found in iPS cells, to provide an easily understandable framework in which the infrastructure and functioning of cellular metabolism might operate as a key molecular constraint controlling the efficiency and kinetics of stemness reprogramming in the re-routing of non-CSC to CSC-like cellular states during cancer genesis and progression ().
Figure 1. Metabolic restructuring and the acquisition of CSC cellular states: Beyond the nuclear-centric view of cancer stemness. The inherent aggressiveness of carcinomas appears to derive not from the pre-existing content of CSC, but rather from the intrinsic proclivity of a given tumor tissue to generate new CSC-like cellular state from non-CSC cell populations. We are accumulating evidence that enabling such cellular plasticity potential in cancer tissues requires an underexplored integration of metabolic stimuli with the epigenetic control of cell fate.Citation122,135 Metabolic-related processes including bioenergetic retuning, redox balance, restructuring of biosynthetic requirements, and relative shifts in metabolic sensors and regulators are not secondary consequences of the acquisition of cancer cell stemness but they exert an important modulatory role.Citation140,141
Metabolism and Cancer Stemness: Lessons from iPS Cells
First lesson: OXPHOS-to-glycolysis bioenergetic retuning
A shift in the balance between mitochondrial oxidative phosphorylation (OXPHOS) and glycolysis that reconfigures cellular anabolic requirements to those commonly found in the Warburgian bioenergetic transformation of cancer tissues (i.e., high glycolytic carbon flux and increased decoupling from ATP production in mitochondria) precedes the acquisition of stemness traits in iPS cells.Citation33-47 Similar to well-recognized genetic and epigenetic factors, OXPHOS-to-glycolysis bioenergetic resetting appears to operate as a crucial enabling regulator of nuclear reprogramming because the self-renewal and pluripotency attributes cannot be efficiently acquired in the presence of an inadequate bioenergetic metabotype. Thus, efficiency of reprogramming is greater the closer the glycolytic/OXPHOS energy metabolism profiles of the starting somatic cells are to those of embryonic stem cells (ESC). Moreover, bioenergetic resetting has an early and active role in reprogramming since manipulations that inhibit glycolysis reduce, whereas augmenting glycolysis enhances, reprogramming efficiency. Therefore, only when a critical, very early step of engagement to the stemness metabotype is correctly initiated can transcriptional regulators of self-renewal and pluripotency induce additional endogenous factors to acquire a bona fide stem cell state. This process implicates a hierarchical role of an adequate bioenergetic competence during nuclear reprogramming. Accordingly, induced pluripotency can be achieved with a combination of only one stemness transcription factor and small molecules able to facilitate the metabolic transition from OXPHOS to glycolysis.Citation48
Although the absence of the reprogramming factors c-Myc and Lin28, which are able to directly regulate metabolism by enhancing glycolysis and repressing mitochondrial respiration,Citation49 does not impede metabolic resetting in iPS cells (i.e., metabolic resetting is a property of pluripotency which is independent of c-Myc and Lin28),Citation36 it should be noted that the OXPHOS/glycolysis signature of iPS cells is closely related to their intrinsic tumor-initiating capacity and differentiation potential. The inclusion of c-Myc, a key signaling node in cancer cell metabolism, significantly potentiates the glycolytic behavior and the tumorigenic incidence of derived iPS cells;Citation50 conversely, removal of c-Myc decreases the tumorigenicity of iPS cells and facilitates OXPHOS-dependent lineage commitment and terminal differentiation. Also, iPS cells drastically limit the activity and cellular content of the mitochondrial H+-ATPase synthase, a molecular feature that correlates directly with OXPHOS activity and inversely with the rate of glucose utilization by aerobic glycolysis in tumor tissues.Citation51-59 Somatic cell reprogramming involves a dramatic downregulation of the catalytic β1-F1-ATPase subunit and a significant increase in the expression of ATPase inhibitor factor 1 (IF1), a marker that is expressed in adult stem cells (ASC) for maintaining the activity of aerobic glycolysis, but is not expressed in equivalent differentiated cells.Citation60
Although little is known about the bioenergetic resetting of CSC, it appears that analogous to iPS cells, a direct link might exist between the occurrence of a metabolic switch from OXPHOS to aerobic glycolysis and the occurrence and maintenance of CSC cellular states. Compared to their more differentiated progeny, a stem cell cellular state might necessarily promote changes in the bioenergetic metabotype because CSC appear to preferentially perform glycolysis over OXPHOS, at least in some cancer types. Accordingly, recent studies have confirmed that a metabolic switch to glucose metabolism is a critical promotional event in the epithelial-to-mesenchymal (EMT)-driven CSC-like phenotype.Citation61,62 Epigenetic silencing of the gluconeogenic enzyme fructose-1,6-biphosphate, which catalyzes the energy-consuming conversion of fructose 1,6-biphosphate to fructose-6-phosphate, is employed by CSC as a mechanism of glucose flux maintenance via glycolysis and other associated biosynthetic pathways. Indeed, because its presence in the cell-culture milieu has been shown to significantly increase the percentage of CSC in the overall cancer cell population, glucose appears to act as an essential nutrient for CSC.Citation63 Conversely, glucose starvation is sufficient to cause a rapid depletion of the CSC subpopulation in vitro. A recent proteomic and targeted metabolomic analysis of the main differences between breast CSC and their differentiated counterparts has identified a metabolic phenotype associated with the stem-like condition, indicating that breast CSC shift from mitochondrial OXPHOS toward fermentative glycolysis.Citation64 Of note, whereas the treatment of fibroblasts undergoing nuclear reprogramming with 2-deoxyglucose (2DG), a general inhibitor of glycolysis, blunted the generation of iPS cells,Citation36 2DG treatment inhibited breast CSC proliferation, thus revealing how the glycolytic requirement for iPS generation may be a potentially effective strategy to target breast CSCs.Citation64
Second lesson: Redox regulation
Beyond the activation of multiple, redundant mechanisms limiting energy production by OXPHOS in preference to glycolysis and its biosynthetic pathway branches (e.g., the pentose phosphate pathway), the cellular redox state also impacts the balance between differentiated and stem cellular states.Citation65 The oxidation-reduction state appears to parallel the stem cell fate since the accumulation of reactive oxygen species (ROS) has been shown to be minimized in iPS cells via a reduction in substrate oxidation and respiratory coupling, which is accompanied by the activation of antioxidant stress genes.Citation38,66,67 In contrast, an oxidative metabolome, which effectively increases the overall cellular oxidation state during differentiation,Citation43,68,69 appears to replace the unsaturated metabolome possessed by stem cellular states, including those of iPS cells.
Since strict regulation of specific redox species, such as the NAD+/NADH ratio, is directly impacted by glycolytic and mitochondrial activities that change during reprogramming, the NAD+/NADH redox state might have a key role in driving the stem cell fate.Citation70 Nicotinamide, a precursor of NAD, significantly lowers the barrier to reprogramming to stemness by accelerating cell proliferation and protecting cells from apoptosis and senescence through alleviating oxidative stress, ROS accumulation, and subsequent mitochondrial potential collapse during iPS generation.Citation71 Nicotinamide's ability to overcome pluripotency deficits and reprogramming barriers has yet to be evaluated in the acquisition and maintenance of CSC cellular states and future studies should establish whether an adequate NAD+ content is important for enhancing the resistance to stress in CSC. Accumulating evidence suggests that, in contrast to differentiated cancer cells where ROS levels are increased, CSC maintain low levels of ROS,Citation72-75 and thus exhibit redox patterns similar to the corresponding normal stem cell. Accordingly, an increased reliance on glucose metabolism reduces the levels of ROS to promote EMT and CSC-like phenotypes.Citation61,62 While the ratio of reduced glutathione (GSH), the primary intracellular antioxidant, to oxidized (GSSH) glutathione decreases, as does the level of NADH, during stem cell differentiation, CSC possess enhanced mechanisms of protection from stress induced by ROS that might render them resistant to chemo- and radiotherapy through an upregulation of GSH synthesis.Citation76-78 Consequently, new strategies aimed to induce ROS via depletion of cellular glutathione can be viewed as promising therapeutic approaches against CSC.Citation79-81
Third lesson: Metabolism of amino acids and fatty acids
The Warburg effect is associated with a survival advantage as well as the generation of substrates such as nucleotides, amino acids, and fatty acids.Citation82-85 In this regard, the stemness feature also takes advantage of the ability of the mitochondrial tricarboxylic acid (TCA) cycle intermediates to be siphoned off into amino acid and lipid biosynthesis, which have been shown to be required for self-renewal and differentiation of ES and iPS cells. In a series of studies employing combinatorial approaches of metabolomics, nutrition, and genetics, the amino acid threonine was identified as an essential nutrient for mouse and human ESC.Citation69 Beyond its recognized role as a protein precursor, threonine dehydrogenase (TDH)-mediated catabolism of threonine provides glycine through one-carbon metabolism for biosynthesis of purines to support DNA replication and the epigenetic modifications required for self-renewal and maintenance of pluripotency.Citation42 The high-flux metabolic state of iPS cells relies not only on a high dependence on threonine catabolism but also on large amounts of methionine; thus, iPS cells display regulatory systems to maintain a constant level of intracellular methionine and S-adenosylmethionine (SAM), a key regulator for maintaining undifferentiated iPS cells and regulating their differentiation.Citation86 Whether the acquisition of CSC cellular states and CSC self-renewal and differentiation similarly relies on TDH-related purine biosynthesis and/or methionine metabolism remains an unexplored area in the field of CSC biology. Nevertheless, the fact that threonine provides a substantial fraction of both cellular glycine and the acetyl-coenzyme A required for SAM synthesis, together with the recently recognized ability of SAM to influence trimethylation of histone H3 lysine 4 (H3K4me3),Citation87 provides a probable epigenetic mechanism by which modulation of a metabolic pathway might directly influence aberrant stemness in cancer tissues.Citation88-90
When the catalytic activities of acetyl-CoA carboxylase (ACACA) and fatty acid synthase (FASN) lipogenic enzymes are inhibited, the efficiency of reprogramming is significantly decreased. Coincidentally, ACACA and FASN are highly expressed in iPS cells.Citation51 Lipids that participate in signaling cascades, such as arachidonic acid, diacylglycerol, and prostaglandins, are also among the most predominant metabolites found in iPS cells.Citation42 It is known that higher expression levels of lipogenic genes and proteins such as FASN are found in CSC subpopulations of breast cancer cell lines, and that upregulation of de novo fatty acid biogenesis is a prerequisite for the formation of premalignant lesions by endowing CSC survival.Citation91,92 Beyond the fact that fatty acids can play a shared role in normal and cancerous stem cell energy generation via fatty acid oxidation, pharmacological inhibition of ACACA and FASN efficiently impedes the formation of mammospheres in a fatty acid-dependent manner, strongly suggesting that the self-renewal and survival of CSC can be directly impacted by de novo lipogenesis, lipid metabolites, and lipid catabolism.Citation93,94 With regard to the latter, overexpression of monoacylglycerol lipase (MAGL) in nonaggressive cancer cells is sufficient to increase their pathogenicity by recapitulating a fatty acid network enriched in oncogenic signaling lipids that promote migration, invasion, survival, and in vivo tumor growth.Citation95,96 Given the unique role of MAGL in providing lipolytic sources of free fatty acids for the synthesis of oncogenic signaling lipids, CSCs might co-opt lipolytic enzymes such as MAGL to translate their lipogenic state of stem cells into an array of protumorigenic signals. The recent discovery that cancer adaptation to antiangiogenic treatments, which generate hypoxic and nutrient-starved tumor microenvironments that suppress cell cycle progression but enrich non-proliferating CSC,Citation97-103 involves a significant upregulation of lipid synthesis that fuels tumor regrowth and metastasis after angiogenic therapy withdrawal,Citation104 strongly supports the notion that key enzymes involved in lipid metabolism such as FASN could play key roles in the reprogramming of CSC cellular states.
Fourth lesson: Nutrient- and energy-sensing pathways
By integrating many metabolic signals, the mammalian target of rapamycin (mTOR)/AMP-activated protein kinase (AMPK) signaling network appears to operate as a critical “metabolic rheostat” that can direct the generation and maintenance of stem cells.Citation26,105 A timely and precise regulation of the activation/deactivation status of mTOR, a kinase with a central role in sensing O2, nutrients (glucose, amino acids), and growth factors, critically determines the successful reprogramming of somatic cells to iPS cells.Citation106-109 Accordingly, an early short burst of mTOR suppression followed by restoration of mTOR activity at a later stage is required for successful reprogramming. The stemness and oncogenic transcription factor, Sox2, exclusively initiates the resetting of the metabolic infrastructure by suppressing mTOR activity in a timely manner before the acquisition of stemness. Continuous mTOR signaling would evoke active cycling and exhaustion of normal stem cells or fail-safe mechanisms (e.g.,, cellular senescence, apoptosis, or terminal differentiation) that may extinguish the evolution of pre-malignant stem cell clones or prevent efficient reprogramming of somatic cells. Conversely, the cells can exploit the proto-oncogenic and reprogramming signaling emanating from active mTOR only by evading the above-mentioned cancer-protecting systems, which is an escape mechanism that depends not only on the genetic make-up of the cells (e.g., loss of tumor suppressor genes) but also on the activation status of energy-sensing regulators and mTOR suppressors, such as AMPK. Consequently, iPS cells can avoid the inhibition of stemness-related anabolic pathways by downregulating the catalytic activity of AMPK,Citation33 a metabolic master switch that senses and decodes intracellular changes in the energy status and that, upon activation, can “switch off” biosynthetic pathways by negatively regulating the Warburg effect. AMPK activation imposes a metabolic flow away from the required pro-immortalizing glycolysis that fuels the induction of stemness and pluripotency, endowing somatic cells with an energetic infrastructure that is protected against reprogramming.Citation110,111 Thus, AMPK activation establishes a metabolic barrier to reprogramming that cannot be bypassed even by a deficiency of key tumor-suppressor genes such as p53, which is a key element that greatly improves the efficiency of stem cell production.
Regarding cross talk between mTOR and Sox2, which regulates early genetic reprogramming and the acquisition of stemness not only in iPS cells but also CSCs, the metabolic barrier evoked by AMPK activation impacts the genetic regulation of normal and cancerous stem cell states by regulating core transcriptional regulators. AMPK activation impedes early reprogramming by preventing transcriptional activation of Oct4, the master regulator of the pluripotent state.Citation110-112 Moreover, AMPK activation facilitates the specific elimination of Oct4-positive teratoma-initiating pluripotent stem cells that are intermixed with non-tumorigenic iPS cell derivatives, strongly suggesting that an adequate functioning of the metabolic infrastructure might be an indispensable component of the CSC machinery.Citation111 Using an in vitro model of de novo generation of CSC-like states through nuclear reprogramming in an established breast cancer cell line, we have recently shown that the transcriptional suppression of mTOR repressors (i.e., PRKAA1, which codes for the catalytic α1 subunit of AMPK; DDIT4/REDD1, a stress response gene that operates as a negative regulator of mTOR; and DEPTOR, a naturally occurring endogenous inhibitor of mTOR activity) appears to be an intrinsic process that occurs during the acquisition of CSC-like properties by differentiated breast cancer cells.Citation17 AMPK and mTOR alone, or through their substrates, operate as interconnected metabolic controllers that fine-tune the stemness activity of core reprogramming factors such as Sox2 and Oct4, rather than acting as on/off reprogramming-to-stemness switches. It is therefore not surprising that the potential oncology applications for biguanides,Citation113 which can activate AMPK leading to inactivation of mTOR, appear to be closely related to their differential metabolic effects during the inducible generation of CSC.Citation114-117
Cancer Metabostemness: A Waddingtonian Perspective of the Metabolic Control of Stemness and Cell Fate in Cancer Tissues
An intuitive perspective of the metabolic control of stemness in cancer tissues can be offered using Waddingtonian landscapes, which accurately model the complex network of molecular barriers governing cell fate transitions. In 1957, Conrad Waddington proposed a metaphoric representation of cell differentiation phenomena in which pluripotent stem cells (or, in the analogy, “balls”) are positioned at the top of a hill, progressively losing potential while journeying downhill into different valleys representing irreversible, fully differentiated cellular states.Citation118 Thus, once the group of balls at the top start to roll downhill and select a route influenced by various genetic and environmental factors, the balls are committed (i.e., they become differentiated cells) and ultimately land at the bottom. Movement upwards will be difficult, inferring therefore that commitment of cells to specific tissues and organs is fundamentally one directional. They cannot roll back uphill to the bifurcation and they cannot climb over the portion of land between the valleys because reversal of the commitment process will trigger undesirable damaging effects (e.g.,, cancer).Citation119,120 In this scenario, the radical change of identity required for cellular reprogramming of non-CSC to CSC states is expected to be not easily accomplished because by the time all the organs are formed most cells are highly committed to their specific tissue and, in many cases, few ASC remain. However, although the one-way process of commitment generally suppresses cancer until late in life, the groundbreaking discovery of Yamanaka and colleagues demonstrating that terminally differentiated cells can be pushed “upwards” to an original pluripotent stem cell state,Citation1 is rapidly and revolutionarily providing strong mechanistic evidence for the convergence and commonality of iPS generation and CSC states. Genetically and microenvironmentally induced pathological reprogramming, by operating as a sideward deviation of the self-organizing property of developmental epigenetic landscapes, radically modifies our current perception of cancer genesis and tumor behavior.
Cancer is beginning to be understood as a disease of cellular reprogramming (or a disease affecting cellular differentiation) in which many driving forces such as the loss of tumoral suppressors or the aberrant activation of certain transcription factors, signaling cascades, or epigenetic regulators, appear to share a permissive role which alleviates the developmentally unfavorable process of acquiring tumor-initiating and/or metastasis-initiating capabilities possessed by CSC.Citation13,20-23 Because the notion that reprogramming can occur through non-strictly genetic cues (i.e., “master stemness genes” can be dispensable for cell fate reprogramming) has been substantiated through the observation that stemness in somatic cells can be achieved using a chemical cocktail that exclusively regulates the signaling pathways and levels of metabolic intermediates,Citation121 we recently coined the term “metabostemness” to refer to the cellular metabotype or metabolic parameters (e.g., OXPHOS-to-glycolysis resetting of bioenergetic metabolites, metabolic cofactors of epigenetic modifiers, bona fide oncometabolites, etc.) that might causally control or, more importantly, functionally substitute the epitranscriptional orchestration of genetic reprogramming that redirects normal and non-CSC tumor cells toward less-differentiated CSC cellular states.Citation122 From a mathematical perspective, a given cellular metabotype might not only dictate the plasticity of the original cell that is targeted to be reprogrammed by early or late genetic and/or epigenetic hits but also, by removing, diminishing, or modifying the nature of “molecular barriers” present in the Waddington epigenetic landscapes, it can operate as a bona fide accelerator of the reprogramming processes, thus allowing not only ASC but also (normal, pre-malignant or tumor) differentiated cells to more easily and rapidly enter, or re-enter, into CSC cellular macrostates ().
Figure 2. Metabolic-driven control of cellular reprogramming: An integrated view of cell fate transitions. Reversible epigenetic barriers that can be overcome given the correct stimuli preserve cellular fate. Thus, once a certain combination of epigenetic changes has been acquired, cells can assume a new identity (iPS cells or CSC-cells). At a cellular level, both induced pluripotency (left panels) and acquired cancer stemness (right panels) should be viewed as multi-step processes that result in a change of cell identity or differentiation potential where nascent iPS cells and non-CSC cells should face the same epigenetic barriers to alter cell identity.Citation142 Moreover, the “end product” is in both cases an immortal cell with tumor-initiating capacity. Figure illustrates the “energy landscape” experienced by cells under reprogramming conditions under different epigenetic perturbations (modified from ref. 143). Energy peaks represent barriers in the reprogramming path, where higher barriers correspond with low conversion rates. The black line represents the energy plot of the path from somatic cells to either iPS cells or CSC-like states in response to reprogramming stimuli (e.g.,, pluripotency-promoting transcription factors, canonically Oct4, Sox2, Klf4, and Myc [abbreviated as OSKM]). Certain cell metabotypes are more susceptible to de novo reprogramming indicating a more permissive epigenetic environment. Certain metabolic shifts may represent an early bona fide reprogramming event (blue line) and, by having direct effects at the epigenetic level, certain metabolic features operates as catalyzers that reduce energy barriers and accelerates reprogramming (red line). Conversely, imposed barriers (dashes black lines) can occur via certain metabolic conditions that might inhibit and impair the epigenetic rewiring during reprogramming (e.g., AMPK activation hampers the reactivation of the stemness factor Oct4).Citation110-112 Indeed, CSC are not irreversibly locked in a tumorigenic state but instead amenable to metabolo-epigenetic reversion into a phenotypically non-CSC state.
![Figure 2. Metabolic-driven control of cellular reprogramming: An integrated view of cell fate transitions. Reversible epigenetic barriers that can be overcome given the correct stimuli preserve cellular fate. Thus, once a certain combination of epigenetic changes has been acquired, cells can assume a new identity (iPS cells or CSC-cells). At a cellular level, both induced pluripotency (left panels) and acquired cancer stemness (right panels) should be viewed as multi-step processes that result in a change of cell identity or differentiation potential where nascent iPS cells and non-CSC cells should face the same epigenetic barriers to alter cell identity.Citation142 Moreover, the “end product” is in both cases an immortal cell with tumor-initiating capacity. Figure illustrates the “energy landscape” experienced by cells under reprogramming conditions under different epigenetic perturbations (modified from ref. 143). Energy peaks represent barriers in the reprogramming path, where higher barriers correspond with low conversion rates. The black line represents the energy plot of the path from somatic cells to either iPS cells or CSC-like states in response to reprogramming stimuli (e.g.,, pluripotency-promoting transcription factors, canonically Oct4, Sox2, Klf4, and Myc [abbreviated as OSKM]). Certain cell metabotypes are more susceptible to de novo reprogramming indicating a more permissive epigenetic environment. Certain metabolic shifts may represent an early bona fide reprogramming event (blue line) and, by having direct effects at the epigenetic level, certain metabolic features operates as catalyzers that reduce energy barriers and accelerates reprogramming (red line). Conversely, imposed barriers (dashes black lines) can occur via certain metabolic conditions that might inhibit and impair the epigenetic rewiring during reprogramming (e.g., AMPK activation hampers the reactivation of the stemness factor Oct4).Citation110-112 Indeed, CSC are not irreversibly locked in a tumorigenic state but instead amenable to metabolo-epigenetic reversion into a phenotypically non-CSC state.](/cms/asset/6402b061-c7ba-40c6-91e0-6a1dee9acec8/kccy_a_1022697_f0002_c.gif)
Metabolo-epigenetic reprogramming of CSC functions
In cancers with a stem cell origin, such as haematopoietic malignancies, certain cellular metabotypes could sculpt the landscape in a manner that leads to previously normal stem cells or early progenitors becoming stuck in close proximity or in the same state space attractor of immature stem-like regions of the landscape,Citation123 thus increasing the number of undifferentiated cells that may be targetable by oncogenic mutations. Indeed, an invaluable model supporting the notion that certain metabotypic alterations might operate as pivotal molecular events rendering stem cells susceptible to the metabolic rewiring necessary for the acquisition of aberrant stemness and, concurrently, of refractoriness not only to apoptosis but also to differentiation, arises from cancers with a stem cell origin in which gain-of-function mutations in isocitrate dehydrogenase (IDH) generates the oncometabolite 2-hydroxyglutarate (2HG).Citation124-130 Because IDH enzymes produce α-ketoglutarate, a cofactor for the TET family of DNA demethylases, neomorphic mutations of IDH that drive the aberrant neosynthesis of the TET inhibitor 2HG can be expected to mostly restrict the “methylation plasticity” that is required for the hierarchical transitions between stem cells and their differentiated progeny. 2HG-induced global hypermethylation prevents the demethylation of genes that are implicated in differentiation, promoting a metabolically-driven increase in the number of stem cells that might occur prior to oncogenic mutations promoting proliferation.Citation21
In the absence of IDH mutations, however, the accumulation of 2HG has been shown also to be part of the c-Myc-driven metabolic reprogramming phenomenon observed in biologically aggressive breast carcinomas that exhibit globally increased DNA methylation.Citation131 The hypermethylation phenotype of 2HG-overexpressing breast carcinomas is characterized by a strong enrichment of a stem cell-like transcriptional signature, a molecular feature that has been similarly observed in other solid tumors with non-stem cell origins such as intrahepatic cholangiocarcinoma, a deadly liver malignancy in which highly prevalent 2HG-producing IDH mutations subvert the hepatocyte differentiation/quiescence program to create a persistent pre-neoplastic state which is primed for transformation into adenocarcinoma by additional oncogenic mutations.Citation132 Given the inherent cell attractor nature of nuclear reprogramming phenomena,Citation119,120,123,133,134 we recently provided an intuitive perspective on how the commonly forgotten ability of metabolism to reshape the topography of Waddingtonian epigenetic landscapes can influence the probability that pathologically reprogrammed cells can stay trapped as developmentally immature, CSC-like states in the “high mountain” regions of the landscape.Citation135 We proposed that refining the cancer attractor hypothesis by merging “pathological nuclear reprogramming” with the specific metabolic remodeling traits of iPS cells offers a new integrative-systems perspective of the stem cell theory of cancer that explains why oncometabolic-like traits (i.e., the specific ways that CSC generate and use metabolic pathways and intermediate metabolites) converge to “encode” an immature, stem-like program in cancer tissue, regardless of the stem cell/non-stem cell source.
For cancers with non-stem cell origins, certain metabotypes can permissively alleviate the “uphill," unfavourable developmental process of “jumping back” from non-CSC differentiated valleys to high-altitude CSC attractors while concomitantly promoting the ground-state character of self-maintaining CSC-like states. Because CSC states can be viewed as inherently inevitable epigenetic deviations of Waddington's landscapes, in which metabolism modifies the probability that not only stem cells/early progenitors but also normal or cancer-differentiated cells can find either pre-existing or de novo occupied, self-organizing attractors encoding dynamically robust CSC signatures, it would be interesting to determine whether the very strict cellular metabotypes that are compatible exclusively with iPS cell generation truly represent a non-genetic dimension of cancer that generates bona fide “aberrant attractors” (i.e., dead-end side valleys) that are necessarily located close to or in the same location of the developmentally immature, stem-like cellular states in these “higher mountains." These aberrant attractors might prevent pathologically reprogrammed undifferentiated cells in cancer tissues from completing their physiological, predestined journey down to the differentiated cell type attractors.
Metabolism-dependent regulation of epigeneticsCitation122,135 plays a key role in directing cancer cell fate, including CSC. The two primary epigenetic codes, DNA methylation and histone modification, and the consequent epigenetic regulation of differentiation genes might be viewed as the pivotal molecular events that successfully integrate the recognized ability of cellular metabolism to competitively inhibit epigenetic regulation of cell differentiation with the process in which the stemness regulatory circuitry is established during nuclear reprogramming. Certain metabolic events and elite metabolites (e.g.,, the Warburg effect, oncometabolites such as 2-HG, sub-cellular compartmentalization of critical metabolic cofactors of epigenetic enzymes, etc.) might markedly lower the “energy barriers” separating non-CSC and CSC attractors, diminish the average time of reprogramming, and increase the size of the basin of attraction of the macrostate occupied by CSC, thus promoting the ground-state character of the self-maintaining CSC cell state. Indeed, from a Waddingtonian perspective, the underexplored topic of differentiation therapy can be viewed in terms of metabolic interventions able to knock CSC cellular states “trapped” in attractors back to the physiological trajectories that lead to non-malignant, more differentiated cells. Although these speculations appear plausible, considerably more direct experimental support will be required before they can be accepted.
Metabostemness: From CSC energetics to micro- and macro-epigenetics (a corollary)
At one time, the cancer genomics era pushed Warburg's metabolic cancer hypothesis into obscurity. The Nobel prized discovery of iPS cells, however, is rapidly delivering mechanistic evidence on how the metabolic control of stemness might causally participate in the generation of CSC cellular states.Citation105,122,135 I here postulate that incorporating certain metabolic observations of iPS cell behavior into pathological nuclear reprogramming phenomena might provide an integrative view of the stem cell theory of cancer that may radically amend the molecular understanding and clinical management of cancer diseases. Looking forward, we will continue to make advances in this area by exploring at least 2 provocative questions:
First, a systematic delineation of CSC-specific expression of metabolic enzymes, metabolites, and certain bioenergetic processes (e.g., Warburgian metabolism versus mitochondrial respiration) should definitively clarify whether the metabolic state rather than phenotypic marker expression is a crucial operational criterion that defines CSC. In this regard, advances in functional metabolomics should reveal how catabolic processes such as autophagyCitation26,136,137 contribute to the maintenance of substrate and metabolite levels that support not only the bioenergetic and biosynthetic needs of CSC, but also the aberrant reprogramming of the epigenome in CSC.
Second, although we are only just beginning to understand how cellular metabolism can maintain the hallmark trait of self-renewal in CSCs, it is likely that metabolism has an indispensable role to directly influence the most relevant epigenetic alterations, from DNA methylation to chromatin organization, which contributes to the regulation of CSC features in tumor progression, metastasis and response to therapies. If we adopt the view that metabolism-dependent regulation of epigenetics plays a role in directing CSC states beyond matching energetic demands of stage-specific cancer cell states, we will be entering an unforeseen dimension of cancer diseases in which the “physiological state” of CSC is governed not only by cell-autonomous cues, but also by local micro-environmental and systemic metabolo-epigenetic interactions.
By deciphering how specific metabolites integrate and mediate the overlap between the CSC-intrinsic “micro-epigenetics” and the “upstream” local and systemic “macro-epigenetics," as recently proposed by Miguel Ramalho-Santos,Citation138 we will pave the way for future developments to target epigenetic regulation of CSC through metabolic modulation including "smart foods" or systemic "metabolic nichotherapies” ().Citation139 The incorporation of bioenergetics and metabolo-epigenetics to the development of therapeutics targeting CSC should offer a new perspective on the frequently forgotten relevance of basic metabolic research in the post-genomic era of cancer research.
Figure 3. Metabostemness: A new therapeutic opportunity in cancer. Further knowledge is warranted to decipher the metabolic-driven control of cancer cell stemness in order to either improve novel discovery technologies or accelerate scanning of existing pharmacopoeia for repositioning candidates as a new therapeutic opportunity in cancer. New anti-metabostemness strategies should involve not only a conventional development of drugs directly targeting CSC metabolism,Citation144-146 but also should consider the metabolic demands associated with the acquisition of CSC-like cellular states and therefore the influence of environmental metabolic factors and their usage in the epigenetic control of cell fate in cancer tissues.

Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by grants from the Ministerio de Ciencia e Innovación (Grant SAF2012-38914), Plan Nacional de I+D+I, Spain and the Agència de Gestió d’Ajuts Universitaris i de Recerca (AGAUR) (Grant 2014 SGR229), Departament d’Economia I Coneixement, Catalonia, Spain to Javier A. Menendez.
References
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006; 126:663-76; PMID:16904174; http://dx.doi.org/10.1016/j.cell.2006.07.024
- Jordan CT, Guzman ML, Noble M. Cancer stem cells. N Engl J Med 2006; 355:1253-61; PMID:16990388; http://dx.doi.org/10.1056/NEJMra061808
- Dalerba P, Cho RW, Clarke MF. Cancer stem cells: models and concepts. Annu Rev Med 2007; 58: 267-84; PMID:17002552; http://dx.doi.org/10.1146/annurev.med.58.062105.204854
- Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997; 3:730-7; PMID:9212098; http://dx.doi.org/10.1038/nm0797-730
- Nguyen LV, Vanner R, Dirks P, Eaves CJ. Cancer stem cells: an evolving concept. Nat Rev Cancer 2012; 12:133-43; PMID:22237392
- Cozzio A, Passegue E, Ayton PM, Karsunky H, Cleary ML, Weissman IL. Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev 2003; 17:3029-35; PMID:14701873; http://dx.doi.org/10.1101/gad.1143403
- Yoo MH, Hatfield DL. The cancer stem cell theory: is it correct? Mol Cells 2008; 26:514-6; PMID:18711315
- Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nat Med 2009; 15:1010-2; PMID:19734877; http://dx.doi.org/10.1038/nm0909-1010
- Scheel C, Weinberg RA. Cancer stem cells and epithelial-mesenchymal transition: concepts and molecular links. Semin Cancer Biol 2012; 22:396-403; PMID:22554795; http://dx.doi.org/10.1016/j.semcancer.2012.04.001
- Marjanovic ND, Weinberg RA, Chaffer CL. Cell plasticity and heterogeneity in cancer. Clin Chem 2013; 59:168-79; PMID:23220226; http://dx.doi.org/10.1373/clinchem.2012.184655
- Tomasson MH. Cancer stem cells: a guide for skeptics. J Cell Biochem 2009; 106:745-9; PMID:19184979; http://dx.doi.org/10.1002/jcb.22050
- Lee AS, Tang C, Rao MS, Weissman IL, Wu JC. Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nat Med 2013; 19:998-1004; PMID:23921754; http://dx.doi.org/10.1038/nm.3267
- Suva ML, Riggi N, Bernstein BE. Epigenetic reprogramming in cancer. Science 2013; 339:1567-70; PMID:23539597; http://dx.doi.org/10.1126/science.1230184
- Ben-David U, Benvenisty N. The tumorigenicity of human embryonic and induced pluripotent stem cells. Nat Rev Cancer 2011; 11:268-77; PMID:21390058; http://dx.doi.org/10.1038/nrc3034
- Riggs JW, Barrilleaux BL, Varlakhanova N, Bush KM, Chan V, Knoepfler PS. Induced pluripotency and oncogenic transformation are related processes. Stem Cells Dev 2013; 22:37-50; PMID:22998387; http://dx.doi.org/10.1089/scd.2012.0375
- Menendez JA, Joven J, Cufí S, Corominas-Faja B, Oliveras-Ferraros C, Cuyàs E, Martin-Castillo B, López-Bonet E, Alarcón T, Vazquez-Martin A. The Warburg effect version 2.0: metabolic reprogramming of cancer stem cells. Cell Cycle 2013; 12:1166-79; PMID:23549172; http://dx.doi.org/10.4161/cc.24479
- Corominas-Faja B, Cufí S, Oliveras-Ferraros C, Cuyàs E, López-Bonet E, Lupu R, Alarcón T, Vellon L, Iglesias JM, Leis O, et al. Nuclear reprogramming of luminal-like breast cancer cells generates Sox2-overexpressing cancer stem-like cellular states harboring transcriptional activation of the mTOR pathway. Cell Cycle 2013; 12:3109-24; PMID:23974095; http://dx.doi.org/10.4161/cc.26173
- Vazquez-Martin A, Cufí S, López-Bonet E, Corominas-Faja B, Cuyàs E, Vellon L, Iglesias JM, Leis O, Martín AG, Menendez JA. Reprogramming of non-genomic estrogen signaling by the stemness factor SOX2 enhances the tumor-initiating capacity of breast cancer cells. Cell Cycle 2013; 12:3471-7; PMID:24107627; http://dx.doi.org/10.4161/cc.26692
- Menendez JA, Alarcón T, Corominas-Faja B, Cuyàs E, López-Bonet E, Martin AG, Vellon L. Xenopatients 2.0: reprogramming the epigenetic landscapes of patient-derived cancer genomes. Cell Cycle 2014; 13:358-70; PMID:24406535; http://dx.doi.org/10.4161/cc.27770
- Friedmann-Morvinski D, Verma IM. Dedifferentiation and reprogramming: origins of cancer stem cells. EMBO Rep 2014; 15:244-53; PMID:24531722; http://dx.doi.org/10.1002/embr.201338254
- Goding CR, Pei D, Lu X. Cancer: pathological nuclear reprogramming? Nat Rev Cancer 2014; 14:568-73; PMID:25030952; http://dx.doi.org/10.1038/nrc3781
- Ohnishi K, Semi K, Yamamoto T, Shimizu M, Tanaka A, Mitsunaga K, Okita K, Osafune K, Arioka Y, Maeda T, et al. Premature termination of reprogramming in vivo leads to cancer development through altered epigenetic regulation. Cell 2014; 156:663-77; PMID:24529372; http://dx.doi.org/10.1016/j.cell.2014.01.005
- Knoepfler PS. Deconstructing stem cell tumorigenicity: a roadmap to safe regenerative medicine. Stem Cells 2009; 27:1050-1056; PMID:19415771; http://dx.doi.org/10.1002/stem.37
- Tung PY, Knoepfler PS. Epigenetic mechanisms of tumorigenicity manifesting in stem cells. Oncogene 2014; PMID:AMBIGUOUS
- Prigione A, Adjaye J. Modulation of mitochondrial biogenesis and bioenergetic metabolism upon in vitro and in vivo differentiation of human ES and iPS cells. Int J Dev Biol 2010; 54:1729-41; PMID:21305470; http://dx.doi.org/10.1387/ijdb.103198ap
- Menendez JA, Vellon L, Oliveras-Ferraros C, Cufí S, Vazquez-Martin A. mTOR-regulated senescence and autophagy during reprogramming of somatic cells to pluripotency: a roadmap from energy metabolism to stem cell renewal and aging. Cell Cycle 2011; 10:3658-77; PMID:22052357; http://dx.doi.org/10.4161/cc.10.21.18128
- Chen CT, Hsu SH, Wei YH. Mitochondrial bioenergetic function and metabolic plasticity in stem cell differentiation and cellular reprogramming. Biochim Biophys Acta 2012; 1820:571-6; PMID:21983491; http://dx.doi.org/10.1016/j.bbagen.2011.09.013
- Folmes CD, Nelson TJ, Terzic A. Energy metabolism in nuclear reprogramming. Biomark Med 2011; 5:715-29; PMID:22103608; http://dx.doi.org/10.2217/bmm.11.87
- Zhang J, Nuebel E, Daley GQ, Koehler CM, Teitell MA. Metabolic regulation in pluripotent stem cells during reprogramming and self-renewal. Cell Stem Cell 2012; 11:589-95; PMID:23122286; http://dx.doi.org/10.1016/j.stem.2012.10.005
- Xu X, Duan S, Yi F, Ocampo A, Liu GH, Izpisua Belmonte JC. Mitochondrial regulation in pluripotent stem cells. Cell Metab 2013; 18:325-32; PMID:23850316; http://dx.doi.org/10.1016/j.cmet.2013.06.005
- Shyh-Chang N, Daley GQ, Cantley LC. Stem cell metabolism in tissue development and aging. Development 2013; 140:2535-47; PMID:23715547; http://dx.doi.org/10.1242/dev.091777
- Ito K, Suda T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat Rev Mol Cell Biol 2014; 15:243-56; PMID:24651542; http://dx.doi.org/10.1038/nrm3772
- Prigione A, Fauler B, Lurz R, Lehrach H, Adjaye J. The senescence-related mitochondrial/oxidative stress pathway is repressed in human induced pluripotent stem cells. Stem Cells 2010; 28:721-33; PMID:20201066; http://dx.doi.org/10.1002/stem.404
- Prigione A, Adjaye J. Modulation of mitochondrial biogenesis and bioenergetic metabolism upon in vitro and in vivo differentiation of human ES and iPS cells. Int J Dev Biol 2010; 54:1729-41; PMID:21305470; http://dx.doi.org/10.1387/ijdb.103198ap
- Prigione A, Lichtner B, Kuhl H, Struys EA, Wamelink M, Lehrach H, Ralser M, Timmermann B, Adjaye J. Human induced pluripotent stem cells harbor homoplasmic and heteroplasmic mitochondrial DNA mutations while maintaining human embryonic stem cell-like metabolic reprogramming. Stem Cells 2011; 29:1338-48; PMID:21732474
- Folmes CD, Nelson TJ, Martinez-Fernandez A, Arrell DK, Lindor JZ, Dzeja PP, Ikeda Y, Perez-Terzic C, Terzic A. Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab 2011; 14:264-71; PMID:21803296; http://dx.doi.org/10.1016/j.cmet.2011.06.011
- Varum S, Rodrigues AS, Moura MB, Momcilovic O, Easley CA 4th, Ramalho-Santos J, Van Houten B, Schatten G. Energy metabolism in human pluripotent stem cells and their differentiated counterparts. PLoS One 2011; 6:e20914; PMID:21698063; http://dx.doi.org/10.1371/journal.pone.0020914
- Zhang J, Khvorostov I, Hong JS, Oktay Y, Vergnes L, Nuebel E, Wahjudi PN, Setoguchi K, Wang G, Do A, et al. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J 2011; 30:4860-73; PMID:22085932; http://dx.doi.org/10.1038/emboj.2011.401
- Shyh-Chang N, Zheng Y, Locasale JW, Cantley LC. Human pluripotent stem cells decouple respiration from energy production. EMBO J 2011; 30:4851-2; PMID:22166995; http://dx.doi.org/10.1038/emboj.2011.436
- Folmes CD, Nelson TJ, Dzeja PP, Terzic A. Energy metabolism plasticity enables stemness programs. Ann N Y Acad Sci 2012; 1254:82-9; PMID:22548573; http://dx.doi.org/10.1111/j.1749-6632.2012.06487.x
- Folmes CD, Dzeja PP, Nelson TJ, Terzic A. Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell 2012; 11:596-606; PMID:23122287; http://dx.doi.org/10.1016/j.stem.2012.10.002
- Panopoulos AD, Yanes O, Ruiz S, Kida YS, Diep D, Tautenhahn R, Herrerías A, Batchelder EM, Plongthongkum N, Lutz M, et al. The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell Res 2012; 22:168-77; PMID:22064701; http://dx.doi.org/10.1038/cr.2011.177
- Folmes CD, Arrell DK, Zlatkovic-Lindor J, Martinez-Fernandez A, Perez-Terzic C, Nelson TJ, Terzic A. Metabolome and metaboproteome remodeling in nuclear reprogramming. Cell Cycle 2013; 12:2355-65; PMID:23839047; http://dx.doi.org/10.4161/cc.25509
- Son MJ, Jeong BR, Kwon Y, Cho YS. Interference with the mitochondrial bioenergetics fuels reprogramming to pluripotency via facilitation of the glycolytic transition. Int J Biochem Cell Biol 2013; 45:2512-18; PMID:23939289; http://dx.doi.org/10.1016/j.biocel.2013.07.023
- Liu W, Long Q, Chen K, Li S, Xiang G, Chen S, Liu X, Li Y, Yang L, Dong D, et al. Mitochondrial metabolism transition cooperates with nuclear reprogramming during induced pluripotent stem cell generation. Biochem Biophys Res Commun 2013; 431:767-71; PMID:23333381; http://dx.doi.org/10.1016/j.bbrc.2012.12.148
- Prigione A, Rohwer N, Hoffmann S, Mlody B, Drews K, Bukowiecki R, Blümlein K, Wanker EE, Ralser M, Cramer T, et al. HIF1α modulates cell fate reprogramming through early glycolytic shift and upregulation of PDK1-3 and PKM2. Stem Cells 2014; 32:364-76; PMID:24123565; http://dx.doi.org/10.1002/stem.1552
- Bukowiecki R, Adjaye J, Prigione A. Mitochondrial function in pluripotent stem cells and cellular reprogramming. Gerontology 2014; 60:174-82; PMID:24281332; http://dx.doi.org/10.1159/000355050
- Zhu S, Li W, Zhou H, Wei W, Ambasudhan R, Lin T, Kim J, Zhang K, Ding S. Reprogramming of human primary somatic cells by OCT4 and chemical compounds. Cell Stem Cell 2010; 7:651-5; PMID:21112560; http://dx.doi.org/10.1016/j.stem.2010.11.015
- Zhu H, Shyh-Chang N, Segrè AV, Shinoda G, Shah SP, Einhorn WS, Takeuchi A, Engreitz JM, Hagan JP, Kharas MG, et al. The Lin28/let-7 axis regulates glucose metabolism. Cell 2011; 147:81-94; PMID:21962509; http://dx.doi.org/10.1016/j.cell.2011.08.033
- Folmes CD, Martinez-Fernandez A, Faustino RS, Yamada S, Perez-Terzic C, Nelson TJ, Terzic A. Nuclear reprogramming with c-Myc potentiates glycolytic capacity of derived induced pluripotent stem cells. J Cardiovasc Transl Res 2013; 6:10-21; PMID:23247633; http://dx.doi.org/10.1007/s12265-012-9431-2
- Vazquez-Martin A, Corominas-Faja B, Cufi S, Vellon L, Oliveras-Ferraros C, Menendez OJ, Joven J, Lupu R, Menendez JA. The mitochondrial H(+)-ATP synthase and the lipogenic switch: new core components of metabolic reprogramming in induced pluripotent stem (iPS) cells. Cell Cycle 2013; 12:207-18; PMID:23287468; http://dx.doi.org/10.4161/cc.23352
- Sánchez-Aragó M, Chamorro M, Cuezva JM. Selection of cancer cells with repressed mitochondria triggers colon cancer progression. Carcinogenesis 2010; 31:567-76; PMID:20080835; http://dx.doi.org/10.1093/carcin/bgq012
- Sánchez-Cenizo L, Formentini L, Aldea M, Ortega AD, García-Huerta P, Sánchez-Aragó M, Cuezva JM. Up-regulation of the ATPase inhibitory factor 1 (IF1) of the mitochondrial H+-ATP synthase in human tumors mediates the metabolic shift of cancer cells to a Warburg phenotype. J Biol Chem 2010; 285:25308-13; PMID:20538613; http://dx.doi.org/10.1074/jbc.M110.146480
- Willers IM, Cuezva JM. Post-transcriptional regulation of the mitochondrial H(+)-ATP synthase: a key regulator of the metabolic phenotype in cancer. Biochim Biophys Acta 2011; 1807:543-51; PMID:21035425; http://dx.doi.org/10.1016/j.bbabio.2010.10.016
- Aldea M, Clofent J, Núñez de Arenas C, Chamorro M, Velasco M, Berrendero JR, Navarro C, Cuezva JM. Reverse phase protein microarrays quantify and validate the bioenergetic signature as biomarker in colorectal cancer. Cancer Lett 2011; 311:210-8; PMID:21880415; http://dx.doi.org/10.1016/j.canlet.2011.07.022
- Formentini L, Sánchez-Aragó M, Sánchez-Cenizo L, Cuezva JM. The mitochondrial ATPase inhibitory factor 1 triggers a ROS-mediated retrograde prosurvival and proliferative response. Mol Cell 2012; 45:731-42; PMID:22342343; http://dx.doi.org/10.1016/j.molcel.2012.01.008
- Sánchez-Aragó M, Formentini L, García-Bermúdez J, Cuezva JM. IF1 reprograms energy metabolism and signals the oncogenic phenotype in cancer. Cell Cycle 2012; 11:2963-4; PMID:22871729; http://dx.doi.org/10.4161/cc.21387
- Sánchez-Aragó M, Formentini L, Cuezva JM. Mitochondria-mediated energy adaption in cancer: the H(+)-ATP synthase-geared switch of metabolism in human tumors. Antioxid Redox Signal 2013; 19:285-98; PMID:22901241; http://dx.doi.org/10.1089/ars.2012.4883
- Sánchez-Aragó M, Formentini L, Martínez-Reyes I, García-Bermudez J, Santacatterina F, Sánchez-Cenizo L, Willers IM, Aldea M, Nájera L, Juarránz A, et al. Expression, regulation and clinical relevance of the ATPase inhibitory factor 1 in human cancers. Oncogenesis 2013; 2:e46; PMID:23608753; http://dx.doi.org/10.1038/oncsis.2013.9
- Sánchez-Aragó M, García-Bermúdez J, Martínez-Reyes I, Santacatterina F, Cuezva JM. Degradation of IF1 controls energy metabolism during osteogenic differentiation of stem cells. EMBO Rep 2013; 14:638-44; PMID:23722655; http://dx.doi.org/10.1038/embor.2013.72
- Dong C, Yuan T, Wu Y, Wang Y, Fan TW, Miriyala S, Lin Y, Yao J, Shi J, Kang T, et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 2013: 23:316-31; PMID:23453623; http://dx.doi.org/10.1016/j.ccr.2013.01.022
- Schieber MS, Chandel NS. ROS links glucose metabolism to breast cancer stem cell and EMT phenotype. Cancer Cell 2013; 23:265-7; PMID:23518342; http://dx.doi.org/10.1016/j.ccr.2013.02.021
- Liu PP, Liao J, Tang ZJ, Wu WJ, Yang J, Zeng ZL, Hu Y, Wang P, Ju HQ, Xu RH, Huang P. Metabolic regulation of cancer cell side population by glucose through activation of the Akt pathway. Cell Death Differ 2014; 21:124-135; PMID:24096870; http://dx.doi.org/10.1038/cdd.2013.131
- Ciavardelli D, Rossi C, Barcaroli D, Volpe S, Consalvo A, Zucchelli M, De Cola A, Scavo E, Carollo R, D'Agostino D, et al. Breast cancer stem cells rely on fermentative glycolysis and are sensitive to 2-deoxyglucose treatment. Cell Death Dis 2014; 5:e1336; PMID:25032859; http://dx.doi.org/10.1038/cddis.2014.285
- Perales-Clemente E, Folmes CD, Terzic A. Metabolic regulation of redox status in stem cells. Antioxid Redox Signal 2014; 21(11):1648-59; PMID:24949895; http://dx.doi.org/10.1089/ars.2014.6000
- Saretzki G, Walter T, Atkinson S, Passos JF, Bareth B, Keith WN, Stewart R, Hoare S, Stojkovic M, Armstrong L, et al. Downregulation of multiple stress defense mechanisms during differentiation of human embryonic stem cells. Stem Cells 2008; 26:455-64; PMID:18055443; http://dx.doi.org/10.1634/stemcells.2007-0628
- Armstrong L, Tilgner K, Saretzki G, Atkinson SP, Stojkovic M, Moreno R, Przyborski S, Lako M. Human induced pluripotent stem cell lines show stress defense mechanisms and mitochondrial regulation similar to those of human embryonic stem cells. Stem Cells 2010; 28:661-73; PMID:20073085; http://dx.doi.org/10.1002/stem.307
- Yanes O, Clark J, Wong DM, Patti GJ, Sánchez-Ruiz A, Benton HP, Trauger SA, Desponts C, Ding S, Siuzdak G. Metabolic oxidation regulates embryonic stem cell differentiation. Nat Chem Biol 2010; 6:411-7; PMID:20436487; http://dx.doi.org/10.1038/nchembio.364
- Wang J, Alexander P, Wu L, Hammer R, Cleaver O, McKnight SL. Dependence of mouse embryonic stem cells on threonine catabolism. Science 2009; 325:435-9; PMID:19589965; http://dx.doi.org/10.1126/science.1173288
- Locasale JW, Cantley LC. Metabolic flux and the regulation of mammalian cell growth. Cell Metab 2011; 14:443-51; PMID:21982705; http://dx.doi.org/10.1016/j.cmet.2011.07.014
- Son MJ, Son MY, Seol B, Kim MJ, Yoo CH, Han MK, Cho YS. Nicotinamide overcomes pluripotency deficits and reprogramming barriers. Stem Cells 2013; 31:1121-35; PMID:23526681; http://dx.doi.org/10.1002/stem.1368
- Kobayashi CI, Suda T. Regulation of reactive oxygen species in stem cells and cancer stem cells. J Cell Physiol 2012; 227:421-30; PMID:21448925; http://dx.doi.org/10.1002/jcp.22764
- Shi X, Zhang Y, Zheng J, Pan J. Reactive oxygen species in cancer stem cells. Antioxid Redox Signal 2012; 16:1215-28; PMID:22316005; http://dx.doi.org/10.1089/ars.2012.4529
- Nagano O, Okazaki S, Saya H. Redox regulation in stem-like cancer cells by CD44 variant isoforms. Oncogene 2013; 32:5191-8; PMID:23334333; http://dx.doi.org/10.1038/onc.2012.638
- Watson J. Oxidants, antioxidants and the current incurability of metastatic cancers. Open Biol 2013; 3:120144; PMID:23303309; http://dx.doi.org/10.1098/rsob.120144
- Tamada M, Nagano O, Tateyama S, Ohmura M, Yae T, Ishimoto T, Sugihara E, Onishi N, Yamamoto T, Yanagawa H, et al. Modulation of glucose metabolism by CD44 contributes to antioxidant status and drug resistance in cancer cells. Cancer Res 2012; 72:1438-48; PMID:22293754; http://dx.doi.org/10.1158/0008-5472.CAN-11-3024
- Croker AK, Allan AL. Inhibition of aldehyde dehydrogenase (ALDH) activity reduces chemotherapy and radiation resistance of stem-like ALDHhiCD44⁺ human breast cancer cells. Breast Cancer Res Treat 2012; 133:75-87; PMID:21818590; http://dx.doi.org/10.1007/s10549-011-1692-y
- Pani G, Galeotti T, Chiarugi P. Metastasis: cancer cell's escape from oxidative stress. Cancer Metastasis Rev 2010; 29:351-78; PMID:20386957; http://dx.doi.org/10.1007/s10555-010-9225-4
- Boivin A, Hanot M, Malesys C, Maalouf M, Rousson R, Rodriguez-Lafrasse C, Ardail D. Transient alteration of cellular redox buffering before irradiation triggers apoptosis in head and neck carcinoma stem and non-stem cells. PLoS One 2011; 6:e14558; PMID:21283807; http://dx.doi.org/10.1371/journal.pone.0014558
- Pei S, Minhajuddin M, Callahan KP, Balys M, Ashton JM, Neering SJ, Lagadinou ED, Corbett C, Ye H, Liesveld JL, et al. Targeting aberrant glutathione metabolism to eradicate human acute myelogenous leukemia cells. J Biol Chem 2013; 288:33542-58; PMID:24089526; http://dx.doi.org/10.1074/jbc.M113.511170
- Kawamura T, Kondoh Y, Muroi M, Kawatani M, Osada H. A small molecule that induces reactive oxygen species via cellular glutathione depletion. Biochem J 2014; 463:53-63; PMID:25011393; http://dx.doi.org/10.1042/BJ20140669
- DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A 2007; 104:19345-50; PMID:18032601; http://dx.doi.org/10.1073/pnas.0709747104
- Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer 2007; 7:763-77; PMID:17882277; http://dx.doi.org/10.1038/nrc2222
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009; 32:1029-33; PMID:19460998; http://dx.doi.org/10.1126/science.1160809
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 2008; 7:11-20; PMID:18177721; http://dx.doi.org/10.1016/j.cmet.2007.10.002
- Shiraki N, Shiraki Y, Tsuyama T, Obata F, Miura M, Nagae G, Aburatani H, Kume K, Endo F, Kume S. Methionine metabolism regulates maintenance and differentiation of human pluripotent stem cells. Cell Metab 2014; 19:780-94; PMID:24746804; http://dx.doi.org/10.1016/j.cmet.2014.03.017
- Shyh-Chang N, Locasale JW, Lyssiotis CA, Zheng Y, Teo RY, Ratanasirintrawoot S, Zhang J, Onder T, Unternaehrer JJ, Zhu H, et al. Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science 2013; 339:222-6; PMID:23118012; http://dx.doi.org/10.1126/science.1226603
- Lu C, Thompson CB. Metabolic regulation of epigenetics. Cell Metab 2012; 16:9-17; PMID:22768835; http://dx.doi.org/10.1016/j.cmet.2012.06.001
- Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer 2013; 13:572-83; PMID:23822983; http://dx.doi.org/10.1038/nrc3557
- Johnson C, Warmoes MO, Shen X, Locasale JW. Epigenetics and cancer metabolism. Cancer Lett 2015; 356:309-14; PMID:24125862; http://dx.doi.org/10.1016/j.canlet.2013.09.043
- Li G, Zhao F, Cui Y. Proteomics using mammospheres as a model system to identify proteins deregulated in breast cancer stem cells. Curr Mol Med 2013; 13:459-463; PMID:23331018
- Pandey PR, Xing F, Sharma S, Watabe M, Pai SK, Iiizumi-Gairani M, Fukuda K, Hirota S, Mo YY, Watabe K. Elevated lipogenesis in epithelial stem-like cell confers survival advantage in ductal carcinoma in situ of breast cancer. Oncogene 2013; 32:5111-22; PMID:23208501; http://dx.doi.org/10.1038/onc.2012.519
- Pandey PR, Okuda H, Watabe M, Pai SK, Liu W, Kobayashi A, Xing F, Fukuda K, Hirota S, Sugai T, et al. Resveratrol suppresses growth of cancer stem-like cells by inhibiting fatty acid synthase. Breast Cancer Res Treat 2011; 130:387-98; PMID:21188630; http://dx.doi.org/10.1007/s10549-010-1300-6
- Corominas-Faja B, Cuyàs E, Gumuzio J, Bosch-Barrera J, Leis O, Martin ÁG, Menendez JA. Chemical inhibition of acetyl-CoA carboxylase suppresses self-renewal growth of cancer stem cells. Oncotarget 2014; 5:8306-16; PMID:25246709
- Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW, Cravatt BF. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 2010; 140:49-61; PMID:20079333; http://dx.doi.org/10.1016/j.cell.2009.11.027
- Louie SM, Roberts LS, Mulvihill MM, Luo K, Nomura DK. Cancer cells incorporate and remodel exogenous palmitate into structural and oncogenic signaling lipids. Biochim Biophys Acta 2013; 1831:1566-72; PMID:23872477; http://dx.doi.org/10.1016/j.bbalip.2013.07.008
- Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 2005; 8:299-309; PMID:16226705; http://dx.doi.org/10.1016/j.ccr.2005.09.005
- Shen R, Ye Y, Chen L, Yan Q, Barsky SH, Gao JX. Precancerous stem cells can serve as tumor vasculogenic progenitors. PLoS One 2008; 3:e1652; PMID:18286204; http://dx.doi.org/10.1371/journal.pone.0001652
- Cabarcas SM, Mathews LA, Farrar WL. The cancer stem cell niche–there goes the neighborhood? Int J Cancer 2011; 129:2315-27; PMID:21792897; http://dx.doi.org/10.1002/ijc.26312
- Cascone T, Herynk MH, Xu L, Du Z, Kadara H, Nilsson MB, Oborn CJ, Park YY, Erez B, Jacoby JJ, et al. Upregulated stromal EGFR and vascular remodeling in mouse xenograft models of angiogenesis inhibitor-resistant human lung adenocarcinoma. J Clin Invest 2011; 121:1313-28; PMID:21436589; http://dx.doi.org/10.1172/JCI42405
- Osawa T, Shibuya M. Targeting cancer cells resistant to hypoxia and nutrient starvation to improve anti-angiogeneic therapy. Cell Cycle 2013; 12:2519-20; PMID:23907114; http://dx.doi.org/10.4161/cc.25729
- Philip B, Ito K, Moreno-Sánchez R, Ralph SJ. HIF expression and the role of hypoxic microenvironments within primary tumours as protective sites driving cancer stem cell renewal and metastatic progression. Carcinogenesis 2013; 34:1699-707; PMID:23740838; http://dx.doi.org/10.1093/carcin/bgt209
- Mathonnet M, Perraud A, Christou N, Akil H, Melin C, Battu S, Jauberteau MO, Denizot Y. Hallmarks in colorectal cancer: Angiogenesis and cancer stem-like cells. World J Gastroenterol 2014; 20:4189-96; PMID:24764657; http://dx.doi.org/10.3748/wjg.v20.i15.4189
- Sounni NE, Cimino J, Blacher S, Primac I, Truong A, Mazzucchelli G, Paye A, Calligaris D, Debois D, De Tullio P, et al. Blocking lipid synthesis overcomes tumor regrowth and metastasis after antiangiogenic therapy withdrawal. Cell Metab 2014; 20:280-94; PMID:25017943; http://dx.doi.org/10.1016/j.cmet.2014.05.022
- Menendez JA, Joven J. Energy metabolism and metabolic sensors in stem cells: the metabostem crossroads of aging and cancer. Adv Exp Med Biol 2014; 824:117-40; PMID:25038997; http://dx.doi.org/10.1007/978-3-319-07320-0_10
- Chen T, Shen L, Yu J, Wan H, Guo A, Chen J, Long Y, Zhao J, Pei G. Rapamycin and other longevity-promoting compounds enhance the generation of mouse induced pluripotent stem cells. Aging Cell 10:908-11; PMID:21615676; http://dx.doi.org/10.1111/j.1474-9726.2011.00722.x
- Easley CA 4th, Ben-Yehudah A, Redinger CJ, Oliver SL, Varum ST, Eisinger VM, Carlisle DL, Donovan PJ, Schatten GP. mTOR-mediated activation of p70 S6K induces differentiation of pluripotent human embryonic stem cells. Cell Reprogram 2010; 12:263-73; PMID:20698768; http://dx.doi.org/10.1089/cell.2010.0011
- He J, Kang L, Wu T, Zhang J, Wang H, Gao H, Zhang Y, Huang B, Liu W, Kou Z, et al. An elaborate regulation of Mammalian target of rapamycin activity is required for somatic cell reprogramming induced by defined transcription factors. Stem Cells Dev 21:2630-41; PMID:22471963; http://dx.doi.org/10.1089/scd.2012.0015
- Wang S, Xia P, Ye B, Huang G, Liu J, Fan Z. Transient activation of autophagy via Sox2-mediated suppression of mTOR is an important early step in reprogramming to pluripotency. Cell Stem Cell 2013; 13:617-25; PMID:24209762; http://dx.doi.org/10.1016/j.stem.2013.10.005
- Vazquez-Martin A, Vellon L, Quirós PM, Cufí S, Ruiz de Galarreta E, Oliveras-Ferraros C, Martin AG, Martin-Castillo B, López-Otín C, et al. Activation of AMP-activated protein kinase (AMPK) provides a metabolic barrier to reprogramming somatic cells into stem cells. Cell Cycle 2012; 11:974-89; PMID:22333578; http://dx.doi.org/10.4161/cc.11.5.19450
- Vazquez-Martin A, Cufi S, Lopez-Bonet E, Corominas-Faja B, Oliveras-Ferraros C, Martin-Castillo B, Menendez JA. Metformin limits the tumourigenicity of iPS cells without affecting their pluripotency. Sci Rep 2012; 2:964; PMID:23236586; http://dx.doi.org/10.1038/srep00964
- Jung JW, Park SB, Lee SJ, Seo MS, Trosko JE, Kang KS. Metformin represses self-renewal of the human breast carcinoma stem cells via inhibition of estrogen receptor-mediated OCT4 expression. PLoS One 2011; 6:e28068; PMID:22132214; http://dx.doi.org/10.1371/journal.pone.0028068
- Pollak M. Potential applications for biguanides in oncology. J Clin Invest 2013; 123: 3693-700; PMID:23999444; http://dx.doi.org/10.1172/JCI67232
- Iliopoulos D, Hirsch HA, Wang G, Struhl K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci U S A 2011; 108:1397-402; PMID:21220315; http://dx.doi.org/10.1073/pnas.1018898108
- Hirsch HA, Iliopoulos D, Struhl K. Metformin inhibits the inflammatory response associated with cellular transformation and cancer stem cell growth. Proc Natl Acad Sci U S A 2013; 110:972-7; PMID:23277563; http://dx.doi.org/10.1073/pnas.1221055110
- Janzer A, German NJ, Gonzalez-Herrera KN, Asara JM, Haigis MC, Struhl K. Metformin and phenformin deplete tricarboxylic acid cycle and glycolytic intermediates during cell transformation and NTPs in cancer stem cells. Proc Natl Acad Sci U S A 2014; 111:10574-9; PMID:25002509; http://dx.doi.org/10.1073/pnas.1409844111
- Del Barco S, Vazquez-Martin A, Cufí S, Oliveras-Ferraros C, Bosch-Barrera J, Joven J, Martin-Castillo B, Menendez JA. Metformin: multi-faceted protection against cancer. Oncotarget 2011; 2:896-917; PMID:22203527
- Waddington CH. The strategy of the genes. London, UK: Allen and Unwin; 1957.
- Huang S. Reprogramming cell fates: reconciling rarity with robustness. Bioessays 2009; 31:546-60; PMID:19319911; http://dx.doi.org/10.1002/bies.200800189
- Huang S. Systems biology of stem cells: three useful perspectives to help overcome the paradigm of linear pathways. Philos Trans R Soc Lond B Biol Sci 2011; 366:2247-59; PMID:21727130; http://dx.doi.org/10.1098/rstb.2011.0008
- Hou P, Li Y, Zhang X, Liu C, Guan J, Li H, Zhao T, Ye J, Yang W, Liu K, et al. Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science 2013; 41:651-4; PMID:23868920; http://dx.doi.org/10.1126/science.1239278
- Menendez JA, Alarcón T. Metabostemness: A new cancer hallmark. Front Oncol 2014; 4:262; PMID:25325014; http://dx.doi.org/10.3389/fonc.2014.00262
- Huang S, Ernberg I, Kauffman S. Cancer attractors: a systems view of tumors from a gene network dynamics and developmental perspective. Semin Cell Dev Biol 2009; 20:869-876; PMID:19595782; http://dx.doi.org/10.1016/j.semcdb.2009.07.003
- Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010; 17:225-34; PMID:20171147; http://dx.doi.org/10.1016/j.ccr.2010.01.020
- Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of a-ketoglutarate-dependent dioxygenases. Cancer Cell 2011; 19:17-30; PMID:21251613; http://dx.doi.org/10.1016/j.ccr.2010.12.014
- Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, Leung IK, Li XS, Woon EC, Yang M, et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep 2011; 12:463-9; PMID:21460794; http://dx.doi.org/10.1038/embor.2011.43
- Yang M, Soga T, Pollard PJ. Oncometabolites: linking altered metabolism with cancer. J Clin Invest 2013; 123:3652-8; PMID:23999438; http://dx.doi.org/10.1172/JCI67228
- Cairns RA, Mak TW. Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer Discov 2013; 3:730-41; PMID:23796461; http://dx.doi.org/10.1158/2159-8290.CD-13-0083
- Krell D, Mulholland P, Frampton AE, Krell J, Stebbing J, Bardella C. IDH mutations in tumorigenesis and their potential role as novel therapeutic targets. Future Oncol 2013; 9:1923-35; PMID:24295421; http://dx.doi.org/10.2217/fon.13.143
- Menendez JA, Alarcón T, Joven J. Gerometabolites: the pseudohypoxic aging side of cancer oncometabolites. Cell Cycle 2014; 13:699-709; PMID:24526120; http://dx.doi.org/10.4161/cc.28079
- Terunuma A, Putluri N, Mishra P, Mathé EA, Dorsey TH, Yi M, Wallace TA, Issaq HJ, Zhou M, Killian JK, et al. MYC-driven accumulation of 2-hydroxyglutarate is associated with breast cancer prognosis. J Clin Invest 2014; 124: 398-412; PMID:24316975; http://dx.doi.org/10.1172/JCI71180
- Saha SK, Parachoniak CA, Ghanta KS, Fitamant J, Ross KN, Najem MS, Gurumurthy S, Akbay EA, Sia D, et al. Mutant IDH inhibits HNF-4a to block hepatocyte differentiation and promote biliary cancer. Nature 2014; 513:110-4; PMID:25043045; http://dx.doi.org/10.1038/nature13441
- Hanna JH, Saha K, Jaenisch R. Pluripotency and cellular reprogramming: facts, hypotheses, unresolved issues. Cell 2010; 143:508-25; PMID:21074044; http://dx.doi.org/10.1016/j.cell.2010.10.008
- Huang S. The molecular and mathematical basis of Waddington's epigenetic landscape: a framework for post-Darwinian biology? Bioessays 2012; 34:149-57; PMID:22102361; http://dx.doi.org/10.1002/bies.201100031
- Menendez JA, Corominas-Faja B, Cuyàs E, Alarcón T. Metabostemness: Metaboloepigenetic reprogramming of cancer stem-cell functions. 2015; 1:803-6. PMID: 25621295
- Cufí S, Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Vellon L, Menendez JA. Autophagy positively regulates the CD44(+) CD24(-/low) breast cancer stem-like phenotype. Cell Cycle 2011; 10:3871-85; PMID:22127234; http://dx.doi.org/10.4161/cc.10.22.17976
- Guan JL, Simon AK, Prescott M, Menendez JA, Liu F, Wang F, Wang C, Wolvetang E, Vazquez-Martin A, Zhang J. Autophagy in stem cells. Autophagy 2013;9:830-49; PMID:23486312; http://dx.doi.org/10.4161/auto.24132
- Stem Cell Energetics. 2014; 15:679-82; http://dx.doi.org/10.1016/j.stem.2014.11.013
- Cuyàs E, Corominas-Faja B, Menendez JA. The nutritional phenome of EMT-induced cancer stem-like cells. Oncotarget 2014; 5:3970-82; PMID:24994116
- Prigione A, Ruiz-Pérez MV, Bukowiecki R, Adjaye J. Metabolic restructuring and cell fate conversion. Cell Mol Life Sci 2015 Jan 14. [Epub ahead of print]; PMID:25586562
- Teslaa T, Teitell MA. Pluripotent stem cell energy metabolism: an update. EMBO J 2015; 34:138-153; PMID:25476451; http://dx.doi.org/10.15252/embj.201490446
- Apostolou E, Hochedlinger K. Chromatin dynamics during cellular reprogramming. Nature 2013; 502:462-71; PMID:24153299; http://dx.doi.org/10.1038/nature12749
- Zviran A, Hanna JH. Lucky iPSCs. Genome Biol 2014; 15:109; PMID:25002192; http://dx.doi.org/10.1186/gb4167
- Teicher BA, Linehan WM, Helman LJ. Targeting cancer metabolism. Clin Cancer Res. 2012; 18:5537-45; PMID:23071355; http://dx.doi.org/10.1158/1078-0432.CCR-12-2587
- Galluzzi L, Kepp O, Vander Heiden MG, Kroemer G. Metabolic targets for cancer therapy. Nat Rev Drug Discov 2013; 12:829-46; PMID:24113830; http://dx.doi.org/10.1038/nrd4145
- DeLaBarre B, Hurov J, Cianchetta G, Murray S, Dang L. Action at a distance: allostery and the development of drugs to target cancer cell metabolism. Chem Biol 2014; 21:1143-61; PMID:25237859; http://dx.doi.org/10.1016/j.chembiol.2014.08.007